
Hypoxia Inducible Factor Stabilization As a Novel Strategy to Treat Anemia
Send Orders of Reprints at [email protected] Current Medicinal Chemistry, 2013, 20, ????-???? 1 Hypoxia Inducible Factor Stabilization As a Novel Strategy to Treat Anemia S. Zhaoa and J. Wu*,a,b a b State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing, Jiangsu 210093, P.R. China; Center for Novel Drug Development of Taizhou, Taizhou, Jiangsu 225300, P.R. China Abstract: Anemia, one of the most common blood disorders, globally affecting ~1.62 billion people, occurs when the level of healthy red blood cells (RBCs) or/and hemoglobin in the body becomes too low. It can cause a variety of complications to human body, some of which are potentially very serious and carry significant risk factors, thus representing a big burden for social and economic development. Current therapeutics methods are efficient in controlling this disease but associated with many problematic issues. One way to circumvent these issues is by targeting HIF-PH (Hypoxia inducible factor prolyl hydroxylases) pathway. HIF is an oxygen-sensitive transcription factor that enables aerobic organisms to adapt to hypoxia through the transcriptional activation of up to 200 genes, many of which are critical to cell survival. Experimental and clinical studies have demonstrated that stabilization of HIF can up-regulate erythropoietin (EPO) expression and in turn increase count of RBCs potentially without causing drug resistance and cardiovascular diseases commonly seen with other therapies, rendering HIF stabilization a promising way to treat anemia. In this review, we highlight the biology of HIF-PH pathway, as well as the recent advances of HIF stabilizers of a natural or synthetic origin and concerns regarding drug development in this field. Keywords: Anemia, FIH inhibitors, HIF-PH pathway, iron chelators, natural products, PHD2 inhibitors, von HippelLindau protein. 1. INTRODUCTION 2. ANEMIA AND CURRENT THERAPIES HIF is a transcriptional activator critical in local and systemic responses to hypoxia through mediation of the transcription of over 200 target genes, including genes of erythropoietin (EPO) and vascular endothelial growth factor (VEGF), etc [1, 2]. The role of HIF in activation of EPO genes under hypoxia was believed to provide an alternative way for the treatment of anemia and other ischemia related diseases. Since currently available therapies for anemia give rise to many problematic issues, novel therapeutic strategies are required to replace these therapies and probably the modulation of HIF pathway will hold that promise [3]. Encouraging results by targeting this pathway have already returned from clinical trials supporting the possibility to pursue an orally bioavailable therapy, in principle significantly less expensive than current treatments and thus generally more accessible for certain types of anemia [3-8]. This review will summarize current knowledge or recent advances in this field with regard to the biology of hypoxia inducible factor pathway, the development of HIF stabilizer as antianemic agents and further concerns about the utilization of this pathway. Specifically, emphases will be focused on agents of a natural or synthetic origin possessing HIF activation or stabilization activity in order to rethink the characteristics of these compounds from an anti-anemic drug development point of view. Anemia is the most common disorder of human blood, globally affecting ~1.62 billion people [9], starving tissues and organs of oxygen which results in fatigue and overall diminished quality of life, and if left untreated can be fatal [10]. Countries with low or mid-income are most heavily affected by this disease, and the prevalence of anemia in these countries is roughly 5 fold of that (43% vs 9%) in developed countries [11]. This disease mainly occurs in pregnant women and young children, with global anemia prevalence estimates of 47% in children younger than 5 years, and 42% in pregnant women [11, 12]. The major concerns regarding health consequences of anemia include increased risk of maternal and child mortality due to severe anemia, loss of productivity from impaired work capacity, impaired cognitive and physical development of children, and increased susceptibility to infection [11], all of which certainly lead to a substantial economic burden. As a result, anemia is regarded as a global public health problem with major consequences for human health as well as social and economic development [11, 13]. Anemias result from numerous causes (e.g., iron deficiency, Vitamin B12 deficiency, bone marrow failure, chronic disease, thalassemia) but all present with a quantitative deficiency of the hemoglobin, often accompanied by a reduced number of red blood cells (World Health Organization criteria hemoglobin concentration 13.5 g/dL in men and 12 g/dL in women) [13, 14]. Among all types of anemias, iron deficiency anemia is the most common one around the world and presumably accounts for 50% of the cases of anemia, while the second leading cause is com- *Address correspondence to this author at the 22 Hankou Road, Nanjing 210093, China; Tel: +86-13913026062; Fax: +86-25-83596143; E-mail: [email protected] 0929-8673/13 $58.00+.00 © 2013 Bentham Science Publishers 2 Current Medicinal Chemistry, 2013, Vol. 20, No. 1 monly found with many chronic diseases (e.g., chronic renal or liver disease, tuberculosis, AIDS) and results from human body’s inability to use iron stores and improper installation of iron into the hemoglobin [13, 14]. Since anemia is a multifactorial disease and considered to be symptomatic of an underlying disease process rather than an illness in their own right, the cause for the anemia must be identified and corrected for efficient management [14]. Currently available treatments of anemia typically rely on nutritional supplements, treatment of inflammation or malignancies, and intravenous (iv) administration of either red blood cells (RBCs) or erythropoiesis-stimulating agents (ESAs) [15]. RBC transfusion is frequently used in patients with severe or lifethreatening anemia due to its rapid effect in practically all patients [16]. However, there are many negative aspects in clinical settings associated with RBC transfusion, namely HCV, HBV or HIV infection, life-threatening immediate hemolytic transfusion reactions, iron overload or suppression of endogenous EPO production [16]. The introduction of ESA in late 1980s has fundamentally changed the way to treat anemia and greatly reduced the need for blood transfusion [17]. Patients with chronic renal failure or different types of malignancy have shown to benefit from it [16]. However, this therapy is still impeded due to several problems: first, they are highly expensive ( quarterly costs for Darbepoetin and Epoetin , two most commonly available recombinant ESAs, are roughly 2,000 [18]). Additionally, they show to cause serious cardiovascular side effects, and suffer from resistance in clinical practice which further exacerbates the disease [19-23]. In response to this situation, newer strategies for correcting anemia have been investigated during the last few years. Hypoxia inducible factor (HIF) stabilization represents one of these strategies to combat anemia by circumventing issues related to current therapies, and a great amount of efforts have been devoted to this area [24]. 3. HYPOXIA INDUCIBLE FACTOR PATHWAY AND ITS IMPLICATION IN ANEMIA AND OTHER ISCHEMIA RELATED DISEASES Development and evolution of animals largely rely on harness of the oxidizing potential of molecular oxygen which represents a crucial factor involved in numerous fundamental processes of life, for instance, cellular respiration, anabolism of metabolites and catabolism of nutrients and drugs [25]. For this reason, human body develops complex systems to sustain oxygen homeostasis and to be able to adapt to hypoxia (a common pathological condition related to anemia due to low oxygen supply) [26]. Hypoxia inducible factor-1 (HIF-1), a transcription factor first discovered in 1992 and later purified in 1995 by Semenza and Wang, appears to be the major regulator of O2 sensing and homeostasis in human body [27, 28]. In their landmark research, this nuclear factor, designated hypoxia inducible factor-1 (HIF-1), showed to activate erythropoietin (EPO) gene transcription in Hep3B cells subjected to hypoxia or cobalt chloride treatment [27]. Their finding was anticipated to open a new avenue for anemia management since Epo is a glycoprotein growth factor that stimulates proliferation and differentiation of erythroid progenitor cells into RBCs, which is also the main function of ESAs. Zhao and Wu HIF-1 is a heterodimer, consisting an (the regulatory subunit) and a subunit [29]. Currently, three related subunits (HIF-1, HIF-2 and HIF-3) and three isoforms of subunit, also known as the aryl hydrocarbon receptor nuclear translocator (Arnt1, Arnt2 and Arnt3) have been characterized [30]. While the presence of subunit is generally constant and in excess, the activity of subunit within the cell shows an oxygen-sensitive pattern (i.e., inversely related to the O2 concentration) [31]. This orchestrating mode was later found to be mainly regulated by a family of prolyl hydroxylase enzymes, namely PHD1, PHD2 and PHD3, which was first disclosed in 2001 [32, 33]. What is currently known regarding their roles (as depicted in Fig. 1) is under normal oxygen conditions, HIF- is constitutively expressed and rapidly degraded via the hydroxylation at proline residues (Pro402 and Pro564 in HIF-1 and Pro405 and Pro531 in HIF-2) by these PHD enzymes, upon which enables its complex with the von Hippel-Lindau protein [3]. This formed ensemble together with elongin B, elongin C, Rbx1 and Cul2 then forms the E3 ubiquitin ligase complex, which subsequently targets HIF-1 for proteolytic degradation by the ubiquitin-proteasome pathway [3]. While under the hypoxia condition, PHD-mediated hydroxylation of HIF- is inhibited such that HIF- will escape from proteasome degradation. The accumulated HIF- will translocate from the cytoplasm to the nucleus, where it dimerizes with HIF- subunits and forms a complex with the transcriptional coactivator, p300/CBP. This complex subsequently binds to the hypoxia-responsive element sequences of target gene promoters to initiate the activation of genes involved in hypoxic responses such as erythropoietin (epo) secretion, anaerobic glycolysis, and angiogenesis [3, 30, 34]. Therefore, the positive manipulation of this pathway may provide a new approach to these diseases, including myocardial infarction, stroke, metabolic disorders and aforementioned anemia. To date, modulation of this pathway is also showing an emerging role in management of inflammatory diseases, cardioprotection, cancer treatment, and neuroprotection, etc [35-38]. HIF-1 is ubiquitously expressed, while HIF-2 and HIF3 have more restricted expression patterns [39]. There has been long-term interest in distinguishing the roles of HIF-1 and HIF-2, while the roles of HIF-3 largely remain unknown [30]. HIF-2 is indicated to be the primary HIF molecule in the response to anemia attributed to its prime role in the regulation of renal erythropoietin (Epo) synthesis [3]. HIF-1, on the other hand, increases angiogenic factors (e.g., VEGF) and enzymes active in glycolysis, appearing to be the primary HIF molecule which responds to local tissue ischemia and hypoxia [3]. However, isolated hypoxia response element (HRE) derived from the EPO locus could bind HIF-1 preferentially according to a DNA-protein binding analysis, suggesting that HIF-1 may also contribute to EPO regulation [40]. Unlike HIF-1 and HIF-2, HIF-3 lacks the DNA-binding domain, and thus cannot affect gene expression. It may act as a negative regulator of HIF-1 and HIF-2 activity [3, 30]. In contrast to PHD3, which only contains the catalytic domain, PHD1 and PHD2 have additional N-terminal domains [30]. PHDs show distinct localization within the cell and have distinct substrate specificity: HIF-1 and HIF-2 are established substrates for PHD2 while PHD1 is more specific for Rpb1, the large subunit of RNA Hypoxia Inducible Factor Stabilization Current Medicinal Chemistry, 2013, Vol. 20, No. 1 3 Fig. (1). Hypoxia-inducible factor (HIF) activity under normoxic and hypoxic conditions. In normoxia, hydroxylation at two proline residues by PHDs enhances HIF- association with VHL and HIF- degradation via the ubiquitin--proteasome pathway, while hydroxylation of an asparagine residue by FIH hinders association with P300/CBP. In hypoxia, these processes are suppressed, and thus HIF- subunits could escape proteolysis, dimerize with HIF-, recruit co-activators and activate transcription via hypoxia response elements (HREs). polymerase II, which carries the fundamental enzymatic activity of the complex synthesizing all cellular mRNAs. These evidences strongly suggest distinct roles of these PHD isoforms in cells [30]. Another means to regulate HIF- stability is through hydroxylation in the C-terminal of HIF- exerted by factor inhibiting HIF (FIH), an asparaginyl hydroxylase [36]. FIH along with PHD1-3 belongs to the family of ferrous iron and 2-oxoglutarate-dependent dioxygenases. As PHDs, FIH binds Fe(II) via a conserved 2His-1Asp/Glu triad of residues although structural study shows significant variation in the 2OG binding site of FIH relative to other dioxygenases [41]. In the presence of oxygen, hydroxylation by FIH, of an asparagine residue (Asn803 and Asn851 in human HIF-1 and HIF-2, respectively) will inhibit the interaction between HIF- and the CH-1 domain of the transcriptional coactivator, p300/CBP, and in turn the activation of target genes will be suppressed [41]. It is beyond the scope of this review to further delineate the details of HIF-PH pathway. For a more thorough description of certain concepts, such as catalytic mechanism, isoforms comparison, and crosstalk between HIFs and other transcription factors, etc, the reader is encouraged to consult the most recent reviews and research articles with respect to this pathway [30, 34, 36, 42-44]. 4. NATURAL PRODUCTS AND IRON CHELATORS AS HIF STABILIZERS The first natural product known to activate HIF-1 is Desferoxamine (DFO) 1 (Fig. 2), a bacterial siderophore produced by the actinobacteria Streptomyces pilosus [45]. DFO is a strong multidentate iron binder used clinically for the treatment of iron overload and toxicity. Prior to the discovery of the PHD enzymes, it was demonstrated by the same group discovering HIF-1 that DFO increases EPO RNA and HIF-1 levels in Hep3B cells [27]. Since then, DFO has been widely used as a reference compound in biological or drug development studies. DFO was believed to exert its action via simply non-selective chelation of iron in the culture medium [46], which leads to the unavailability of iron to the prolyl hydroxylase active sites and in turn inactivates PHDs. Agents with this type of action mode, were viewed as noncompetitive inhibitors of PHDs [25]. Nagle and Zhou recently in their article reviewed natural product-derived small molecule activators or stabilizers of HIF-1 with different scaffolds, such as alkaloids, phenolic compounds, steroids, carbohydrates, etc [45]. It is well recognized that natural products have been provided promising lead compounds and drug candidates and also serve as useful molecular probes to elucidate signal transduction pathways for many decades [45]. Potent natural product-derived HIF-1 activators or stabilizers that exhibit a low level of toxicity and side effects thus hold promise as new treatment options for anemia and a variety of debilitating ischemia-associated diseases [45]. Here, we will only focus on those newly characterized natural products showing HIF-1 activation activity during these years, and no further attention will be directed to previous reported natural product-derived HIF-1 activators or stabilizers. Beturetol 2 (Fig. 3) and isosakuranetin 3 (Fig. 3) were isolated from propolis, a natural resinous hive product produced by honeybees from various plant sources. They show to induce all HIF-1 target genes, such as GLUT1, HK2 and 4 Current Medicinal Chemistry, 2013, Vol. 20, No. 1 Zhao and Wu VEGFA mRNA, both under hypoxic and normoxic conditions in HCT116 cells [47]. Beturetol 2 and isosakuranetin 3 fall into the category of flavonoids, and their structures are quite similar to quercetin 4 (Fig. 3), another flavonoid known to activate HIF-1 in HeLa cells and murine brain endothelial cells (MBEC) that overexpress HIF-1 [45]. The ability of flavonoids to induce accumulation of HIF-1 is attributed to their 5-hydroxyl 4-oxo group, which behaves as an iron chelating moiety and thus may inhibit HIF-1 prolyl hydroxylases (PHDs), the key regulator for HIF-1 degradation, resulting in stabilization of HIF-1 in normoxia [48]. However, discrepant results were obtained with regard to quercetin and some other flavonoids under low oxygen pressure. These flavonoids were shown to reduce HIF-1dependent reporter activity under hypoxic condition, indicating that the HIF-1 modulating effects of the flavonoids are oxygen concentration-dependent [49]. So it would be interesting to know that flavonoids 2 and 3 still can enhance HIF1 transcriptional activity even under hypoxic condition, if considering PHDs are inactivated under hypoxic condition. One possible explanation could be the inducing effects on HIF-1 transcriptional activities in hypoxia by 2 and 3 may be not due to the inhibition of PHDs but to some direct effects on the structure or functions of HIF-1 protein [47]. Flavonoids also can naturally exist as polymers. Cinnamon, a spice produced from the bark of trees from the genus Cinnamomum, has been used as a treatment for diabetes [50]. The active water soluble components of cinnamon were found to contain double-linked procyanidin type-A polymers (e.g., 5, Fig. 3), mainly trimers and tetramers of flavonoids [50]. In a study aimed to investigate the molecular mechanism accounting for the beneficial effects of cinnamon in diabetes, Cinnulin PF® (CPF, a commercial cinnamon extract containing the bioactive double-linked procyanidin type-A trimers and tetramers) was shown to improve angiogenesis by increasing HIF-1 expression in keratinocytes and by the upregulation of VEGF-A, VEGF-B and VEGF-C observed in the cell lines through genome-wide mRNA-Seq analysis [50]. Taken together, flavonoid seems to be an interesting and unique scaffold of HIF-1 modulator, and it is worth further investigation of this scaffold based on aforementioned information. O O N O OH H3CO HO OH OH OH Type A double link O OH OH OCH3 O H N OH OH O H OH OH O OH OH OH 1 HO OH HO O 3 O OH HO OH O Fig. (2). Desferoxamine (DFO). Polyphenol gallic acid 6 (3,4,5-trihydroxybenzoic acid) (Fig. 4) naturally exists either as a free compound or combined with other compounds, particularly as a common feature in a majority of catechins, such as (-)-epigallocatechin gallate 7 (EGCG) (Fig. 4) and (-)-epicatechin gallate 8 (ECG) (Fig. 4), etc [51]. The interesting merits of these catechins, for instance, EGCG and ECG, are that they can induce the content of HIF-1 in human cell lines, whereas other O O 2 HO O N OH HO N O H2N OCH3 OH N H OH catechins without a gallate moiety have a low activity of inducing HIF-1 [52]. This discrepancy indirectly indicates that the action of the catechins on the HIF-1 expression is ascribed to the gallate moiety [52]. In a study, nPG 9 (npropyl gallate) (Fig. 4), a food additive, was found to increase the content of HIF-1 in human heart muscle cells through inhibition of the protein degradation presumably via PHD inhibition as a strong iron chelator chelating the active site Fe2+ ion [52]. However, it was revealed that the PHD inhibitory activity of nPG was not caused by its own but by gallate, which is the hydrolyzed product of nPG in the tested cells but retains the polyphenol structure and has a halfmaximum inhibition concentration of about 0.3 mM [52]. Molecular modeling also shows that gallate binds to the PHD by mimicking the structure of 2-OG in the PHD active site, which cannot be met by nPG. This study suggests the actions of gallate-conjugated catechins to increase the HIF-1 content are mediated by the gallate released within the cells by hydrolysis [52]. Nonetheless, this concept seems to be an oversimplification and cannot be utilized to explain other experimental outcomes related to catechins. In another study, mass spectrometric experiments confirmed that EGCG formed covalent modifications with PHD2 at both physiological and alkaline pHs [53]. Consequently, EGCG could behave like CP94 14 (see Fig. 8) (will be discussed later) to effectively reduce higher iron oxidation states (Fe4+ back to Fe3+ ) by the virtue of its higher affinity for Fe3+ to disrupt PHD function. This catalytic redox cycle that results in the formation of a ferryl (Fe4+) intermediate has been reported to be responsible for the hydroxylation and subsequent degradation of HIF-1 under normoxia [54]. 3D docking models also show EGCG could bind covalently within the catalytic domain of PHD2 at Cys323, Arg383 or Cys283 from a relative short distance. These findings in Hep3B cell cultures experiments suggest that EGCG regulation of HIF-1 levels is primarily directed by PHD inhibitory activity within its iron binding pocket [54]. 5 trimer OH OH O 4 Fig. (3). Flavonoids. Hinokitiol 10 (-thujaplicin) (Fig. 5) is a natural mono terpenoid found in the heartwood of cupressaceous plants [55]. It contains a -diketone structure that facilitates the Hypoxia Inducible Factor Stabilization Current Medicinal Chemistry, 2013, Vol. 20, No. 1 5 OH OH OH HOOC HO O OH HO OH OH OH O OH O 6 OH O OH O O OH O OH 7 OH OH O OH 8 OH OH 9 Epigallocatechin gallate OH Epicatechin gallate OH Fig. (4). Structures of gallic acid and related compounds. formation of iron chelates and thus confers its biological actions. It shows to trigger apoptosis via activation of caspase-3 as an iron chelating compound, and exert a wide range of biological effects including anti-inflammatory, antibacterial, antioxidant capacities, and anti-tumor activity, etc [56]. When HepG2 and HeLa cells were exposed to hinokitiol in normoxic conditions, HIF-1 was found to be stabilized in both cells in a dose-dependent manner, which appeared a little less potent than clioquinol [56]. Further study determined that hinokitiol had inhibitory effects only on the prolyl hydroxylase activity, but not on the HIF-1– VBC (VHL-Elongin B-Elongin C) interaction [56]. Inhibitory effects exerted by hinokitiol on HIF-specific hydroxylases can be reversed by addition of 2-OG and Fe(II), suggesting that 2-OG overcompetes hinokitiol in the binding to PHD2. This also indicats hinokitiol likely works as a competitive antagonist of 2-OG to elicit transcriptional activation of VEGF, which might be beneficial in therapeutic angiogenesis for ischemic diseases [56]. O HO 10 Fig. (5). Hinokitiol. L-Mimosine 11 (Fig. 6), isolated from Mimosa pudica, is a fully characterized iron chelator, reversibly blocking mammalian cell proliferation at late G1 phase [57]. This plant amino acid exhibits neuroprotective effects partly correlated with its ability to induce the transcription factors HIF-1 and ATF-1/CREB and the up-regulation of glycolytic enzymes, p21waf1/cip1, and erythropoietin in embryonic cortical neuronal cultures [57]. L-Mimosine has also been reported to act as a prolyl 4-hydroxylase inhibitor and has similar effects on the hypoxic induction of HIF-1 protein in human and rodent cells [57]. HO N wide applications in health improvement, nutritional supplementation as well as prevention and treatment of diseases for centuries [58]. In recent years, these plant extracts have furnished numerous promising leads for modern drug discovery [59], and consequently attracted increasing attention from industry and academia around the world. Currently, there is a great deal of efforts ongoing to elucidate the therapeutic mechanisms and effects of these extracts [60-63]. Rhodiola, the root and rhizome of Rhodiola crenulata (Hook. f. et Thoms.) H. Ohba, has been used in traditional medical system to increase the body resistance to mountain sickness in preventing hypoxia [40]. Salidroside 12 (Fig. 7), the chemical marker to evaluate the quality of Rhodiola, is one of the major phenylpropanoid glycosides derived from Rhodiola [40]. Salidroside showed to induce EPO mRNA expression in a dose-dependent manner in cultured HEK293T and HepG2 cells (the maximal induction rate: ~100% increase at 10 μM of salidroside after 24 h of treatment in HEK293T cells, and ~120% increase at 30 μM of salidroside in HepG2 cells) [64]. Salidroside at a concentration of 100 M induced HIF-1 expression at an increase of ~115% and 110% in HEK293T and HepG2 cells, respectively, while the HIF-2 expression level was not significantly altered, indicating that HIF-1 protein, not HIF-2 protein, was a critical mediator in the salidroside-induced EPO expression [64]. Meanwhile, in the same study, it was found that HIF-1 mRNA expression level was not altered, suggesting the possibility that the accumulation of HIF-1 is due to reduction of HIF-1 degradation, which finally causes EPO gene activation [40, 64]. However, the exact role of salidroside in HIF degradation blocking is still not clear. In addition to the aforementioned hematopoietic function, salidroside was also shown to induce the expression of VEGF and to prevent hypoxia-induced cell death in cultured cardiomyocytes via the expression of HIF-1 [64]. HO COOH 11 Fig. (6). L-Mimosine. Plant extracts derived from traditional medicines, for example, Traditional Chinese Medicines (TCMs), have found O HO O OH 12 NH2 O OH HO Fig. (7). Salidroside. As we can see, many natural products behave like iron chelators to inhibit PHDs through their chelating groups. So it is obvious that many iron chelators are derivatives of certain natural products. Iron chelators are involved in the 6 Current Medicinal Chemistry, 2013, Vol. 20, No. 1 treatment of potentially fatal conditions, namely iron overload disease, such as -thalassemia, via avidly coordinating with intracellular and extracellular iron [65]. The redundant iron could cause cell death or damage to other tissues and organs by converting superoxide (O2.-) and hydrogen peroxide (H2O2) into highly reactive, toxic hydroxyl radicals (OH. ) in a sequence of reactions known as the Haber–Weiss reaction [65]. However, the underlying mechanisms of overall protective effects by iron chelators are not simply suppressing hydroxyl radical formation. Accumulating evidence have demonstrated that iron chelators are capable of inducing HIF-1 or HIF-2 and other hypoxia-regulated gene expression in a variety of cell lines by inhibiting prolyl hydroxylases through non-selective or selective iron chelation or by other undetermined mechanisms. A close derivative of commercially available Deferiprone 13 (Fig. 8), named CP94 14 (1,2-diethyl-3-hydroxypyridin4-one) (Fig. 8), is a bidentate hydroxypyridinone belonging to a class of low molecular weight Fe3+ chelators of considerable clinical interest as orally active compounds for the treatment of iron overload [66, 67]. CP94 was recently characterized as a novel molecule able to inhibit PHD activity and stabilize HIF [68]. CP94 could disrupt the catalytic redox cycle in PHD iron-binding pocket via iron chelation in the active site to exert its inhibitory activity (As we will see later, Deferiprone, the parent compound of CP94, shows to inhibit PHD2 through iron chelation in the active site [69]). It is also found that rats treated with CP94 led to a marked increase in HIF-1 protein in both kidneys as well as muscle tissues after seven days treatment [68]. Target gene expressions were up-regulated, for EPO (4 fold), VEGF (1.5 fold) and PDK-1 (2 fold) after 24 h treatment [68]. The level of HIF-2, as measured by Western blot analysis, parallels that of EPO gene expression in the kidney. However, there was little or no observable changes in hematological profiles, particularly hematocrit, nor in the iron levels in plasma from these rats. In HEK293 human embryonic kidney cells as well as bovine aortic endothelial cells, CP94 can increase the levels of HIF-1 protein in a dose dependant manner [68]. O O OH OH N Deferiprone 13 N CP94 14 Fig. (8). Deferiprone and CP94. Although chelation of iron with iron chelators appears to be an effective means to inhibit PHD enzymes, those inhibiting PHDs through non-selective iron chelation such as desferrioxamine seem to have little success in vivo [70]. This strategy would probably not be viable in intact organisms since the amount of iron in the labile iron pool that would have to be chelated would be relatively large [70]. Moreover, non-selective iron chelation would interfere with other cellular process such as electron transport and may inhibit other non-heme, iron-containing enzymes and cause side effects [70]. So it is necessary to take into consideration to improve the chelation selectivity of these iron chelators to the PHD active site when developing HIF stabilizers based on them. Zhao and Wu 5. PHD2 INHIBITORS In the HIF-PH axis, prolyl hydroxylase is described as a key enzyme in the degradation of HIF. Hence, inhibition of PHD enzymes becomes an extensively used strategy by which to potentiate the transcriptional activity of HIF in a therapeutically relevant manner during the last several years. Among three PHD isoforms, PHD2 appears to take the main role in the degradation of HIF- in many cell types, particularly for HIF-1 [71]. Therefore, current anemia drug development in this area is mainly focused on the stabilization of HIF-1 through inhibition of PHD2 mediated hydroxylation of HIF-1. Recent advances regarding PHD2 inhibitor development through structure based drug design and other methodologies will be summarized in this section. Meanwhile, concerns regarding PHD2 inhibitor development will also be discussed in the late part of this section. 5.1. Structure Based Rational Drug Design Two groups independently reported X-ray cocrystal structures of a truncated form of PHD2 with two structurally related isoquinoline inhibitors in 2006, both of which clearly mimic the endogenous cofactor 2-OG in the active site [7274]. In this site as shown in McDonough’s case, isoquinoline A (Fig. 9b) binds to the Fe(II) via bidentate coordination through nitrogen of its isoquinoline ring and oxygen of the amide carbonyl forming a ca. planar 5 membered chelate ring (Fig. 9a) [73]. Its carboxylate side chain is bound in a predominantly hydrophobic pocket with the exception of the Arg383 and Tyr329 side chains, with which it forms electrostatic and H-bonding interactions. Using this information as the starting point, scaffolds as hydroxyquinoline (e.g., compound A, Fig. 9b), imidazol[1,2-a]pyridine (e.g., 15, Fig. 9b), pyrazolopyridine (e.g., 16, Fig. 9b), and 8-hydroxyqui nolines (e.g., 17, Fig. 9b), etc., which can maintain the chelation with Fe(II) and salt bridge with Arg383 were proposed in many recent published literatures and patents through structure based drug design [25, 31, 74-76]. Generally, their iron chelating moieties ligate the iron atom in a bidentate manner with side chains participating in electrostatic and Hbonding interactions with Arg383 and making favorable van der waals contacts with the hydrophobic residues lining the PHD active site. These efforts have already led to the introduction of four compounds into clinical trials for the treatment of various anemias [25]. Among them, FG-2216 and FG-4592 are most notable, and both are intended as orally acting drugs for anemia. Available data from clinical studies demonstrated that these two compounds can up-regulate other genes besides Epo that are important in erythropoiesis including EpoR, transferring, transferring receptor, ferroportin, and the divalent metal transporter 1[6-8]. In addition, they are safe and well tolerated in the clinical studies conducted to date [6-8]. The significant problems with Epo analogs are primarily due to decreased availability of iron because of inflammation, increased morbidity, and mortality associated with higher doses of Epo causing high levels of erythropoietin in the blood. Increasing hemoglobin by stabilization of HIF may be safer, because of the diverse downstream effects of HIF stabilization, for example, increased Epo, increased available iron, and a relatively small increase in erythropoietin blood levels [8]. Therefore, normalization of hemoglobin with PHD inhibitors will circumvent those Hypoxia Inducible Factor Stabilization Current Medicinal Chemistry, 2013, Vol. 20, No. 1 concomitant risks related to Epo therapy. These promising findings suggest that an oral PHD inhibitor could be effective for the treatment of anemia. Arg 383 Tyr 303 Tyr 329 O OH O HN His 313 N O Cl Fe(II) Asp 315 OH H 2O His 374 to FIH. A cocrystal of JNJ-42041935 in the active site of PHD2181–417 was thus obtained supporting the competitive nature of the inhibition observed in the functional assay [70]. In the cocrystal, JNJ-42041935 binds in a way similar to that reported for compound A (see Fig. 9a), except that the shared water bridge interaction is replaced by direct hydrogen bound with Tyr303 for compound A. Meanwhile, JNJ42041935 not only caused a concentration-dependent elevation of cellular HIF-1 and erythropoietin release in Hep3B cells, but also corrected the microcytic nature of the inflammation-induced anemia as demonstrated by increases in MCV, MCH, and the cellular hemoglobin content of mature red blood cells, which cannot be attained by exogenous erythropoietin. These results suggest that JNJ-42041935 or other PHD inhibitors may be applied to treat anemia of various origin, in which current treatments are ineffective or not optimal. R1 N R1 O N N H R2 COOH O N N N Cl A N H R2 COOH COOH R3 ii i COOH 15 HN R3 HN N N R4 O N H 7 Cl N N N O N Cl 16 N N H N N COOH OH O 17 F3CO N H COOH JNJ-42041935 18 Fig. (9). a. Schematic diagram of key PHD2/isoquinoline A interactions based on x-ray cocrystal ; b. Structures of compound A and several known PHD2 inhibitors. A most recent study of using structure based drug design can be seen with the recently disclosed JNJ-42041935 18 (Fig. 10) [31, 70]. In that study, an initial series of compounds i (Fig. 10) were reported to possess measurable inhibitory activity in PHD2 enzymatic assay [31]. However, these compounds were not able to significantly stimulate epo release from Hep3B cells at concentrations up to 100 μM. Isosteric replacement of glycine in i (Fig. 10) with pyrazole ring leads to a number of benzimidazole-2-pyrazoles ii (Fig. 10), among which some were proved to be potent inhibitors of PHD2 relative to the benzimidazole-2-carboxamides i, displaying pIC50 values of 7.0 or more. Compounds of this set displayed low iron chelation ability in solution, indicating that the increased inhibitory activity was not a result of iron sequestration. Interestingly, these compounds showed to cause a robust increase of epo secretion from Hep3B cells measured after 24 h of incubation, and this epo secretion was ascribed to PHD inhibition. JNJ-42041935, a thoroughly characterized compound from series ii, was described as the most potent inhibitor of PHD2181–417 with a pIC50 value of 7.0 ± 0.03 compared to 3,4-EDHB, desferrioxamine, clioquinol, ciclopirox and DMOG [70]. JNJ-42041935 was also found to inhibit full-length PHD1, PHD2, and PHD3 enzymes with pKI values 7.91 ± 0.04, 7.29 ± 0.05, and 7.65 ± 0.09, respectively, consistent with the high degree of homology of the active site of the PHD enzymes. This study confirmed that JNJ-42041935 behaves as 2-OG competitive inhibitors of PHD, showing highly selective for PHD relative Fig. (10). JNJ-42041935 and general structures of series i & ii. 5.2. PHD2 Inhibitors Derived From Other Methodologies A virtual screening on a data set of compounds was performed to identify novel target-specific PHD inhibitors by combining pharmacophore model generation, docking and manual interpretation altogether [77]. A 3D pharmacophore comprising three H-bond acceptor, one negative ionizable group and one ring aromatic feature was developed according to custom-based pharmacophore generation protocol, which was further validated by decoy set method and used as a 3D query in database screening to retrieve 189 hits from 500,000 compounds. These 189 compounds were further subjected to molecular docking study and analyzed based on the docking score, binding modes, and molecular interactions with essential active site residues. Finally, 18 compounds with different scaffolds having good docking score and ADME properties were chosen. They were supposed to be used as potential leads in designing new PHD inhibitors. However, none enzymatic or cellular assays had ever been carried out on these compounds to determine their inhibitory activity. A phenotypic cell-based high-throughput screen was conducted to identify activators of the HIF pathway on the NIH’s Molecular Libraries Small Molecule Repository, which contains more than 300 K compounds [78]. The subsequent medicinal chemistry optimization of a triazine hit led to the identification of a new molecular probe ML228 19 (Fig. 11), which represents a novel chemotype lacking the acidic functional group almost universally present in PHD inhibitors, although no direct evidence shows that ML228 is 8 Current Medicinal Chemistry, 2013, Vol. 20, No. 1 Zhao and Wu a PHD2 inhibitor. This feature may be important for certain disease applications. ML228 was demonstrated to behave like iron chelator to potently activate HIF in vitro as well as its downstream target VEGF. used for assigning structures to the ligands that bind preferentially without denaturing proteins and thus very time efficient [82]. In other words, support ligands bearing certain dynamic functional groups will co-incubate with target proteins and library compounds bearing reactive groups which could reversibly interact with above dynamic functional groups. The complex thus formed is consisted of the target proteins and conjugated products between support ligands and library compounds, which will further be analyzed by mass spectrometry to differentiate the binding ability of these conjugate products (as depicted in Fig. 13) [82]. This technology has recently been successfully applied in the identification of a series of potent PHD2 inhibitors through the boronic acid/boronate ester dynamic systems [82]. Support ligands 24 (Fig. 13) and 25 (Fig. 13) were used in this DCMS. Either of them reacted with forty diols from four sets in the presence of PHD2·Fe(II) under the incubation conditions. ESI-MS was then used to characterize which of the in situ formed boronate esters (structure depicted as 26, Fig. 13) bind preferentially to PHD2·Fe(II). The mass shifts peaks were corresponded to complexes of PHD2 with boronate esters of 26, which were derived from 7 diols (e.g., 27, 28, 29, Fig. 13). And this result was validated by ESI-MS analysis of mixtures of 24 with the individual diols. However, because 25 shows to clash with active site according to docking study, when 25 was employed, no binding of boronate esters was observed in the corresponding assay. Then synthesis of stable analogues that mimic the structure of the identified hits (boronate esters derived from 24 and 7 diols) was carried out to determine the potential of the boronic acid/boronate ester-based DCMS for identification of PHD2 inhibitors. Analogues 30 (Fig. 13) and 31 (Fig. 13) were therefore prepared, and they function as 6/5- and 6/6-fused bicyclic mimics of the catechols boronate ester moiety in the boronate ester derived from 24 and catechol 27. They showed to have fairly good affinity, suggesting both ring systems are accommodated in the protein binding pocket. Analysis of PHD2 structures suggests hydroxy group of Tyr303 might be used to promote inhibitor binding. Thus, a derivative of inhibitor 32 (Fig. 13), another compound to mimic the hits from DCMS containing a hydroxy group at the C3 position, 33 (Fig. 13), was prepared. As predicted, compound 33 showed significantly higher potency and affinity (IC50=13 nM, KD=0.8 mM) than 32, and further crystal study corroborates the existence of the desired hydrogen bonding interaction. Overall, this study has demonstrated the value of DCMS in the discovery of potent PHD2 inhibitors. N N N N NH ML 228 19 Fig. (11). ML 228. A viewpoint paper by William A. Denny recently highlighted the work done by Vachal et al. [79]. Vachal and coworkers reported their synthesis and evaluation of a novel class of 1,3,8-triazaspiro[4.5]decane-2,4-dione based paninhibitors of PHD1-3 [80]. Not like other cases, they utilized an affinity selection mass spectrometry technique (AS-MS) as an initial high-throughput screen, where PHD2 enzyme was incubated with mixtures of compounds, and proteinligand complexes were separated by size exclusion chromatography and identified by mass spectrometry. This resulted in a set of spiroindole hits of novel structure (e.g., 20, Fig. 12) [80]. In an effort to minimize the pharmacophore of the spiroindolone, a spirohydantoin class of compounds was obtained (exemplified by 21 in Fig. 12, exhibiting improved PK properties and in vivo elevating erythropoietin levels in a mouse pharmacodynamic erythropoietin assay). However, hydantoins are potentially associated with multiple biological activities [79]. To avoid this issue, Vachal et al. tried to derivatize the unsubstituted hydantoins into N,Ndisubstituted analogues such as 22 (Fig. 12), which has a much better PK profile than 21. Final attempt to modify the hydantoin N-heterocycle unit led to the discovery of the clinical candidate 23 (Fig. 12), which showed a good PK profile (rat PK: Cmax= 390 nM, t1/2= 1.1 h, F = 47%) and substantial elevation of erythropoietin levels (19 μg/L at a dose of 100 mg/kg iv) at 4 h, with no concomitant elevation in liver enzyme levels [80]. 23 was thus believed as a very promising clinical candidate. Dynamic-combinatorial mass spectrometry (DCMS) is a novel technique for the discovery of small molecular ligands for target proteins [81]. It has the advantage of being efficient and providing information on mass shifts, which can be Another group reported their work to realize inhibition of a prolyl hydroxylase domain (PHD) by substrate analog pep- N N N N N N N N N O HO O O O N N N HN N N N N N O AS-MS hit 20 21 N O O Ph N Ph 22 Fig. (12). Design evolution from high-throughput screen hit 20 to clinical candidate 23. Clinical candidate 23 COOH Hypoxia Inducible Factor Stabilization Current Medicinal Chemistry, 2013, Vol. 20, No. 1 HO OH HO B 9 OH B H N N H N COOH N 24 O COOH O 25 R1 OH OH HO O O B R1 diols OH Y H N N R1 B H N COOH NH4OAc buffer pH 7.5 O N OH B Y COOH COOH O O 24 H N N Y = O, OC(O) R1 = Various Dynamic Combinatorial Library (DCL) of boronate esters 26 PHD2 Complex OH HOOC Protein MS OH OH HOOC OH OH 27 OH 29 28 MeO X O H N N 30 COOH H N N 31 O H N COOH N O COOH O 32 X = H 33 X = OH Fig. (13). Schematic representation of the dynamic combinatorial mass spectrometry (DCMS) method and structures of selected compounds used in this work. tides [83]. In this manner, they can target the C-terminal oxygen dependent domain (CODD) binding site of PHD2 active pocket to exert inhibitory activity acting independently of the 2-OG/Fe(II) interaction, and thus provide selectivity for PHD2 over FIH. To that end, 20-mer peptides were synthesized, which consist of amino acids identical to those in the CODD556–575 except the 564 proline residue (at position of X, Fig. 14) by using the protocol for the conventional Fmoc pepetide synthesis. The 564 proline residue was intentionally converted into proline-derived amino acids such as 4-fluoroproline, 4-hydroxy-proline, piperidine-2-carboxylic acid, 3,4-dehydroproline and 4-thioproline to prevent hydroxylation reaction by PHD2. Among these peptides, DhP 34 (Fig. 14) and Thz 35 (Fig. 14) were found to be inhibitors of PHD2 without disrupting the 2-OG-Fe(II) interaction at the active site of PHD2 and therefore could selectively inhibit PHD2 over FIH, which met the initial purpose of this study. DLDLEALA-X-YIPADDDFQLR 34 DhP 35 Thz Fig. (14). Synthesized peptides. L-3,4-dehydroproline L-4-thioproline 5.3. Other Concerns About PHD2 Inhibitor Development 5.3.1. Iron Chelating Groups Almost all current known PHD2 inhibitors feature iron chelating groups which are required to bidentately chelate the Fe(II) at the active site of PHD2 enzyme as shown in several published crystal structures. Despite the successes in targeting this enzyme by focusing on modifying those frameworks with some iron chelating moieties, current availability of this small group of chelating moieties may restrict therapeutic development towards an effective PHD2 inhibitors. Flagg’s group’s work is a good attempt to find alternate chelating moieties as leads for selective inhibition of HIF hydroxylases, namely FIH and PHD2 [69]. Since both PHD2 and FIH share a similar Fe(II) binding site containing a His2Asp facial triad and incorporate same cofactor to hydroxylate certain amino acid residues on HIF-1, FIH obviously would be the other potential target of these chelating structures. They screened a small library of chelating structures for their ability to inhibit FIH and PHD2, with particular emphasis on bidentate chelating moieties as structural mimics of 2-OG. Three classes of iron chelating groups [pyridines (e.g., 36, Fig. 15), hydroxypyrones/hydroxy pyridinones (e.g., 37, 38, Fig. 15) and catechols (e.g., 39, Fig. 15)] were tested as inhibitors for FIH and PHD2. Most compounds tested were more effective inhibitors of PHD2 than 10 Current Medicinal Chemistry, 2013, Vol. 20, No. 1 of FIH, with a preference of aromatic chelating groups to bind to PHD2 and bulky compounds such as N-derivatized NOG to FIH. This trend reflects the difference of the volume of the active site between these two enzymes, rendering selective targeting of them possible. Dose response curves at moderate [2-OG] showed that the hydroxypyrones/hyd roxypyridinones were selective inhibitors, with IC50 in the μM range, and that the catechols were generally strong inhibitors of both FIH and PHD2, with IC50 in the low μM range, while pyridine-based compounds exhibited a wide range of IC50 values, ranging from 3 to >1000 μM. They also applied electron paramagnetic resonance (EPR) spectroscopy to monitor ligand binding to the active site of both PHD2 and FIH. The EPR data indicated that the representative inhibitors bound to the metal center in both PHD2 and FIH as a 2-OG mimic, supporting binding at the active site of each enzyme as the mode of inhibition. Finally, they came to a conclusion that hydroxypyrones and hydroxypyridones (HPOs) may be promising chelates for further elaboration of selective inhibitors toward FIH or PHD2. Because they have wide variation in IC50 in response to functional group polarity and bulk and wide application as the foundation for metalloenzyme inhibitors and medicinal inorganic compounds, such as matrix metalloproteases and other 2-OG oxygenases. Hydroxypyrones and hydroxypyridones (HPOs) are typically chelating groups found in siderophores which are lowmolecular-weight compounds secreted by grasses and microorganisms possessing high-affinity and selectivity for iron(III) and are widely used in therapies dealing with iron(III) or Al(III) overload [84, 85]. It is well known that the quality of leads and structural moieties arising from natural products is better and often more biologically friendly, due to their co-evolution with the target sites in biological systems [86]. Therefore, it is of great significance and value to elaborate these natural siderophile moieties into effective PHD2 inhibitors. HPOs have attracted extensive attention in iron chelator development, and it would be interesting to see how these siderophiles will function in this anemia setting as PHD2 inhibitors. In this study, HOP-COOH 37 (Fig. 15) was the most effective HOP inhibitor for both PHD2 and FIH (PHD2 IC50: 30 M; FIH IC50: 50 M), which structurally resembles a cis-isomer of 2-OG. Compared to other HOP inhibitors, HOP-COOH would be able to form a salt-bridge with the buried basic residue found in both PHD2 (Arg383) and FIH (Lys214). The dimethyl-substituted HOPO, also known as Deferiprone (see Fig. 8), has stronger inhibitory activity (PHD2 IC50: 40 M; FIH IC50: 30 M) than the methyl-substituted HOP-Me 38 (Fig. 15) (PHD2 IC50: 1000 M; FIH IC50: 70 M) for each enzyme, reflecting the inherently stronger binding of the HOPO framework to divalent metals as compared to the HOP framework. In a word, Flagg’s work provides us with new aromatic or pseudoaromatic groups as effective chelating groups of PHD2 and FIH, thereby expanding the number of chelating structures used for therapeutic development. 5.3.2. PHD Subtype Selectivity During these years, some groups reported their discovery of potent PHD inhibitors possessing subtype selectivity. Murray and coworkers are the first to report their work toward identifying quinolone based derivatives which inhibit Zhao and Wu PHD1, 2, and 3 with altered selectivity with the purpose of elucidating different in vivo roles of these three prolyl hydroxylases [87]. Sequence comparisons and modeling studies indicate that the 2-OG binding pocket (all 14 hydrophobic residues) is highly conserved among the three PHD isoforms [73], suggesting that substrate specificity of the PHDs, in part, is determined by regions relatively remote from the iron center [88]. By extending the molecule out of the 2-OG binding site into a putative hydrophobic pocket within the HIF1 peptide binding site, several quinolone based compounds (e.g., 40, Fig. 16) show to possess higher potency for PHD1 and PHD3 over PHD2, and they could potentially be developed into useful compounds for the further understanding of HIF biology [87]. In another study, 41 (Fig. 16) and JNJ42041935 (see Fig. 10) display a respective 14 and 5-fold higher apparent potency against the PHD3 subtype compared to the PHD1 and PHD2 subtype [89] (the statement about JNJ-42041935 is controversial in another study saying JNJ42041935 is not selective for PHD isoforms [70]). However, apart from these findings, information concerning the specificity of different PHD inhibitors with respect to PHD subtype selectivity is still very limited. The influence of suppressing different PHD subtypes was mainly appreciated through gene knock down assay in mice. Noticeably, a recent study by Minamishima and Kaelin suggests that inhibition of all three PHD isozymes is required to reactivate hepatic erythropoietin production [90]. As known to us, erythropoietin is produced mainly by the kidney in adults, but in fetus and for the first few months after birth, the liver is the primary source. Therefore, pan-PHD inhibitors are supposed to be more effective in treating anemia resulting from chronic kidney disease compared to PHD2-selective inhibitors [70]. O OH N NOP 36 O O OH HOOC O Hop-COOH 37 OH OH O Hop-Me 38 O2N OH 4NCat 39 Fig. (15). Inhibitor classes tested. NOP (2-Hydroxypyridine 1oxide), Hop-COOH (5-Hydroxy-4-oxo-4H-pyran-2-carboxylic acid), Hop-Me (3-Hydroxy-2-methyl-4-pyrone), 4NCat (4Nitrocatechol). 5.3.3. Endogenous 2-oxoglutarate Levels A further issue needs to be taken into consideration when developing prolyl hydroxylase inhibitors for clinical use is the cellular concentration of 2-OG in the tissue of interest. Majority of the current PHD2 inhibitors compete with the natural substrate, 2-OG to bind to the active site. 2-OG is a major constituent of the citric acid cycle occurring in the mitochondria and takes its place in many important physiological processes [89]. The cellular concentration of 2-OG is generally high, as evidenced by the fact that cytosolic 2OG concentration in liver and hepatocytes is in the range of 0.8–1.2 mM and more recently defined relatively high concentration ranging from 640 M in cortical tissue to 3300 M in tissues from substantia nigra [89]. The cellular concentration of 2-OG in PC12 and SH-SY5Y cells were found to be Hypoxia Inducible Factor Stabilization Current Medicinal Chemistry, 2013, Vol. 20, No. 1 O H N O NH OH O N H O OH N H N 11 O COOH O S N O But O N S COOH 41 40 Fig. (16). Compounds showing PHD isoform selectivity. 2020 M and 2300 M in a recent study, respectively [89]. It can therefore be envisioned that high level of 2-OG would largely impact the potency of these PHD2 inhibitors, which was confirmed by the determination of the EC50 value of compound 41 (Fig. 16) in a cellular assay measuring HIFreporter activity. Compound 41 possesses an EC50 value of 18,000 nM which is approximately three orders of magnitude higher compared to the IC50 values, whereas the EC50 value of 7500 M in the cellular assay for the indirect PHD inhibitor FG-0041 (Fig. 17) is within the same magnitude as the IC50 values of 10,500–48,000 nM for the PHD subtypes in the enzymatic assay. Thus a competitive inhibitor with a low Ki should be developed in response to this issue [89]. O HOOC N H N FG-0041 Fig. (17). FG-0041. uitination, were obtained respectively in an attempt to suggest possible modifications for improved inhibitor design [95]. From the observed binding mode of CQ in FIH-1, it was propose that CQ can be modified to improve the specificity of CQ derivatives, without losing the ability to coordinate Fe(II). In another study, eight novel inhibitors (e.g., 47, 48, Fig. 18) of FIH-1 were identified through a computeraided drug design protocol involving the structure-based virtual screening with docking simulations under consideration of the effects of ligand solvation in the scoring function. It is noteworthy that six of the eight inhibitors include a thioxothiazolidinone moiety in the middle of their molecular structures, as represented by 47 (Fig. 18) [96]. Because the inhibitory activities of these hits are moderate with the IC50 values ranging from 30 to 80 M, further development by structure–activity relationship studies or de novo design methods is highly valued. Generally, the aforementioned FIH inhibitors exert their activity through Fe(II) chelation at the active site, the same way adopted by a series of cyclic 2oxoglutarate analogues (e.g., 49, 50, 51, Fig. 18) to bind to FIH [91]. 6. AGENTS STABILIZE HIF BY INHIBITING FIH 7. AGENTS STABILIZE HIF BY TARGETING HIF-1 AND VHL INTERACTION FIH is another pivotal regulator of HIF- and can serve as a high potential target compared to PHDs for the development of therapeutic agents for anemia or ischemia because FIH is more active on HIF- than the other hydroxylases under hypoxic conditions [91]. Nevertheless, development of selective FIH inhibitors is challenging because FIH is closely related to the JmjC domain histone demethylase family [91], the reason why only a few inhibitors of FIH have been reported so far. Recent work has also demonstrated that FIH is able to hydroxylate a number of ankyrin repeat domain (ARD) proteins such as Notch and Gankyrin [92]. Although the precise role of FIH-mediated ARD hydroxylation is currently far from clear, competition between the ARD pool and HIF for FIH could serve to fine tune the HIF response [92]. This situation would render the pursuit of an effective FIH inhibitor even more challenging. N-oxalyl-D-Phe 42 (Fig. 18) derived from a series of N-oxalyl amino acids was reported to be a potent FIH selective inhibitor [93]. Several oxocarboxylic acid based compounds (e.g., 43, 44, Fig. 18) also demonstrated moderate FIH inhibitory activity [94]; however, there is no information showing they were selective for FIH only. In 2010, the cocrystal structures of human FIH-1 in complex with 5-chloro-7-iodo-8-hydroxy-quinoline 45 (Clioquinol, CQ) (Fig. 18) and 8-hydroxyquinoline 46 (HQ) (Fig. 18), two compounds shown to stabilize the transactive HIF-1 by blocking both Asn-hydroxyation and ubiq- A series of compounds were reported to target the von HippelLindau protein (VHL), the substrate recognition subunit of an E3 ligase, which upon binding to hydroxylated HIF-1 could lead to its ubiquitination and subsequent degradation by proteasome [97]. This is a first workable example of disrupting the interactions between hydroxylated HIF1 and VHL of an E3 ligase, which is an attractive drug target due to its exquisite substrate specificity and also an extraordinarily challenging one as modulation of E3 ligase activities requires the targeting of proteinprotein interactions consisting large contact surfaces and shallow grooves or flat interfaces involved. These inhibitors may avoid the HIFindependent off-target effects observed with PHD inhibitors. Since residue Hyp564 on HIF-1 makes key interactions with VHL and is crucial for HIF-1 binding, hydroxyproline (Hyp) was used as a starting point in the design of smallmolecule inhibitors of the VHL/HIF-1 interaction. The authors employed the de novo design software BOMB to guide their selection of plausible hydroxyproline analogues. 52 (Fig. 19) and 53 (Fig. 19) were two interesting hits featuring an isoxazole moiety positioned to interact with a crystallographic water observed in the structure of VHL bound to the HIF-1 peptide (549582) and a benzyl group stacked along the side chain of Tyr98. Further SAR study based on 52 led to the discovery of 54 (Fig. 19), which was found to 12 Current Medicinal Chemistry, 2013, Vol. 20, No. 1 H N HOOC Zhao and Wu COOH O O HOOC HOOC O N H R N H N-oxalyl-D-phenylalanine 44 43 42 S O OH OH I O N N H2N S NH N S O O Cl 46 45 N N MeO 47 N O S N OMe N N HO R HOOC 48 49 R = COOH 50 R = NHOH COOH 51 Fig. (18). Structures of FIH inhibitors. bind with a 4.1 μM IC50 value. The cocrystal structure of 54 and VHL confirmed that 54 mimics the binding mode of the transcription factor HIF-1 on VHL and that the hydroxyproline ring recapitulates the interactions seen in the HIF-1 peptide:VHL complex. CONFLICT OF INTEREST HO The authors confirm that this article content has no conflicts of interest. N NHR ACKNOWLEDGEMENTS O O O N O N 54 52 R = of more than 200 genes is under its control. Only when the long-term safety of targeting this pathway is established, can these novel agents hold great promise in the treatment of anemia and ischemia related diseases as well. R= Cl This paper was supported by Research Fund for the Doctoral Program of Higher Education of China (No. 201100 91120044), Natural Science Foundation of Jiangsu BK2011572, National Natural Science Foundation (No. 81202474) and Postdoctoral Foundation (2012M521051). OH 53 R= REFERENCES [1] Fig. (19). VHL Ligands. 8. CONCLUSION In summary, anemia represents a big burden for social and economic development due to its astounding prevalence and ensued consequences. HIF signaling pathway is the central player in the regulation of cell responses to hypoxia and normoxia, and thus provides us with many promising therapeutic targets to treat anemia and other ischemia related diseases. Positive modulation of this pathway has undergone significant advancement over these years showing the possibility of overcoming drawbacks associated with current therapeutic methods. It can be speculated that the development of novel and specific inhibitors of PHD, FIH or agents disrupting protein-protein interactions in this pathway may provide an innovative therapeutic means of therapy for anemia of chronic disease and anemia of chronic kidney disease (CKD). However, the other side of the coin is the safety issue about manipulation of this system, since the transcription [2] [3] [4] [5] [6] [7] Loor, G.; Schumacker, P.T. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ., 2008, 15(4), 686-690. Semenza, G.L. HIF-1: mediator of physiological and pathophysio logical responses to hypoxia. J. Appl. Physiol., 2000, 88(4), 14741480. Muchnik, E.; Kaplan, J. HIF prolyl hydroxylase inhibitors for anemia. Expert Opin. Invest. Drugs, 2011, 20(5), 645-656. Beuck, S.; Schaenzer, W.; Thevis, M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test. Anal., 2012, 4(11), 830-845. Bakris, G.L.; Yu, K.-H.P.; Leong, R.; Shi, W.; Lee, T.; Saikali, K.; Henry, E.; Neff, T.B. Effects of a Novel Anemia Treatment, FG4592, an Oral Hypoxia-inducible Factor Prolyl Hydroxylase Inhibitor (HIF-PHI) on Blood Pressure and Cholesterol in Patients with Chronic Kidney Disease. J. Clin. Hypertens., 2012, 14(7), 489. Besarab, A.; Belo, D.; Diamond, S.; Martin, E.; Sun, C.; Lee, T.; Saikali, K.; Franco, M.; Leong, R.; Neff, T.; Yu, P. Evaluation of Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitor FG-4592 for Hemoglobin Correction and Maintenance in Nondialysis Chronic Kidney Disease Patients for 16 and 24 Weeks. Nephrol. Dial. Transpl., 2012, 27(suppl 2), ii133-ii145. Provenzano, R.; Goodkin, D.; Klaus, S.; Linde, P.; Kazazi, F.; Lee, T.; Neff, T.; Yu, P. Evaluation of FG-4592, A Novel Oral Hypoxia- Hypoxia Inducible Factor Stabilization [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31] Inducible Factor Prolyl Hydroxylase Inhibitor, to Treat Anemia in Hemodialysis Patients. Am. J. Kidney Dis., 2011, 57(4), B80. Klaus, S.; Langsetmo, I.; Neff, T.; Lin, A.; Liu, D. Beneficial Pharmacodynamic Effects Resulting from 'Complete Erythro poiesis' Induced by Novel HIF Prolyl Hydroxylase Inhibitors FG2216 and FG-4592 J. Am. Soc. Nephrol., 2008, 19, 524A. McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de, B.B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993-2005. Public Health Nutr, 2009, 12(4), 444-454. Melnikova, I. Anaemia therapies. Nat. Rev. Drug Discovery, 2006, 5(8), 627-628. Balarajan, Y.; Ramakrishnan, U.; Ozaltin, E.; Shankar, A.H.; Subramanian, S.V. Anaemia in low-income and middle-income countries. Lancet, 2011, 378(9809), 2123-2135. Tolentino, K.; Friedman, J.F. An update on anemia in less developed countries. Am J Trop Med Hyg, 2007, 77(1), 44-51. Benoist, B.d.; McLean, E.; Egli, I.; Cogswell, M. Worldwide prevalence of anaemia 1993-2005: WHO global database on anaemia. World Health Organization: Switzerland, 2008. In: Quick look nursing: Pathophysiology; Madara, B.; PomaricoDenino, V., Ed.; Jones and Bartlett Publishers: Sudbury, 2008, 81104. In: Pharmacology; Finkel, R.; Clark, M.A.; Cubeddu, L.X., Ed.; Lippincott Williams & Wilkins: Philadelphia, 2009; pp. 229-248. Ludwig, H.; Oesterborg, A. In: Current Clinical Oncology: Biology and Management of Multiple Myeloma; Berenson, J.R., Editor; Humana Press Inc.: Totowa, 2004; pp. 303-318. Melosky, B.L. Erythropoiesis-stimulating agents: benefits and risks in supportive care of cancer. Curr Oncol, 2008, 15, S10-S15. Reichardt, B. Cost comparison of epoetin alpha, epoetin beta and darbepoetin alpha for cancer patients with anaemia in the clinical practice setting. J Clin Pharm Ther, 2006, 31, 503-512. Drueke, T.B.; Locatelli, F.; Clyne, N.; Eckardt, K.-U.; Macdougall, L.C.; Tsakiris, D.; Burger, H.-U.; Scherhag, A. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N. Engl. J. Med., 2006, 355, 2071-2084. Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D. Correction of anemia with epoetin alfa in chronic kidney disease. N. Engl. J. Med., 2006, 355, 2085-2098. Pfeiffer, M.A.; Burdmann, E.A.; Chen, C.-Y.; Cooper, M.E.; De, Z.D.; Eckardt, K.-U.; Feyzi, J.M.; Ivanovich, P.; Kewalramani, R.; Levey, A.S.; Lewis, E.F.; McGill, J.B.; McMurray, J.J.V.; Parfey, P.; Parving, H.-H.; Remuzzi, G.; Singh, A.K.; Solomon, S.D.; Toto, R. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N. Engl. J. Med., 2009, 361, 2019-2032. Kanbay, M.; Perazella, M.A.; Kasapoglu, B.; Koroglu, M.; Covic, A. Erythropoiesis stimulatory agent- resistant anemia in dialysis patients: review of causes and management. Blood Purif, 2010, 29, 1-12. Johnson, D.W.; Pollock, C.A.; MacDougall, I.C. Erythropoiesisstimulating agent hyporesponsiveness. Nephrology, 2007, 12, 321330. Macdougall, I.C. New anemia therapies: translating novel strategies from bench to bedside. Am J Kidney Dis, 2012, 59(3), 444-451. Rabinowitz, M.H.; Barrett, T.D.; Rosen, M.D.; Venkatesan, H. Inhibitors of HIF prolyl hydroxylases. Annu. Rep. Med. Chem., 2010, 45, 123-139. Beall, C.M. High-altitude adaptations. Lancet, 2003, 362 Suppl, s14-s15. Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol., 1992, 12(12), 5447-5454. Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem., 1995, 270(3), 12301237. Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxiainducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5510-5514. Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 versus HIF-2 - Is one more important than the other? Vasc. Pharmacol., 2012, 56(5-6), 245-251. Rosen, M.D.; Venkatesan, H.; Peltier, H.M.; Bembenek, S.D.; Kanelakis, K.C.; Zhao, L.X.; Leonard, B.E.; Hocutt, F.M.; Wu, X.; Current Medicinal Chemistry, 2013, Vol. 20, No. 1 [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [48] 13 Palomino, H.L.; Brondstetter, T.I.; Haugh, P.V.; Cagnon, L.; Yan, W.; Liotta, L.A.; Young, A.; Mirzadegan, T.; Shankley, N.P.; Barrett, T.D.; Rabinowitz, M.H. Benzimidazole-2-pyrazole HIF Prolyl 4-Hydroxylase Inhibitors as Oral Erythropoietin Secretagogues. ACS Med. Chem. Lett., 2010, 1(9), 526-529. Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von, K.A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Targeting of HIF- to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (Washington, DC, U. S.), 2001, 292(5516), 468-472. Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIF targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science (Washington, DC, U. S.), 2001, 292(5516), 464-468. Smirnova, N.A.; Hushpulian, D.M.; Speer, R.E.; Gaisina, I.N.; Ratan, R.R.; Gazaryan, I.G. Catalytic mechanism and substrate specificity of HIF prolyl hydroxylases. Biochemistry (Moscow), 2012, 77(10), 1108-1119. Hirota, S.A.; Beck, P.L.; MacDonald, J.A. Targeting hypoxiainducible factor-1 (HIF-1) signaling in therapeutics: implications for the treatment of inflammatory bowel disease. Recent Pat. Inflammation Allergy Drug Discovery, 2009, 3(1), 1-16. Ong, S.-G.; Hausenloy, D.J. Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol. Ther., 2012, 136(1), 69-81. Li, Y.; Ye, D. Cancer therapy by targeting hypoxia-inducible factor-1. Curr. Cancer Drug Targets, 2010, 10(7), 782-796. Zhang, Z.; Yan, J.; Chang, Y.; Yan, S.S.D.; Shi, H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr. Med. Chem., 2011, 18(28), 4335-4343. Hirota, K.; Semenza, G.L. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem. Biophys. Res. Commun., 2005, 338(1), 610-616. Zheng, K.Y.-Z.; Zhang, Z.-X.; Guo, A.J.-Y.; Bi, C.W.-C.; Zhu, K.Y.; Xu, S.L.; Zhan, J.Y.-X.; Lau, D.T.-W.; Dong, T.T.-X.; Choi, R.C.-Y.; Tsim, K.W.-K. Salidroside stimulates the accumulation of HIF-1 protein resulted in the induction of EPO expression: A signaling via blocking the degradation pathway in kidney and liver cells. Eur. J. Pharmacol., 2012, 679(1-3), 34-39. Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain: A hypoxic switch. Science (Washington, DC, U. S.), 2002, 295(5556), 858-861. Greer, S.N.; Metcalf, J.L.; Wang, Y.; Ohh, M. The updated biology of hypoxia-inducible factor. EMBO J., 2012, 31(11), 2448-2460. Yang, M.; Ge, W.; Chowdhury, R.; Claridge, T.D.W.; Kramer, H.B.; Schmierer, B.; McDonough, M.A.; Gong, L.; Kessler, B.M.; Ratcliffe, P.J.; Coleman, M.L.; Schofield, C.J. Asparagine and Aspartate Hydroxylation of the Cytoskeletal Ankyrin Family Is Catalyzed by Factor-inhibiting Hypoxia-inducible Factor. J. Biol. Chem., 2011, 286(9), 7648-7660. Dayan, F.; Monticelli, M.; Pouyssegur, J.; Pecou, E. Gene regulation in response to graded hypoxia: The non-redundant roles of the oxygen sensors PHD and FIH in the HIF pathway. J. Theor. Biol., 2009, 259(2), 304-316. Nagle, D.G.; Zhou, Y.-D. Natural product-derived small molecule activators of hypoxia-inducible factor-1 (HIF-1). Curr. Pharm. Des., 2006, 12(21), 2673-2688. Wanner, R.M.; Spielmann, P.; Stroka, D.M.; Camenisch, G.; Camenisch, I.; Scheid, A.; Houck, D.R.; Bauer, C.; Gassmann, M.; Wenger, R.H. Epolones induce erythropoietin expression via hypoxia-inducible factor-1 activation. Blood, 2000, 96(4), 15581565. Hattori, H.; Okuda, K.; Murase, T.; Shigetsura, Y.; Narise, K.; Semenza, G.L.; Nagasawa, H. Isolation, identification, and biological evaluation of HIF-1-modulating compounds from Brazilian green propolis. Bioorg. Med. Chem., 2011, 19(18), 53925401. Jeon, H.; Kim, H.; Choi, D.; Kim, D.; Park, S.-Y.; Kim, Y.-J.; Kim, Y.M.; Jung, Y. Quercetin activates an angiogenic pathway, hypoxia inducible factor (HIF)-1-vascular endothelial growth factor, by inhibiting HIF-prolyl hydroxylase: a structural analysis of quercetin for inhibiting HIF-prolyl hydroxylase. Mol. Pharmacol., 2007, 71(6), 1676-1684. 14 Current Medicinal Chemistry, 2013, Vol. 20, No. 1 [49] [50] [51] [52] [53] [54] [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] Hasebe, Y.; Egawa, K.; Yamazaki, Y.; Kunimoto, S.; Hirai, Y.; Ida, Y.; Nose, K. Specific inhibition of hypoxia-inducible factor (HIF)-1 activation and of vascular endothelial growth factor (VEGF) production by flavonoids. Biol. Pharm. Bull., 2003, 26(10), 1379-1383. Rafehi, H.; Ververis, K.; Balcerczyk, A.; Ziemann, M.; Ooi, J.; Hu, S.; Kwa, F.A.A.; Loveridge, S.J.; Georgiadis, G.T.; El-Osta, A.; Karagiannis, T.C. Investigation of the biological properties of Cinnulin PF in the context of diabetes: mechanistic insights by genome-wide mRNA-Seq analysis. Pathobiol. Aging Age-Relat. Dis., 2012, 2, 11905. Zhou, Y.-D.; Kim, Y.-P.; Li, X.-C.; Baerson, S.R.; Agarwal, A.K.; Hodges, T.W.; Ferreira, D.; Nagle, D.G. Hypoxia-Inducible Factor1 Activation by (-)-Epicatechin Gallate: Potential Adverse Effects of Cancer Chemoprevention with High-Dose Green Tea Extracts. J. Nat. Prod., 2004, 67(12), 2063-2069. Tsukiyama, F.; Nakai, Y.; Yoshida, M.; Tokuhara, T.; Hirota, K.; Sakai, A.; Hayashi, H.; Katsumata, T. Gallate, the component of HIF-inducing catechins, inhibits HIF prolyl hydroxylase. Biochem. Biophys. Res. Commun., 2006, 351(1), 234-239. Mecinovic, J.; Chowdhury, R.; Flashman, E.; Schofield, C.J. Use of mass spectrometry to probe the nucleophilicity of cysteinyl residues of prolyl hydroxylase domain 2. Anal. Biochem., 2009, 393(2), 215-221. Chowdhury, R.; McDonough, M.A.; Mecinovic, J.; Loenarz, C.; Flashman, E.; Hewitson, K.S.; Domene, C.; Schofield, C.J. Structural Basis for Binding of Hypoxia-Inducible Factor to the Oxygen-Sensing Prolyl Hydroxylases. Structure (Cambridge, MA, U. S.), 2009, 17(7), 981-989. Nakano, H.; Ikenaga, S.; Aizu, T.; Kaneko, T.; Matsuzaki, Y.; Tsuchida, S.; Hanada, K.; Arima, Y. Human metallothionein gene expression is upregulated by -thujaplicin: possible involvement of protein kinase C and reactive oxygen species. Biol. Pharm. Bull., 2006, 29(1), 55-59. Lee, M.J.; Kim, J.W.; Yang, E.G. Hinokitiol activates the hypoxiainducible factor (HIF) pathway through inhibition of HIF hydroxylases. Biochem. Biophys. Res. Commun., 2010, 396(2), 370-375. Chung, L.-C.; Tsui, K.-H.; Feng, T.-H.; Lee, S.-L.; Chang, P.-L.; Juang, H.-H. L-Mimosine blocks cell proliferation via upregulation of B-cell translocation gene 2 and N-myc downstream regulated gene 1 in prostate carcinoma cells. Am J Physiol Cell Physiol, 2012, 302(4), C676-685. Lee, J.-J.; Hsu, W.-H.; Yen, T.-L.; Chang, N.-C.; Luo, Y.-J.; Hsiao, G.; Sheu, J.-R. Traditional Chinese medicine, Xue-Fu-Zhu-Yu decoction, potentiates tissue plasminogen activator against thromboembolic stroke in rats. J Ethnopharmacol, 2011, 134(3), 824-830. Graziose, R.; Lila, M.A.; Raskin, I. Merging traditional chinese medicine with modern drug discovery technologies to find novel drugs and functional foods. Curr. Drug Discovery Technol., 2010, 7(1), 2-12. Gao, Q.T.; Cheung, J.K.H.; Choi, R.C.Y.; Cheung, A.W.H.; Li, J.; Jiang, Z.Y.; Duan, R.; Zhao, K.J.; Ding, A.W.; Dong, T.T.X.; Tsim, K.W.K. A chinese herbal decoction prepared from Radix Astragali and Radix Angelicae sinensis induces the expression of erythropoietin in cultured Hep3B cells. Planta Med., 2008, 74(4), 392-395. Bi, C.W.C.; Xu, L.; Zhang, W.L.; Zhan, J.Y.X.; Fu, Q.; Zheng, K.Y.Z.; Chen, V.P.; Lau, D.T.W.; Choi, R.C.Y.; Wang, T.J.; Dong, T.T.X.; Tsim, K.W.K. Fo Shou San, an ancient herbal decoction prepared from Angelicae Sinensis Radix and Chuanxiong Rhizoma, induces erythropoietin expression: a signaling mediated by the reduced degradation of hypoxia-inducible factor in cultured liver cells. Planta Med., 2012, 78(2), 122-127. Chang, M.S.; Kim, D.R.; Ko, E.B.; Choi, B.J.; Park, S.Y.; Kang, S.A.; Park, S.K. Treatment with Astragali Radix and Angelicae Radix Enhances Erythropoietin Gene Expression in the Cyclophosphamide-Induced Anemic Rat. J. Med. Food, 2009, 12(3), 637-642. Gaddipati, J.P.; Madhavan, S.; Sidhu, G.S.; Singh, A.K.; Seth, P.; Maheshwari, R.K. Picroliv - a natural product protects cells and regulates the gene expression during hypoxia/reoxygenation. Mol. Cell. Biochem., 1999, 194(1&2), 271-281. Zhang, J.; Liu, A.; Hou, R.; Zhang, J.; Jia, X.; Jiang, W.; Chen, J. Salidroside protects cardiomyocyte against hypoxia-induced death: Zhao and Wu [65] [66] [67] [68] [69] [70] [71] [72] [73] [74] [75] [76] [77] [78] [79] A HIF-1-activated and VEGF-mediated pathway. Eur. J. Pharmacol., 2009, 607(1-3), 6-14. Kalinowski, D.S.; Richardson, D.R. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev., 2005, 57(4), 547-583. Liu, Z.D.; Khodr, H.H.; Liu, D.Y.; Lu, S.L.; Hider, R.C. Synthesis, Physicochemical Characterization, and Biological Evaluation of 2(1'-Hydroxyalkyl)-3-hydroxypyridin-4-ones: Novel Iron Chelators with Enhanced pFe3+ Values. J. Med. Chem., 1999, 42(23), 48144823. Dobbin, P.S.; Hider, R.C.; Hall, A.D.; Taylor, P.D.; Sarpong, P.; Porter, J.B.; Xiao, G.; van, d.H.D. Synthesis, physicochemical properties, and biological evaluation of N-substituted 2-alkyl-3hydroxy-4(1H)-pyridinones: orally active iron chelators with clinical potential. J. Med. Chem., 1993, 36(17), 2448-2458. Baek, J.H.; Reiter, C.E.N.; Manalo, D.J.; Buehler, P.W.; Hider, R.C.; Alayash, A.I. Induction of hypoxia inducible factor (HIF-1) in rat kidneys by iron chelation with the hydroxypyridinone, CP94. Biochim. Biophys. Acta, Gene Regul. Mech., 2011, 1809(4-6), 262268. Flagg, S.C.; Martin, C.B.; Taabazuing, C.Y.; Holmes, B.E.; Knapp, M.J. Screening chelating inhibitors of HIF-prolyl hydroxylase domain 2 (PHD2) and factor inhibiting HIF (FIH). J. Inorg. Biochem., 2012, 113, 25-30. Barrett, T.D.; Palomino, H.L.; Brondstetter, T.I.; Kanelakis, K.C.; Wu, X.; Haug, P.V.; Yan, W.; Young, A.; Hua, H.; Hart, J.C.; Tran, D.-T.; Venkatesan, H.; Rosen, M.D.; Peltier, H.M.; Sepassi, K.; Rizzolio, M.C.; Bembenek, S.D.; Mirzadegan, T.; Rabinowitz, M.H.; Shankley, N.P. Pharmacological characterization of 1-(5chloro-6-(trifluoromethoxy)-1H-benzoimidazol-2-yl)-1H-pyrazole4-carboxylic acid (JNJ-42041935), a potent and selective hypoxiainducible factor prolyl hydroxylase inhibitor. Mol. Pharmacol., 2011, 79(6), 910-920. Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1 in normoxia. EMBO Journal, 2003, 22(16), 4082-4090. Warshakoon, N.C.; Wu, S.; Boyer, A.; Kawamoto, R.; Renock, S.; Xu, K.; Pokross, M.; Evdokimov, A.G.; Zhou, S.; Winter, C.; Walter, R.; Mekel, M. Design and synthesis of a series of novel pyrazolylpyridines as HIF 1- prolyl hydroxylase inhibitors. Bioorg. Med. Chem. Lett., 2006, 16, 5687-5690. McDonough, M.A.; Li, V.; Flashman, E.; Chowdhury, R.; Mohr, C.; Lienard, B.M.R.; Zondlo, J.; Oldham, N.J.; Clifton, I.J.; Lewis, J.; McNeill, L.A.; Kurzeja, R.J.M.; Hewitson, K.S.; Yang, E.; Jordan, S.; Syed, R.S.; Schofield, C.J. Cellular oxygen sensing: crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9814-9819. Warshakoon, N.C.; Wu, S.; Boyer, A.; Kawamoto, R.; Sheville, J.; Renock, S.; Xu, K.; Pokross, M.; Zhou, S.; Winter, C.; Walter, R.; Mekel, M.; Evdokimov, A.G. Structure-based design, synthesis, and SAR evaluation of a new series of 8-hydroxyquinolines as HIF-1 prolyl hydroxylase inhibitors. Bioorg. Med. Chem. Lett., 2006, 16, 5517-5522. Tegley, C.M.; Viswanadhan, V.N.; Biswas, K.; Frohn, M.J.; Peterkin, T.A.N.; Chang, C.; Buerli, R.W.; Dao, J.H.; Veith, H.; Rogers, N.; Yoder, S.C.; Biddlecome, G.; Tagari, P.; Allen, J.R.; Hungate, R.W. Discovery of novel hydroxy-thiazoles as HIF- prolyl hydroxylase inhibitors: SAR, synthesis, and modeling evaluation. Bioorg. Med. Chem. Lett., 2008, 18, 3925-3928. Frohn, M.; Viswanadhan, V.; Pickrell, A.J.; Golden, J.E.; Muller, K.M.; Buerli, R.W.; Biddlecome, G.; Yoder, S.C.; Rogers, N.; Dao, J.H.; Hungate, R.; Allen, J.R. Structure-guided design of substituted aza-benzimidazoles as potent hypoxia inducible factor1 prolyl hydroxylase-2 inhibitors. Bioorg. Med. Chem. Lett., 2008, 18, 5023-5026. Teli, M.K.; Rajanikant, G.K. Identification of novel potential HIFprolyl hydroxylase inhibitors by in silico screening. Mol. Diversity, 2012, 16(1), 193-202. Theriault, J.R.; Felts, A.S.; Bates, B.S.; Perez, J.R.; Palmer, M.; Gilbert, S.R.; Dawson, E.S.; Engers, J.L.; Lindsley, C.W.; Emmitte, K.A. Discovery of a new molecular probe ML228: An activator of the hypoxia inducible factor (HIF) pathway. Bioorg. Med. Chem. Lett., 2012, 22(1), 76-81. Denny, W.A. Giving Anemia a Boost with Inhibitors of Prolyl Hydroxylase. J. Med. Chem., 2012, 55(7), 2943-2944. Hypoxia Inducible Factor Stabilization [80] [81] [82] [83] [84] [85] [86] [87] [88] Current Medicinal Chemistry, 2013, Vol. 20, No. 1 Vachal, P.; Miao, S.; Pierce, J.M.; Guiadeen, D.; Colandrea, V.J.; Wyvratt, M.J.; Salowe, S.P.; Sonatore, L.M.; Milligan, J.A.; Hajdu, R.; Gollapudi, A.; Keohane, C.A.; Lingham, R.B.; Mandala, S.M.; DeMartino, J.A.; Tong, X.; Wolff, M.; Steinhuebel, D.; Kieczykowski, G.R.; Fleitz, F.J.; Chapman, K.; Athanasopoulos, J.; Adam, G.; Akyuz, C.D.; Jena, D.K.; Lusen, J.W.; Meng, J.; Stein, B.D.; Xia, L.; Sherer, E.C.; Hale, J.J. 1,3,8-Triazaspiro[4.5]decane2,4-diones as Efficacious Pan-Inhibitors of Hypoxia-Inducible Factor Prolyl Hydroxylase 1-3 (HIF PHD1-3) for the Treatment of Anemia. J. Med. Chem., 2012, 55(7), 2945-2959. Bugaut, A.; Jantos, K.; Wietor, J.-L.; Rodriguez, R.; Sanders, J.K.M.; Balasubramanian, S. Exploring the differential recognition of DNA G-quadruplex targets by small molecules using dynamic combinatorial chemistry. Angew. Chem., Int. Ed., 2008, 47, 26772680. Demetriades, M.; Leung, I.K.H.; Chowdhury, R.; Chan, M.C.; McDonough, M.A.; Yeoh, K.K.; Tian, Y.-M.; Claridge, T.D.W.; Ratcliffe, P.J.; Woon, E.C.Y.; Schofield, C.J. Dynamic Combinatorial Chemistry Employing Boronic Acids/Boronate Esters Leads to Potent Oxygenase Inhibitors. Angew. Chem., Int. Ed., 2012, 51(27), 6672-6675, S6672/1-S6672/53. Kwon, H.S.; Choi, Y.K.; Kim, J.W.; Park, Y.K.; Yang, E.G.; Ahn, D.-R. Inhibition of a prolyl hydroxylase domain (PHD) by substrate analog peptides. Bioorg. Med. Chem. Lett., 2011, 21(14), 4325-4328. Amelia, S.M. Hydroxypyridinone complexes with aluminum. In vitro/vivo studies and perspectives. Coord. Chem. Rev., 2002, 228(2), 187-203. Zhou, T.; Winkelmann, G.; Dai, Z.-Y.; Hider, R.C. Design of clinically useful macromolecular iron chelators. J. Pharm. Pharmacol., 2011, 63(7), 893-903. Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem., 2011, 46(10), 47694807. Murray, J.K.; Balan, C.; Allgeier, A.M.; Kasparian, A.; Viswanadhan, V.; Wilde, C.; Allen, J.R.; Yoder, S.C.; Biddlecome, G.; Hungate, R.W.; Miranda, L.P. Dipeptidyl-Quinolone Derivatives Inhibit Hypoxia Inducible Factor-1 Prolyl Hydroxylases-1, -2, and -3 with Altered Selectivity. J. Comb. Chem., 2010, 12, 676-686. Smirnova, N.A.; Rakhman, I.; Moroz, N.; Basso, M.; Payappilly, J.; Kazakov, S.; Hernandez-Guzman, F.; Gaisina, I.N.; Kozikowski, Received: ????? Revised: ????? Accepted: ????? [89] [90] [91] [92] [93] [94] [95] [96] [97] 15 A.P.; Ratan, R.R.; Gazaryan, I.G. Utilization of an In Vivo Reporter for High Throughput Identification of Branched Small Molecule Regulators of Hypoxic Adaptation. Chem. Biol. (Cambridge, MA, U. S.), 2010, 17(4), 380-391. Thirstrup, K.; Christensen, S.; Moller, H.A.; Ritzen, A.; Bergstroem, A.-L.; Sager, T.N.; Jensen, H.S. Endogenous 2oxoglutarate levels impact potencies of competitive HIF prolyl hydroxylase inhibitors. Pharmacol. Res., 2011, 64(3), 268-273. Minamishima, Y.A.; Kaelin, W.G., Jr. Reactivation of Hepatic EPO Synthesis in Mice After PHD Loss. Science (Washington, DC, U. S.), 2010, 329(5990), 407. Conejo-Garcia, A.; McDonough, M.A.; Loenarz, C.; McNeill, L.A.; Hewitson, K.S.; Ge, W.; Lienard, B.M.; Schofield, C.J.; Clifton, I.J. Structural basis for binding of cyclic 2-oxoglutarate analogues to factor-inhibiting hypoxia-inducible factor. Bioorg. Med. Chem. Lett., 2010, 20(20), 6125-6128. Cockman, M.E.; Webb, J.D.; Ratcliffe, P.J. FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Ann. N. Y. Acad. Sci., 2009, 1177(Hypoxia and Conseq uences), 9-18. McDonough, M.A.; McNeill, L.A.; Tilliet, M.; Papamicaeel, C.A.; Chen, Q.-Y.; Banerji, B.; Hewitson, K.S.; Schofield, C.J. Selective Inhibition of Factor Inhibiting Hypoxia-Inducible Factor. J. Am. Chem. Soc., 2005, 127(21), 7680-7681. Banerji, B.; Conejo-Garcia, A.; McNeill, L.A.; McDonough, M.A.; Buck, M.R.G.; Hewitson, K.S.; Oldham, N.J.; Schofield, C.J. The inhibition of factor inhibiting hypoxia-inducible factor (FIH) by oxocarboxylic acids. Chem. Commun. (Cambridge, U. K.), 2005(43), 5438-5440. Moon, H.; Han, S.; Park, H.; Choe, J. Crystal structures of human FIH-1 in complex with quinol family inhibitors. Mol. Cells, 2010, 29(5), 471-474. Ko, S.; Lee, M.K.; Shin, D.; Park, H. Structure-based virtual screening approach to the discovery of novel inhibitors of factorinhibiting HIF-1: Identification of new chelating groups for the active-site ferrous ion. Bioorg. Med. Chem., 2009, 17(22), 77697774. Buckley, D.L.; Van, M.I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1 Interaction. J. Am. Chem. Soc., 2012, 134(10), 4465-4468.

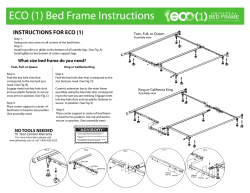








© Copyright 2026