
HEALTH EFFECTS OF LOW DOSES OF RADIATION
NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED NCRP DRAFT SC 1-21 COMMENTARY (Council and Board Draft) (3-31-15) HEALTH EFFECTS OF LOW DOSES OF RADIATION: INTEGRATING RADIATION BIOLOGY AND EPIDEMIOLOGY March 31, 2015 Note: Copyright permission is being sought for the figures and tables requiring such permission prior to their use in the final NCRP publication. National Council on Radiation Protection and Measurements 7910 Woodmont Avenue, Suite 400, Bethesda, Maryland 20814 1 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2 NOT TO BE DISSEMINATED OR REFERENCED Preface 3 4 In order to gain a greater understanding of the biological interactions and health effects of low 5 doses of ionizing radiation, the National Council on Radiation Protection and Measurements 6 (NCRP) has increased its activities on this subject in recent years. The NCRP 2008 Annual Meeting 7 was held on the subject of Low Dose and Low Dose-Rate Radiation Effects and Models (NCRP, 8 2009a). In December 2008 NCRP held a workshop that involved 30 participants with expertise in the 9 effects of low doses of radiation. After the workshop summary was completed,1 a decision was made 10 to convene a panel of experts to provide advice on a new NCRP Commentary related to critical 11 issues and research needs for gaining a better understanding of effects of low doses of radiation. The 12 advisory panel met in August 2010, and NCRP began the preparation of a Commentary in 2012. The 13 focus of the Commentary is on integration of results of basic science studies, including biomarkers 14 and bioindicators of cancer and other diseases, with epidemiologic studies on health effects of low 15 doses of radiation. The Committee members have expertise and experience in the following areas 16 related to low doses of ionizing radiation: epidemiology, radiation biology, radiation oncology, 17 biostatistics, health physics and dosimetry. 18 19 This Commentary was prepared by Scientific Committee 1-21 on Multiplatform National 20 Program for Providing Guidance on Integrating Basic Science and Epidemiological Studies on Low- 21 Dose Radiation Biological and Health Effects. 22 23 Serving on Scientific Committee 1-21 were: 24 25 26 27 28 29 30 31 32 Sally A. Amundson, Co-Chair Columbia University Medical Center New York, New York Jonine L. Bernstein, Co-Chair Memorial Sloan-Kettering Cancer Center New York, New York 1 NCRP (2008). National Council on Radiation Protection and Measurements. Developing a Framework for Incorporating New Information in the Radiation Sciences to Understanding Potential Health Risks for Low Doses of Ionizing Radiation: Summary of NCRP Workshop held on December 1-2, 2008 (National Council on Radiation Protection and Measurements, Bethesda, Maryland). 2 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED Members 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 John D. Boice, Jr. Vanderbilt University Nashville, Tennessee Raymond A. Guilmette Lovelace Respiratory Research Institute Albuquerque, New Mexico Amy Kronenberg Lawrence Berkeley National Laboratory Berkeley, California Mark P. Little National Cancer Institute Bethesda, Maryland William F. Morgan Pacific Northwest National Laboratory Richland, Washington Jac A. Nickoloff Colorado State University Fort Collins, Colorado Simon N. Powell Memorial Sloan-Kettering Cancer Center New York, New York Daniel O. Stram University of Southern California Los Angeles, California Consultant R. Julian Preston U.S. Environmental Protection Agency Research Triangle Park, North Carolina NCRP Secretariat Marvin Rosenstein, Staff Consultant (2013 –) Terry Pellmar, Staff Consultant (2012) Cindy L. O’Brien, Managing Editor Laura J. Atwell, Office Manager David A. Smith, Executive Director (2014 –) James R. Cassata, Executive Director (2012 – 2014) The Council expresses appreciation to the Committee members for the time and effort 51 devoted to the preparation of this Commentary. NCRP also gratefully acknowledges the financial 52 support provided by the Centers for Disease Control and Prevention. 53 54 John D. Boice, Jr. 55 President 56 3 NCRP SC 1-21 Draft of March 31, 2015 [MR] 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 NOT TO BE DISSEMINATED OR REFERENCED Contents Preface ............................................................................................................................................2 Executive Summary ......................................................................................................................6 1. Introduction ................................................................................................................................9 1.1 Low Doses and Low Dose Rates ......................................................................................11 1.2 Use of Radiation Biology Data to Reduce Uncertainty in Risk Estimates ........................12 2. General Approaches to Risk Assessment ..............................................................................19 2.1 Risk Assessment Process and Associated Uncertainties ...................................................19 2.2 Modeling Uncertainties: Dosimetric, Epidemiologic and Biologic ...................................20 2.3 Uncertainties Related to Transfer of Radiation Risk between Populations and Interactions with Carcinogens ...........................................................................................22 2.4 Extrapolating Low-Dose Effects from In Vitro to In Vivo; Animal to Human ................23 3. Studies Integrating Biology and Epidemiology ....................................................................27 3.1 Epidemiologic Studies Focused on Cancer Outcomes .....................................................27 3.1.1 Chromosome-Aberration Studies ............................................................................27 3.1.2 Atomic-Bomb Survivors ..........................................................................................28 3.1.3 Pelvic Irradiation Studies .........................................................................................30 3.1.4 Occupational Studies ...............................................................................................31 3.1.5 Environmental Background Studies ........................................................................33 3.1.6 Transgenerational Studies ........................................................................................34 3.1.7 Gene and Radiation Interaction: The Women’s Environmental Cancer and Radiation Epidemiology Study ................................................................................35 3.1.8 Retinoblastoma ........................................................................................................36 3.2 Epidemiologic Studies Focused on Noncancer Outcomes ...............................................37 4. Bioindicators from Radiation Biology Studies .....................................................................39 4.1 Deoxyribonucleic Acid Damage and Repair ....................................................................39 4.2 Cellular Signaling at Low Doses ......................................................................................40 4.3 Chromosome Alterations ..................................................................................................41 4.4 Mutations ..........................................................................................................................42 4.5 Radiation-Induced Genomic Instability ............................................................................46 4.6 Modulators of Response ...................................................................................................49 4.6.1 Adaptive Responses .................................................................................................49 4.6.2 Bystander Effects .....................................................................................................51 4.6.3 Genetic Susceptibility and Interactions with Radiation ...........................................52 4.6.4 Interactions with Environmental and Lifestyle Factors ...........................................53 4 NCRP SC 1-21 Draft of March 31, 2015 [MR] 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 NOT TO BE DISSEMINATED OR REFERENCED 5. Recommendations for Closing the Gaps towards an Integrated Approach ......................55 5.1 Targeted Research to Identify Adverse Outcome Pathways and Bioindicators of Response ...........................................................................................................................55 5.2 Biologically-Based Modeling ...........................................................................................57 5.2.1 Biologically-Based Dose-Response Approach ........................................................58 5.2.2 Systems Biology ......................................................................................................60 5.3 Registries of Medical-Imaging Exposures Linked to Outcomes and Biological Materials ............................................................................................................................62 5.4 Facilities and Interdisciplinary Training ...........................................................................63 5.4.1 Facilities ....................................................................................................................63 5.4.2 Interdisciplinary Training ........................................................................................63 Glossary .......................................................................................................................................64 Symbols, Abbreviations and Acronyms ....................................................................................70 References ....................................................................................................................................71 5 NCRP SC 1-21 Draft of March 31, 2015 [MR] 116 NOT TO BE DISSEMINATED OR REFERENCED Executive Summary 117 118 For decades, epidemiologic studies have assessed the health effects of radiation exposure from 119 multiple sources: occupational, accidental, environmental and medical. These efforts have been most 120 successful in evaluating radiation-related adverse effects (particularly cancer) at or above ~100 mGy 121 (absorbed dose). Since there are greater uncertainties inherent in epidemiologic studies of exposed 122 individuals at lower doses, it is generally agreed that the effects observed at 100 mGy and above are 123 the more reliable than those observed at <100 mGy. Despite a history of over 70 y, the field of 124 radiation risk estimation has not, or perhaps more correctly, has not been able to make much direct 125 use of radiation biology data from laboratory animal, cellular and molecular studies. Rather, such 126 data have been used extensively in a supportive role for the epidemiology-based risk estimates. 127 128 The current state of knowledge on the effects of low-dose and low dose-rate radiation (defined 129 for the purpose of this Commentary as a dose <100 mGy and a dose rate <5 mGy h–1) is incomplete 130 and uncertain with respect to understanding the shape of the dose-response relationship and the level 131 of risk at low doses. These deficiencies have long-term and profound consequences with regard to 132 radiation protection guidance, compensation programs, and environmental contamination issues. 133 There is a need to identify novel approaches to reduce these uncertainties. 134 135 The focus of this Commentary is on identifying further means to integrate results of basic 136 science studies in radiation biology, including biomarkers and bioindicators of cancer and other 137 diseases, with epidemiologic studies on health effects of low doses of radiation. A biomarker is a 138 biological phenotype (e.g., chromosome alteration, DNA adduct, gene expression change, specific 139 metabolite) that can be used to indicate a response at the cell or tissue level to an exposure. A 140 bioindicator is a cellular alteration that is on the pathway to the disease endpoint itself (a key event), 141 such as a specific mutation in a target cell that is associated with tumor formation. Towards this end, 142 the Commentary reviews general approaches to risk assessment (Section 2), existing epidemiologic 143 studies that have incorporated biological endpoints (Section 3), and areas of radiation biology with 144 potential relevance to low-dose risk (Section 4). 145 6 NCRP SC 1-21 Draft of March 31, 2015 [MR] 146 NOT TO BE DISSEMINATED OR REFERENCED Given major advances in our understanding of the etiology of diseases, host susceptibility, and 147 the cellular processes affected by radiation, coupled with the rapid development of new 148 technologies, there now is the opportunity to integrate information from multiple disciplines in risk 149 assessment. For example, radiation biology data can be incorporated into the process of 150 extrapolating from epidemiologic data at higher doses to predict responses at low doses (<100 mGy) 151 and low dose rates (<5 mGy h–1). The key to successfully doing this is to identify and select 152 appropriate endpoints that inform the process of extrapolation of risk from the higher doses to low 153 doses. Such biological endpoints utilized for extrapolation purposes need to be predictive of the 154 apical endpoint for which risk is being estimated (i.e., a bioindicator predictive specifically of 155 radiation-related cancer or noncancer outcome). While some approaches have been suggested, a key 156 problem is the lack of bioindicators (key events) that are specific for radiogenic disease. It is not yet 157 feasible to assess the robustness of key events in the radiation risk assessment process because too 158 little reliable information has been developed. 159 160 Biologically-based dose-response models have been suggested as a way forward for low-dose 161 risk estimation. However, developing such models requires a fuller understanding of the relevant 162 biology than we currently have, as well as the collection of sufficient data for parameterization of the 163 models. Awareness of such modeling needs should be integrated with experimental design, to ensure 164 the collection of relevant data. 165 166 Radiation-induced genomic instability is one example of a specific biological response that may 167 have relevance to modeling of low-dose radiation risk. It is important to determine if specific types 168 of genomic instability are capable of transforming a cell from a normal to a cancer cell, or furthering 169 the progression of a premalignant cell towards a more aggressive cancer phenotype. Chromosome 170 instability has been shown to persist for as long as a year, but there is little information concerning 171 how cells pass the unstable phenotype through succeeding somatic cell generations. 172 173 Another gap in our understanding of radiation risk is the role of individual variation in genetic 174 susceptibility and interaction with radiation. Clinical studies over the years have identified genetic 175 disorders and a few high penetrance alleles that convey increased risk of radiation-related cancer and 176 other endpoints. Such alleles are often demonstrated in studies of second cancers, but the magnitude 7 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 177 of effect is unknown and likely to vary. As each person is exposed to a different array of lifestyle 178 and environmental factors in addition to any low-dose radiation exposure, the potential impact of 179 interactions between these factors, genetic factors, and radiation exposure must also be considered. 180 181 The nascent field of systems biology, in which global information from multiple levels (e.g., 182 transcriptomic, proteomic, metabolomic) is integrated to develop a clearer picture of a process, for 183 example, response to radiation, may also prove informative for risk prediction. A systems level 184 understanding of radiation response could lead to a more comprehensive understanding of the 185 mechanisms involved, and the interactions between mechanisms, thus supporting more robust 186 biologically-based models of risk. 187 188 The following recommendations are made for closing the gaps towards an integrated approach of 189 basic science studies in radiation biology with epidemiologic studies on health effects of low doses 190 of radiation: 191 192 focus on key events (i.e., bioindicators) and modifying factors in adverse outcomes of 193 ionizing radiation exposure [as outlined in the parallelogram approach presented in 194 Figure 1.1 (for mechanistic studies) and Figure 1.3 (specific for bioindicators)], rather than 195 simple biomarkers of exposure; 196 197 198 develop biologically-based dose-response models to provide a path forward in low-dose radiation risk assessment; assemble registries of patients with extensive exposure to current and emerging medical- 199 imaging procedures (particularly those associated with higher patient doses) linked with 200 biological specimens and outcome data, in order to provide for standardized research 201 planning and data collection for prospective risk assessment; 202 203 204 maintain and develop the specialized facilities required for studies conducted at low doses and low dose rates; and promote and expand interdisciplinary training and integrated cross-professional research 205 programs devoted to understanding and quantifying radiation health effects at low doses in 206 order to meet the growing needs of radiation science for the next generation of radiation 207 researchers. 8 NCRP SC 1-21 Draft of March 31, 2015 [MR] 208 NOT TO BE DISSEMINATED OR REFERENCED 1. Introduction 209 210 We live in an era of increased public exposure to ionizing radiation, from the proliferation of its 211 use in diagnostic and interventional medical procedures to its nonmedical use for security screening. 212 With an increasing need for nuclear power, we also see continuing concern surrounding the public 213 exposures that may result from accidents, as dramatically illustrated by the recent Fukushima- 214 Daiichi disaster. In such an environment, meaningful risk assessment for low-dose and low dose-rate 215 ionizing radiation exposures is more critical than ever for the improvement of radiation protection, 216 environmental protection and remediation, and compensation. A fundamental issue in deriving risks 217 at low levels of dose and dose rate is the shape of the dose response for endpoints relevant to human 218 health outcomes and possible modifications by dose rate. This Commentary outlines a framework to 219 integrate data from biological studies with epidemiologic data to illuminate this issue. This 220 framework is broadly based on a parallelogram approach for mechanistic studies shown in 221 Figure 1.1 and made more specific by the use of bioindicators of response described in Figure 1.3 222 (Section 1.2). The bioindicators proposed are developed from an adverse outcome pathway/key 223 events approach initially described in Section 1.2 (Preston, 2015). 224 225 While epidemiologic studies remain the gold standard for determining risk and setting exposure 226 limits, a number of practical considerations limit the usefulness of such studies in the low-dose 227 range. In recent years, however, considerable progress has been made in low-dose radiation biology 228 at the cellular and molecular levels. Ideally, such work should contribute to our understanding of risk 229 and should help shape radiation protection guidance. In order to effectively integrate the results of 230 radiobiology studies into epidemiology and risk analysis, it is necessary to make multiple 231 comparisons, from cell and molecular studies to in vivo studies, and from animal to human on both 232 the cell and molecular level, and on the in vivo level (Figure 1.1). 233 234 Conceptually, this approach has proven useful particularly at the cellular level (e.g., Brewen 235 et al., 1973). The intercomparisons illustrated in Figure 1.1 are critical for developing a robust 236 analysis of the risk of human exposure to low-dose radiation. Studies with biological models that 237 more closely approximate tissue architecture are of particular use as a bridge between simple cellular 238 studies in vitro and organismal studies (Bissell et al., 1997). Application of extrapolation to the 9 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 239 240 241 242 Fig. 1.1. A parallelogram approach for utilizing laboratory animal and in vitro cell data to estimate human cancer and noncancer risks for ionizing radiation. 243 10 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 244 assessment of human risk has been reviewed in NCRP Report No. 150 (NCRP, 2005), which 245 concluded that an understanding of the molecular mechanisms of radiation carcinogenesis in 246 different animal species was needed for the most robust extrapolation. Although species-specific 247 differences in critical biological processes need to be kept in mind, a focus on conservation of 248 mechanism will be useful when making intercomparisons to inform risk in humans. 249 250 The major goal of this Commentary is to develop a framework to support the useful 251 incorporation of biological endpoints into epidemiologic studies in order to enhance our 252 understanding of the risks of low-dose and low dose-rate ionizing radiation exposures. 253 254 Towards this end, this Commentary reviews general approaches to risk assessment, areas of 255 radiation biology with potential relevance to low-dose risk, and existing epidemiologic studies that 256 have incorporated biological endpoints. The Commentary suggests that focusing on key events and 257 modifying factors for adverse outcomes pathways from ionizing radiation exposure, rather than 258 simple biomarkers of exposure, in concert with the development of biologically-based dose-response 259 models will provide the best path forward in low-dose radiation risk assessment. Finally, we identify 260 the major gaps in knowledge that need to be addressed to enable the integration of basic biology and 261 epidemiologic studies for the advancement of low-dose ionizing radiation risk assessment and 262 radiation protection. 263 264 1.1 Low Doses and Low Dose Rates 265 266 Studies have been conducted over a broad range of radiation doses and dose rates referred to as 267 low, and what constitutes a low dose and a low dose rate is often due to the context in which dose 268 and dose rate are being considered (e.g., radiobiological research, epidemiologic studies, medical 269 exposure, environmental exposure). For the purposes of this Commentary, the context is the levels 270 experienced by individuals or populations to various sources of ionizing radiation, and single values 271 of low dose and low dose rate are useful for general discussion. 272 273 In assessing human health effects (in particular cancer), ICRP (1991) applied a DDREF of two to 274 obtain risk estimates for absorbed doses below 200 mGy and from higher doses when the dose rate is 11 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 275 <100 mGy h–1. The U.K. National Radiological Protection Board (NRPB) concluded that for the 276 purposes of applying a dose-rate effectiveness factor in radiation protection, dose rates of 277 <0.1 mGy min–1 (when averaged over ~1 h) and acute doses of <100 mGy may be regarded as low 278 (Muirhead et al., 1993). This dose rate is equivalent to 6 mGy h–1. Wakeford and Tawn (2010) more 279 recently agreed with the NRPB guidance and suggested that a low dose is <100 mGy delivered 280 acutely and a low dose rate is <5 mGy h–1. The rationale for selecting such dose rates appears to be 281 driven by the desire to link them to what is believed to be the linear portion of a dose-response 282 relationship. A dose of <100 mGy and a dose rate of <5 mGy h–1 are used for low doses and low 283 dose rates in this Commentary. 284 285 It is recognized that these specified values for low dose and low dose rate are most relevant to 286 exposure scenarios related to occupations in which sources of radiation and radioactive material are 287 used. In comparison, ubiquitous background dose rates tend to occur at levels of ~1 µGy h–1, which 288 is a factor of 5,000 less than the value of 5 mGy h–1 used here. Additionally, regulatory limits for 289 doses to members of the public from environmental radiation sources are typically set at levels that 290 equate to a fraction of the ubiquitous background dose rate [e.g., the NRC limit for members of the 291 public from nuclear reactor sites is 1 mSv y–1 (total effective dose equivalent) or 0.11 µSv h–1]. 292 These dose rates assume chronic exposure to relatively constant radiation fields. In any case, it is 293 difficult to discern whether differences in dose rate on the order of 103 to 104 can be understood in 294 terms of mechanisms of radiation action, given our current state of knowledge. 295 296 1.2 Use of Radiation Biology Data to Reduce the Uncertainty in Risk Estimates 297 298 There is an extensive literature on the health effects of radiation exposures (occupational, 299 accidental, environmental and medical) in human populations. Many of these involve relatively high 300 acute doses, although associations at lower doses (around a few tens of milligray) and lower dose 301 rates have been reported. Given the greater uncertainties inherent in epidemiologic studies of 302 exposed individuals at low doses than at higher doses, it is generally agreed that effects observed at 303 ~100 mGy and above are the more reliable than those observed at < 100 mGy. In this regard, despite 304 a history of over 70 y, the field of risk estimation has not, or perhaps more correctly, has not been 305 able to make much direct use of radiation biology data from laboratory animal, cellular and 12 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 306 molecular studies. Rather such data have been used in a more supportive role for the epidemiology- 307 based risk estimates. 308 309 Risk estimates for radiation-related cancer and noncancer effects at low doses (chronic and 310 acute) have relied almost exclusively on epidemiologic data. Exceptions are for the estimation of 311 DDREF and relative biological effectiveness of high-LET radiation that have relied quite 312 significantly on data from biological studies, although also supported by epidemiologic data. Given 313 the major advances in our understanding of the etiology of chronic diseases (especially cancer, 314 although with an increasing emphasis also on certain noncancer diseases) and of the fundamental 315 cellular processes affected by radiation (both targeted and nontargeted), it is prudent to consider how 316 this information can be incorporated into the risk assessment process. In particular, it is critical that 317 the use of data from biological studies be incorporated into the process for extrapolating from 318 epidemiologic data at medium/high doses to predict responses at low doses (<100 mGy) and low 319 dose rates (<5 mGy h–1) (Section 1.1). Given these limitations, two issues to be addressed herein are: 320 321 322 323 324 how can informative endpoints be selected that can enhance the process of extrapolation of “risk” from high/medium to low doses; and how can data from laboratory animal, cellular and molecular studies best be integrated with epidemiologic studies to better predict risk. 325 326 Assistance with the task of estimating the health effects of radiation exposures at low doses and 327 low dose rates can be found in the approach being developed for estimating risks from exposures to 328 chemicals (e.g., Boobis et al., 2006; EPA, 2005; Julien et al., 2009; Preston, 2015; Seed et al., 2005; 329 Thomas et al., 2012; Vinken, 2013). The use of mechanistic and other biological data is necessary 330 for the risk assessment process for chemicals because of the lack of epidemiologic data for almost all 331 chemicals (environmental or occupational exposure). The process is built upon a framework of 332 adverse outcome pathways and key events for the development of adverse health effects following 333 exposure to a chemical or chemical mixture. An adverse outcome pathway for both cancer and 334 noncancer endpoints is defined as a sequence of key events and processes starting with interaction of 335 an agent with a cell, proceeding through operational and anatomical changes, and resulting in 336 formation of an adverse outcome (EPA, 2005). It should be noted that this adverse outcome 13 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 337 pathway/key event approach has not been used for an entire risk assessment process for any 338 chemical. A worked example has recently been developed by Adeleye et al. (2014). 339 340 For ionizing radiation, for example, cancer is considered to be produced by a DNA-reactive 341 mode of action. A key event is defined as an empirically observable precursor step that is itself a 342 necessary element of the mode of action or is a biologically based marker for such an element (EPA, 343 2005). Preston (2015) has recently presented a proposed adverse outcome pathway and associated 344 key events for low doses and low dose rates of ionizing radiation. It is more difficult to utilize such a 345 general approach for noncancer effects given the wide range of mechanisms whereby adverse 346 outcomes (e.g., neurological, reproductive, cardiovascular) can be produced. However, an initial 347 approach is described by Seed et al. (2005) for a range of chemical classes and noncancer endpoints. 348 349 Preston and Williams (2005) developed a set of key events by which a DNA-reactive chemical 350 can produce a metastatic cancer. A modified version of this scheme can be considered as being 351 applicable to ionizing radiation. As a starting point for the development of an adverse outcome 352 pathway for radiation-related cancer, an initial key event is considered to be a DNA double-strand 353 break, if in turn it can be converted into a mutation. The set of key events in Figure 1.2 is a linear 354 progression from the initial key event to a malignant tumor (i.e., an adverse outcome pathway) 355 (Preston, 2015). It is possible to expand this linear approach to include radiation effects on 356 preneoplastic lesions that may have developed prior to exposure. Another approach considers 357 radiation effects on any of the critical processes that are considered hallmarks of cancer development 358 (Boss et al., 2014; Hanahan and Weinberg, 2011). 359 360 For the purpose of maximizing the use of biological data in the dose-response assessment for 361 adverse health outcomes, it is informative to distinguish between biomarkers and bioindicators. A 362 biomarker is a biological phenotype (e.g., chromosome alteration, DNA adduct, gene expression 363 change, specific metabolite) that can be used to indicate a response to an exposure at the cell or 364 tissue level. In this regard, it is generally a measure of the potential for development of an adverse 365 outcome such as cancer (e.g., a predictor of exposure level). A bioindicator is defined as a cellular 366 alteration that is on the pathway to the disease endpoint itself, such as a specific mutation in a target 367 cell that is associated with tumor formation. Thus, a bioindicator can be perceived as informing on 14 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 368 Adverse Outcome Pathway Steps Interaction with Radiation Energy Deposition Macro-Molecular Alterations Key Events Exposure of Target Tissue Single, double and multiple DNA breaks Base Modifications Protein Oxidation Free Radical Formation Chromosome Alterations Gene Activation Protein Production Altered Signaling Cell killing and Tissue Disruption Organ Responses Altered Physiology Disrupted Homeostasis Altered Tissue Development/Function Adverse Outcome Impaired Development Impaired Reproduction Cancer and Noncancer Effects Cellular Responses 369 370 Fig. 1.2. Schematic representation of an adverse outcome pathway for ionizing radiation-related 371 cancer and noncancer diseases showing each step along the proposed pathway and the associated key 372 events (Preston, 2015). 373 15 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 374 the shape of dose-response curve for the disease outcome or on cancer frequency itself, and 375 therefore, is equivalent to a key event. In a logical way, the more proximate a key event/bioindicator 376 is to the apical endpoint (e.g., cancer), the more predictive of this outcome it will be. Thus, for the 377 purposes of risk assessment, it is most pertinent to design research studies to assess bioindicators of 378 response rather than simply biomarkers of an exposure. In addition, it is essential to describe the 379 predicted human relevance of key events/bioindicators developed in nonhuman assay systems 380 (Boobis et al., 2006). Figure 1.1 can be modified (Figure 1.3) to show how bioindicators can be used 381 as the data input for a typical parallelogram approach (Sobels, 1993). The types of informative 382 bioindicators for cancer risk assessment include: cancer-specific (target tissue) chromosome 383 alterations and gene mutations, target tissue cell killing and regenerative cell proliferation, and target 384 tissue inflammatory responses. Genovese et al. (2014) has provided data that suggest that “a subset 385 of genes that are mutated in patients with myeloid cancers is frequently mutated in apparently 386 healthy persons; these mutations may represent characteristic early events in the development of 387 hematologic cancers.” This type of DNA sequencing approach can be extended to investigate other 388 389 390 cancer types. 391 and/or modulating factors can influence the nature of the overall response for both cancer and 392 noncancer outcomes. Consideration of such events and factors is particularly important for 393 394 395 quantitative assessments of dose response. 396 host factors that can modulate the dose-response relationship of one or more key events, thus altering 397 the probability or magnitude of the adverse outcome. As such, some modulating factors might be 398 considered as “inevitable” (fixed) because all humans are subject to their influence; examples are 399 sex, age, and ethnicity. Other modulating factors are more aptly described as “optional” 400 (controllable): these include smoking, diet, and occupation. It is relatively straightforward to 401 incorporate the inevitable modulators into a risk assessment process but much more problematic to 402 adequately account for the optional ones without considering individual risk values. Laboratory 403 animal and cellular and molecular data might be developed that can account for the magnitude of the 404 influence of modulating factors on risk but currently this is very rarely done and frequently it is not 405 possible. Key events provide the critical input for dose-response modeling, so-called associated events A modulating factor can be considered as a biological feature, including control mechanisms or 16 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 406 407 408 409 410 Fig. 1.3. Modified Figure 1.1, showing how bioindicators can be used as the data input for a parallelogram approach (Sobels, 1993). 411 17 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 412 413 An associative event is a biological process that is not itself a key event but is a reliable 414 biomarker for a key event(s). Thus, an associative event can be a biomarker for exposure or closely 415 linked to the mechanism of disease formation. Recent research has identified two novel classes of 416 events, epigenetic changes and nontargeted events [e.g., bystander effects (Section 4.6.2) and 417 genomic instability (Section 4.5)] that are sometimes considered as associative events. However, at 418 present it is not clear whether epigenetic events are associative, but based on the evidence to date it 419 appears unlikely that nontargeted events are associative, since not all radiation exposures are 420 associated with instability in human subjects (Tawn et al., 2000; 2011; 2015; Whitehouse and Tawn, 421 422 423 2001). 424 health effects, it is necessary to utilize or develop some form of a biologically-based dose-response 425 model. Several approaches that have been described suggest the viability of this type of modeling 426 (Eidemuller et al., 2011; Kaiser et al., 2014; Little, 2010; Luebeck et al., 2013; Moolgavkar and 427 Knudson, 1981; Moolgavkar and Luebeck, 1992; Moolgavkar and Venzon, 1979; Shuryak et al., 428 2010), although perhaps limited in their value to reliably predict cancer risk because they do not 429 incorporate any biological data other than the generalized mutational evidence supporting the 430 multistage model. Some recent approaches have perhaps provided a greater reliance on auxiliary 431 biological data (Heidenreich and Rosemann, 2012; Heidenreich et al., 2013). The further 432 development of truly informative models along these lines has to be a research priority given the 433 importance of accurate estimates of radiation-related health effects at low doses and low dose rates. To fully use such key event/bioindicator data to inform dose-response assessment for adverse 434 435 The discussion herein describes an approach for integrating data from radiation biology and 436 epidemiology in the dose-response assessment phase of risk assessment for radiation-related cancer 437 and noncancer effects (Morgan and Bair, 2013). It proposes the development and application of 438 mode of action/key events/bioindicators as providing a link between epidemiology and radiation 439 biology data at the animal, cell and molecular levels. 440 18 NCRP SC 1-21 Draft of March 31, 2015 [MR] 441 442 443 NOT TO BE DISSEMINATED OR REFERENCED 2. General Approaches to Risk Assessment 2.1 Risk Assessment Process and Associated Uncertainties 444 445 Risk estimates for radiation-related cancer mortality and incidence and those for noncancer 446 diseases can play a critical role in the development of dose limits used for radiation protection 447 purposes. A recent example of the use of risk estimates for cancer is provided by the approach for 448 establishing the standards used by NASA for limits on days in space for astronauts (Cucinotta et al., 449 2013; NA/NRC, 2012). The estimates of radiation risk currently used by international and national 450 bodies such as the International Commission on Radiological Protection (ICRP), the United Nations 451 Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) , the National 452 Academies/National Research Council (NA/NRC), National Council on Radiation Protection and 453 Measurements (NCRP), and the U.S. Environmental Protection Agency (EPA) rely heavily upon 454 epidemiologic data on cancer and noncancer diseases in a variety of exposed populations 455 (particularly atomic-bomb survivors and people with estimated or known doses from occupational, 456 medical and environmental exposures). The general approach to risk assessment is to extrapolate 457 from health effects measured at medium to high doses to predict those expected at low doses. 458 Currently, for cancer this extrapolation utilizes a linear-nonthreshold model (ICRP, 2007; NA/NRC, 459 2006; NCRP, 2001). The estimates obtained in this way are corrected for a DDREF to allow for 460 calculated reductions in effect from high to low dose and from high to low dose rate. There are 461 challenges to this approach largely because of the reliance on the linear-nonthreshold model and the 462 uncertainty in a chosen value for DDREF. For noncancer diseases, the approach employed by ICRP 463 (2007; 2012) is to calculate a dose for a practical threshold level of disease response (<1 % chance of 464 occurrence) (ICRP, 2012) based on effects at medium to high doses. Because of the reliance on 465 epidemiologic data, it is extremely important to utilize the most recent and relevant data for 466 developing risk estimates and to take advantage of enhancements in computational modeling 467 approaches. However, at the same time, it is essential to appreciate that any approach used will 468 inevitably have associated uncertainties that should be considered when applying risk estimates for 469 protection purposes. NCRP Report No. 171 (NCRP, 2012) considers the types and magnitude of the 470 several uncertainties that are a component of the risk assessment process for cancer, noncancer 471 effects and heritable effects following radiation exposure. Consideration of this issue is timely 19 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 472 because new data have recently become available for cancer incidence, noncancer occurrence 473 (particularly for cataracts and cardiovascular disease) and heritable effects (reviewed in ICRP, 2007; 474 2012; UNSCEAR, 2001). NCRP (2012) builds upon the analyses in other recent NCRP reports 475 (NCRP, 2007; 2009b; 2009c) of the sources and magnitude of uncertainties in the estimation of 476 organ doses from exposure to external and internal sources of radiation. Given the heavy reliance on 477 epidemiologic studies, a significant component of uncertainty associated with these risk estimates 478 will be accounted for by comprehensive assessments of uncertainties in these epidemiological 479 studies. It is of note that little direct use has been made of the extensive data from ~70 y of 480 radiobiological study in estimating radiation-related health risks (ICRP, 2007; NA/NRC, 2006; 481 NCRP, 2005); the data have generally provided support for the epidemiology-based approach 482 currently used. The most significant uses of laboratory animal and cellular data have been for 483 developing a value for DDREF and values for quality factors and radiation weighting factors used in 484 radiation protection. However, even with the use of these experimental data, uncertainties associated 485 with these factors are large, and contribute significantly to the overall uncertainty in risk estimates 486 (Cucinotta et al., 2013). Sections 2.2 through 2.4 consider the types of uncertainty, their magnitude 487 488 489 490 491 and potential approaches for reducing these uncertainties. 492 (2012), the precision of epidemiologic risk estimates is driven primarily by the: 2.2 Modeling Uncertainties: Dosimetric, Epidemiologic and Biologic Epidemiology is by its nature observational rather than experimental. As discussed in NCRP 493 494 range and distribution of doses; 495 sample size; 496 duration of and ages at observation; 497 baseline frequencies of the health endpoint of interest; 498 “true” strength of the radiation-disease association; 499 various types of dose uncertainties; and 500 degree of accuracy of ascertainment (or, conversely, misclassification or underascertainment) 501 of the disease of interest. 502 20 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 503 However, the degree of systematic errors (i.e., bias) of the epidemiologic risk estimates depends on a 504 different set of factors, such as: 505 506 507 systematic personal reporting errors (with respect to dose-related items, disease experience, or risk-factor covariates); 508 insufficient statistical adjustment for other risk factors; 509 dose-related inequalities in disease ascertainment; 510 errors in assigning average values for shared dosimetry factors (e.g., an incorrect estimate of 511 the magnitude of a radioactive release); 512 failure to correct for individual measurement errors; or 513 failure to adjust for the effects of disease-related covariates. 514 515 The importance of these sources of uncertainty varies from study to study. 516 517 Uncertainties and biases resulting from design and methodologic issues in epidemiologic studies 518 have been considered by a number of scientific committees (NCRP, 2012; UNSCEAR, 2008a). Of 519 special importance are biases, that is, any process at any stage in the conduct of the study that tends 520 to produce results or conclusions that differ systematically from the truth (Sackett, 1979), and 521 include: 522 523 follow-up bias: 524 ascertainment bias: 525 recall bias; and 526 confounding. 527 528 Statistical methods to deal with the complexities of measurement and ascertainment error corrections 529 are still evolving (Carroll et al., 2006). A new generation of epidemiologic studies that incorporate 530 biologic measures for estimating radiation risk will help sharpen assessments and correct for some 531 uncertainties and biases. 532 21 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 533 2.3 Uncertainties Related to Transfer of Radiation Risk between Populations 534 535 536 and Interactions with Carcinogens Despite the relatively large number of data on radiation risk, the question of how to transfer risk 537 estimates derived from one population to a different population remains unanswered. It should not 538 be surprising that the relationship between radiation-related and underlying risk in different 539 populations is not consistent for different cancer sites. The available data suggest that there is no 540 541 542 simple solution to the problem (Muirhead et al., 1993; UNSCEAR, 1994), 543 transfer of risk between populations, or indeed the use of some sort of hybrid approach, such as that 544 employed by Little et al. (1999a) and Muirhead and Darby (1987), may be appropriate. The principal 545 scientific committees implicitly assume that risk transfer is intermediate between additive and 546 multiplicative (ICRP, 2007; NA/NRC, 2006). The subject is discussed in NCRP (2012), highlighting 547 the complexity of the evidence, so that for example for breast cancer a more nearly additive transfer 548 549 550 was suggested, whereas for liver cancer relative risk transfer was more likely to be valid. 551 exposed group to another, it is necessary to also consider the influence of factors other than radiation 552 on the cancer rates. Thus, much of environmental, nutritional and occupational cancer epidemiology 553 is concerned with identifying risk factors that might account for some part of the variation of site- 554 specific underlying cancer rates among populations. While there has been much progress, the 555 problem is complex and there is only limited information on the interaction between radiation dose 556 and lifestyle, or constitutional factors, in terms of cancer risk. Thus it is likely that, for the 557 foreseeable future, the most useful information relevant to transferring radiation-related risk 558 coefficients from one population to another will come from multinational comparisons of site- 559 specific radiation-related risk, rather than from investigations of underlying cancer risk factors and 560 their interactions with radiation dose. There has been some work comparing aggregate radiation- 561 associated cancer mortality risks in humans and in various animals (Carnes et al., 2003), suggesting 562 that a simple scaling of radiation-related mortality may be appropriate. However, without more 563 detailed investigation taking account of the different spectrum of tumors in humans and in these 564 experimental systems, and their distinct etiology, this approach may not be very productive. The available information suggests that, depending on circumstances, relative or absolute When considering the issue of how to transfer site-specific cancer rates from one population or 22 NCRP SC 1-21 Draft of March 31, 2015 [MR] 565 566 NOT TO BE DISSEMINATED OR REFERENCED Sometimes a meta-analysis, based on published findings from several studies, may be performed. 567 Where feasible, as noted below, it is preferable to combine the original data and analyse them using 568 a common format, in other words to perform a pooled analysis. Pooled analyses have been 569 conducted of various cohorts of radiation workers (Cardis et al., 2007) to assess: 570 571 572 effects of radon daughter exposure in relation to lung cancer risk in underground miners (UNSCEAR, 2000); 573 thyroid cancer risk in various (mainly medically exposed) cohorts (Ron et al., 1995); and 574 breast cancer risk in various populations (Howe and McLaughlin, 1996; Little and Boice, 575 1999; Preston et al., 2002). 576 577 Less commonly, analyses combining cohort and case-control data, for example in relation to 578 579 580 581 582 leukaemia risk (Little et al., 1999b), have been conducted. 583 events that occur in response to acute moderate to high doses of radiation, including activation of 584 DNA damage checkpoints, DNA repair pathways, mutagenesis, genomic instability, cell 585 transformation, and the regulation of cell death pathways (Hall and Giaccia, 2011). The radiation- 586 induced genetic changes observed in cells in vitro are similar to those observed in radiation-induced 587 tumors in animal models. As a result, studies of model genetic loci are frequently extrapolated to 588 cancer relevant genetic changes seen in animals and humans. Such genetic events include oncogene 589 activation via chromosome translocation or gene amplification and inactivation of tumor suppressor 590 genes via point mutations or larger scale changes such as deletions, insertions and inversions. 2.4 Extrapolating Low-Dose Effects from In Vitro to In Vivo; Animal to Human Several decades of laboratory research have given us a detailed view of molecular and cellular 591 592 The complexities of extrapolation between animal models and humans has been reviewed in 593 depth in NCRP Report No. 150 (NCRP, 2005), which concluded that studies of life shortening in 594 animals may provide reliable estimates of radiation risks to humans. The strength of life shortening 595 data is that it integrates more completely the detrimental effects of radiation in the entire animal, 23 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 596 rather than focusing on just one endpoint such as cancer induction. NCRP (2005) further concluded 597 that risk extrapolation from animal models to humans will: 598 599 600 require robust, well annotated databases that must be widely disseminated to investigators interested in radiation risk estimation; 601 include experimental data from animal studies and data from human epidemiologic studies; 602 further develop our understanding of molecular mechanisms of cancer and other disease 603 604 605 induction; and account for effects of age and genetic heterogeneity (or lack thereof) of animal models versus humans. 606 607 While the benefits of life shortening studies are clear, the most recent study that validated risk 608 estimates for humans using life shortening measures in mice and dogs demonstrated that there was 609 little effect on life span with acute radiation doses <1 Gy (Carnes et al., 2003). Earlier studies of life 610 shortening in mice indicated that acute radiation doses reduced life span by ~28 d Gy–1 (Grahn, 611 1960). However, with protracted, low dose-rate exposures (<0.1 Gy d–1), life shortening was greatly 612 reduced to ~4 d Gy–1 (Grahn and Sacher, 1968). Therefore, despite the advantages of life shortening 613 as an integrated endpoint of the detrimental effects of radiation, this approach does not appear to 614 have sufficient sensitivity to detect low-dose or low dose-rate effects on animal life span, and even 615 with strong tools to extrapolate risk from animal models to humans (Carnes et al., 2003) this lack of 616 sensitivity poses problems when attempting to use these animal data to extrapolate human risk to 617 618 619 low-dose or low dose-rate radiation exposures. 620 many challenges due to significant environmental and genetic differences among cells in culture 621 versus cells in animals or humans. For example, long-term studies of cells in culture have 622 historically focused on cancer cell lines, which are not only different from the normal cells from 623 which they were derived, but are often genetically unstable. Studies of normal (primary) cells in 624 culture are typically limited to ~50 cell divisions, and immortalization by expression of telomerase 625 reverse transcriptase (TERT, hTERT in humans) to maintain telomeres, introduces another departure 626 from normal tissues. Cells in culture are generally studied as monolayers, which is far different from Extrapolations of in vitro cell culture results to animals and to human cancer risks also pose 24 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 627 their in vivo three dimensional (3D) organization. The development of 3D artificial tissue models 628 has provided an important experimental bridge between traditional monolayer models and animal 629 models, and there is clear evidence of differential signaling and other cellular responses to radiation 630 in two dimensional (2D) monolayer versus 3D tissue models (Barcellos-Hoff and Costes, 2006). For 631 example, 3D tissue models have been used to investigate transcriptome and secretome changes in 632 response to low- versus high-dose radiation (Mezentsev and Amundson, 2011; Zhang et al., 2014), 633 and there is evidence of reduced high-dose radiation cytotoxicity in long-term 3D cultures versus 2D 634 cultures, reflecting differential apoptotic responses in 2D versus 3D cultures despite similar effects 635 on checkpoint delay (Sowa et al., 2010). In addition, cell signaling in vitro may also differ markedly 636 from the behavior of the parent cell in its normal tissue environment within an animal, where 637 638 639 communication among cells of different types may contribute to the response to radiation. 640 understanding low-dose radiation effects in humans, but inbred mice may not accurately reflect 641 effects in outbred human populations with diverse histories of radiation (and other genotoxin) 642 exposures, diet and lifestyles. There is clear evidence of variation in radiation responses in human 643 cells [e.g., cells that are heterozygous for ataxia-telangiectasia mutated (ATM), or the retinoblastoma 644 susceptibility gene (Fernet et al., 2004; Kato et al., 2009; Wilson et al., 2010a)], but at least in mice, 645 ATM heterozyosity does not enhance radiation carcinogenesis (Mao et al., 2008). The idea that low- 646 dose ionizing radiation may have significantly different effects in outbred populations is further 647 supported by a study of metabolic markers in outbred mice exposed to 100 mGy ionizing radiation 648 (Lee Do et al., 2012), and a study of mammary tissue responses to four weekly doses of 75 mGy 649 (Snijders et al., 2012). Similarly, Okayasu et al. (2000) and Yu et al. (2001) demonstrated clear 650 differences in (high-dose) radiation carcinogenesis in different strains of mice, tracing the effects to 651 hypomorphic mutations in the DNA-PKcs gene, a key factor in nonhomologous end joining repair of 652 ionizing radiation-induced double-strand breaks (DSBs). As rodents express DNA-PKcs at ~20-fold 653 lower levels than humans, this highlights yet another point to be considered when extrapolating 654 655 656 experimental results in mice to assess risk in humans. 657 mechanism. For instance, transgenerational effects have been seen in progeny of whole-body 658 irradiated male mice (Barber et al., 2002; Carls and Schiestl, 1999) but there is little evidence of Animal studies represent a critical translational bridge between in vitro studies and Extrapolation of risk from animal models to humans depends on conservation of the underlying 25 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 659 transgenerational effects in exposed human populations (Dauer et al., 2010; Little et al., 2013), 660 suggesting that at least for this endpoint, there are fundamental mechanistic differences between 661 animal models and humans. Particularly pertinent to extrapolation of risk from animals to humans 662 with regard to effects of low doses and low dose rates are inverse dose-rate effects, which have been 663 described in several species (mouse, rat, human cells) and several endpoints (cytotoxicity, 664 mutagenesis, tumor induction) (Brenner, 1994; Lubin et al., 1995; Spiess and Mays, 1973; Ullrich, 665 1984), characterized by greater biological effects of equal doses delivered at low versus high dose 666 rates. While data on inverse dose-rate effects are limited, these effects have been explained in terms 667 of differential checkpoint responses at low- versus high-dose rates. Thus, low dose rates do not 668 activate checkpoint arrest, and low doses absorbed by cycling cells result in greater biological effects 669 per unit dose than higher dose rates which cause arrest and thereby reduce the number of cells in 670 sensitive S and G2 phases of the cell cycle (Hall and Giaccia, 2011). The generality of inverse dose- 671 rate effects across species and endpoints argues for conserved mechanisms, and such effects should 672 be further investigated in animal models to refine risk estimates of low-dose and low dose-rate 673 radiation exposures in humans. 674 26 NCRP SC 1-21 Draft of March 31, 2015 [MR] 675 NOT TO BE DISSEMINATED OR REFERENCED 3. Studies Integrating Biology and Epidemiology 676 677 There are numerous epidemiologic studies documenting radiation-associated cancer 678 (UNSCEAR, 2008a) and certain noncancer effects (UNSCEAR, 2008b). Section 3.1 provides 679 examples of a few studies that successfully incorporate biological measures, sound epidemiologic 680 methods and radiation measures, which is the main focus of this Commentary. Section 3.2 discusses 681 studies of various noncancer diseases and effects, concentrating in particular on circulatory disease 682 and cataracts. Emerging epidemiologic studies focused on chronic low-dose radiation, such as the 683 Million Worker Study (Boice, 2012; 2014a; Bouville et al., 2015), incorporate radiation dosimetry 684 with biological and epidemiologic data, and are being designed to examine both cancer and 685 noncancer outcomes. Although currently in preliminary phases, the integrative approach combining 686 multiple measures, including individual dosimetry measures, and the planned analyses of multiple 687 outcomes promise improvements over prior studies designed in singular contexts. 688 689 3.1 Epidemiologic Studies Focused on Cancer Outcomes 690 691 3.1.1 Chromosome-Aberration Studies 692 693 A small number of radiation epidemiologic studies have also incorporated measures of 694 chromosome aberrations on a subset of the study population, in particular for support of dose 695 estimation. 696 697 Chromosome aberrations in blood lymphocytes are the most thoroughly studied biological 698 sentinel of radiation-induced cellular injury. Unstable aberrations such as rings and dicentrics are 699 used to estimate exposures in the short term, and stable aberrations such as symmetrical 700 translocations are used to estimate exposures many years after they occurred (Ainsbury et al., 2011; 701 Kleinerman et al., 2006; Tucker, 2008; Tucker and Luckinbill, 2011). Epidemiologic observations 702 that have incorporated chromosome-aberration evaluations include atomic-bomb survivors (Kodama 703 et al., 2001), patients treated with radiation for ankylosing spondylitis (Buckton, 1983), cervical 704 cancer (Kleinerman et al., 1989), benign gynecological disorders (Kleinerman et al., 1994), enlarged 705 thymus gland or tonsils (Kleinerman et al., 1990); patients receiving diagnostic radiation for 27 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 706 tuberculosis monitoring (Kleinerman et al., 1990; Littlefield et al., 1991) or given Thorostrast for 707 cerebral angiography (Littlefield et al., 1997); workers in radiation occupations (Bhatti et al., 2008; 708 Little et al., 2014a; Tucker et al., 1997a); and populations living in areas of high natural background 709 radiation (Wang et al., 1990). 710 711 While chromosome aberrations in lymphocytes are indirect indicators of radiation dose and thus 712 correlated with cancer risk, there is evidence that chromosome aberrations may predict human 713 cancer risk independently of such exposure and thus be relevant to the mechanistic and biological 714 understanding of the carcinogenic process (Bonassi et al., 2008; Hagmar et al., 2004; Tucker et al., 715 1997b). Examples are given below for several epidemiologic investigations where concurrent studies 716 of chromosome aberrations have provided insights into the possible mechanisms of carcinogenesis 717 and other human health detriments. 718 719 3.1.2 Atomic-Bomb Survivors 720 721 The principal atomic-bomb survivor cohorts consist of the LSS, which provides follow-up for 722 mortality and cancer incidence of about 90,000 survivors with dose estimates, and the Adult Health 723 Study (AHS), which provides biennial medical follow-up for a subcohort of initially about 20,000 724 LSS members. These two studies are primary sources for investigating the relationship between 725 radiation exposure and cancer and noncancer outcomes. It is from the LSS, for example, that we 726 know that most types of cancer can be associated with radiation exposure, and that increased cancer 727 risk from an acute radiation exposure continues for more than 60 y (through 2009) from initial 728 exposure (Grant et al., 2014; Hsu et al., 2013; Ozasa et al., 2012; Preston et al., 2007). 729 730 There are few known biological effects or lifestyle variables apart from cancer, mortality from 731 various causes, and the standard socio-demographic variables (e.g., age, sex and city) determined in 732 the full LSS cohort. Much more comprehensive biological and lifestyle information was collected in 733 the AHS. The AHS includes a large fraction of LSS members with moderate to high radiation 734 exposures, as well as, for control purposes, survivors who were essentially unexposed. Within this 735 study group a large number of effects and biomarkers have been assessed, many of which have 736 focused on noncancer outcomes. The following presents an overview of those atomic-bomb survivor 28 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 737 studies that have incorporated chromosome aberrations as a biological endpoint and/or a biomarker 738 of exposure. A significant fraction of the AHS members (~4,000 total) have been assessed for stable 739 chromosome aberrations, evaluated in lymphocytes either by traditional Giemsa methods or more 740 recently by fluorescent in situ hybridization. Stable chromosome aberrations (primarily 741 translocations) are clearly related to radiation dose but there is large inter-individual variability in 742 response to dose (Kodama et al., 2001). 743 744 Significant excesses of leukemia have been observed among children and adults exposed to the 745 Hiroshima and Nagasaki atomic bombs (Hsu et al., 2013), but not among survivors who were 746 exposed in utero (Jablon and Kato, 1970). Studies of chromosome aberrations mirror these 747 epidemiologic observations. There was little evidence of dose response for chromosome aberrations 748 among atomic-bomb survivors exposed in utero, although there was a small but significant increase 749 in chromosome-aberration rate at <100 mGy (weighted fetal dose)2, whereas dose-response 750 relationships of the expected magnitude were seen for translocation frequencies in lymphocytes of 751 the mothers of those exposed in utero (Ohtaki et al., 2004). Murine experimental data (using 752 B6C3F1 mice) confirmed the human observational data, indicating that chromosome aberrations in 753 lymphocytes do not persist after an in utero dose of 1 to 2 Gy (x rays) (Nakano et al., 2007). A 754 slightly different, although essentially compatible result was observed in a Sprague-Dawley rat 755 model, where mammary cells from animals irradiated in utero with a dose of 2 Gy (gamma rays) 756 appeared to show the normal chromosome-aberration dose response, but hemato-lymphoid spleen 757 cells did not (Nakano et al., 2014). 758 759 The lack of a dose response at high doses for chromosome aberrations among those exposed in 760 utero may provide insights into the differences seen in excess risks among those exposed in utero 761 and during early childhood for leukemia. However, it should be noted that for solid cancers there is 762 an appreciable risk following in utero exposure, manifest in adulthood, which is similar to that 763 among those survivors exposed in early childhood (Preston et al., 2008). As one possible explanation 764 for these human and experimental observations, Nakano et al., (2007) suggested that the abnormal 765 cells in the fetus were replaced by normal fetal stem cells during the postnatal growth period. 766 Another possibility, suggested by Nakano et al. (2014) is that following relatively high doses, the 2 The estimated fetal dose from gamma rays plus 10 times the estimated fetal dose from neutrons, presented in milligray. 29 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 767 high degree of induced cell sterilization will leave too small a pool of hematopoietic stem cells for 768 any malignancy (i.e., leukemia) to be observed, given the relatively low expected mutation rate (10–5 769 to 10–4 per gene per gray). If this phenomenon is true in general, it might suggest that the fetus is less 770 vulnerable to the oncogenic effect of high-dose radiation exposure than the child, but that this may 771 not be the case at lower doses. However, the apparent contrast between the null findings in relation 772 to leukemia following in utero exposure in the atomic-bomb survivors and in the various studies of 773 leukemia following diagnostic obstetric exposure (Bithell and Stewart, 1975; Harvey et al., 1985; 774 Monson and MacMahon, 1984; Stewart et al., 1956) should not be overstated, since the results are in 775 fact statistically compatible (Wakeford and Little, 2003). 776 777 In addition to the atomic-bomb survivors’ cohort, other studies (Sections 3.1.3 through 3.1.8) 778 have incorporated chromosome aberration measures to evaluate further whether these markers are 779 predictive of dose and possibly cancer risk. 780 781 3.1.3 Pelvic Irradiation Studies 782 783 Significant excesses of leukemia have been observed in women treated with pelvic radiation 784 therapy for cervical cancer (Boice et al., 1987) and for nonmalignant gynecological bleeding 785 disorders (Sakata et al., 2012). Following radiation treatments for cervical cancer, the leukemia risk 786 increased up to doses of ~4 Gy, and then decreased at higher levels. A similar pattern was seen for 787 radiation-induced chromosomal aberrations suggesting that cell killing might predominate over 788 transformation at very high doses (Kleinerman et al., 1989; Littlefield et al., 1991). Excess leukemia 789 was also reported following lower dose partial-body radiation therapy for benign gynecological 790 disease. However, the level of risk was nearly the same as seen among the cervical cancer patients, 791 about twofold, despite a tenfold difference in average dose to active bone marrow (8 versus 0.7 Gy, 792 respectively) (Kleinerman et al., 1994). High-dose cell killing was again postulated as one 793 explanation for this apparent inconsistency. Remarkably, the rate of stable aberrations, which 794 reflects nonlethal damage in surviving stem cells, was also similar among the cancer patients and 795 benign gynecological disease patients. Apparently, the lower-dose treatments for benign disorders 796 resulted in much higher chromosomal-aberration yields per unit dose than seen following radiation 797 therapy for cervical cancer. Assuming that cytogenetically aberrant stem cells that survive radiation 30 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 798 therapy contribute to the leukemogenic process, the chromosome-aberration data in these two 799 irradiated populations are consistent with the leukemia outcomes in the two populations. In these 800 circumstances of partial-body radiation therapy, stable aberrations many years after exposure appear 801 to be indicators of effective risk rather than biomarkers of radiation dose. 802 803 3.1.4 Occupational Studies 804 805 Studies of chromosome aberrations among working populations with accurate measures of 806 chronic radiation exposures can provide insights into the ability of human cellular systems to repair 807 radiation damage incurred at a low dose rate. Understanding human biological responses to radiation 808 exposure has implications concerning radiation protection guidelines where a DDREF is applied to 809 scale the high dose-rate exposure risks among atomic-bomb survivors to low dose-rate exposure 810 circumstances (ICRP, 2007). Studies of workers have indicated that stable chromosome aberrations 811 can be detected and related to the amount of radiation experienced over time (Evans et al., 1979; 812 Little et al., 2014a; Tucker et al., 1997a). 813 814 The ability to detect a biological effect at the chromosome level does not mean that there will be 815 a biological consequence in the population exposed, and it may be informative to contrast 816 chromosome findings in studies for which effects have and have not been demonstrated. In studies of 817 Chernobyl cleanup workers from Estonia, increases in chromosome aberrations (Littlefield et al., 818 1998) and in leukemia have not been observed (Rahu et al., 2013), suggesting that the population 819 size may have been too small and the doses too low to demonstrate effects following protracted 820 exposure had there been any. Other studies of low-dose occupational exposure and chromosome 821 aberrations suggested a consistency with the cytogenetic data among atomic-bomb survivors (Little 822 et al., 2014a) and there is also evidence for an increase in leukemia risk in the radiologic 823 technologist population, albeit based only on number of years worked before 1950, when doses were 824 highest (Liu et al., 2014). 825 826 Studies of workers at Sellafield nuclear facilities have revealed increased chromosome 827 aberrations that are dose related (Tucker et al., 1997a; Tawn et al., 2004; 2006). A separate study 828 found increased leukemia occurrences (Muirhead et al., 2009) that are also dose related. A similar 31 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 829 dose-related excess of chromosome aberrations has been observed in the Mayak nuclear workers 830 (Burak et al., 2001), at a rate lower than, albeit statistically compatible with, those in the Japanese 831 atomic-bomb survivors (Kodama et al., 2001). This parallels a dose-related excess risk of leukemia 832 (Shilnikova et al., 2003) in that cohort, also compatible with that in the LSS (Hsu et al., 2013). The 833 cytogenetic data, however, indicated that DDREF for chromosome aberrations was about 6, 834 suggesting to the authors that “radiation-induced double-strand breaks may be repaired more 835 efficiently at low dose rates, either because there are fewer breaks competing for a limited number of 836 repair enzymes or because chronic exposure results in a higher steady state of induction of these 837 same enzymes.” However, the parallels between the independent studies of dose-related 838 chromosome aberrations and of dose-related leukemia in these studies are only suggestive, as there 839 is no linkage between the two; there is no evidence that the leukemia cases themselves had elevated 840 chromosome aberration rates before disease development. Inferences on DDREF for a disease 841 endpoint in humans should not be drawn from these data. 842 843 Studies of Chernobyl clean-up workers provide limited evidence for chromosome damage 844 among workers whose mean whole-body doses were on the order of 100 mGy (Jones et al., 2002; 845 Littlefield et al., 1997; Moore and Tucker, 1999). This may simply reflect the fact that excess stable 846 translocations cannot be detected at much lower doses than 100 mGy (Tucker and Luckinbill, 2011). 847 Related to this, among liquidators exposed at higher doses there is unambiguous evidence of excess 848 translocations (Sevankaev et al., 2005). The evidence for a leukemia excess in large cohorts of 849 Chernobyl recovery workers also remains equivocal. While recent reports suggest an increase in the 850 incidence of leukemia among the recovery operation workers (UNSCEAR, 2011; Zablotska et al., 851 2013), the limitations of these studies are severe and include low participation rates, proxy 852 interviews for dose reconstruction, uncertainties in dose reconstruction and ascertainment of cases, 853 and internal inconsistencies. An example is a high radiation risk for chronic lymphocytic leukemia, a 854 malignancy that is not generally found to be increased in other exposed populations (UNSCEAR, 855 2008a), although there is significant (p < 0.05) excess in the latest LSS incidence data (Hsu et al., 856 2013), a possibly fragile finding based on 12 cases. This suggests potential biases or confounding 857 factors (UNSCEAR, 2011). The chromosome-aberration data, although limited because of the low 858 doses involved, add additional caution to the interpretation of the reports of leukemia among 32 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 859 Chernobyl recovery workers, and confirm that the worker exposure was generally relatively low and 860 the risk of exposure-related cancer accordingly low (Jones et al., 2002). 861 862 3.1.5 Environmental Background Studies 863 864 There are excess chromosome translocations in populations exposed to discharges from the 865 Mayak nuclear plant, living along the Techa River (Vozilova et al., 2012), at a rate lower than, albeit 866 statistically compatible with those in the Japanese atomic-bomb survivors (Kodama et al. 2001). 867 This group also exhibits excess risk of leukemia (Krestinina et al., 2013), consistent (per unit dose) 868 with those in the LSS (Hsu et al. 2013) and as observed also in various chronically exposed groups 869 (Ivanov et al., 2012, Zablotska et al., 2013). 870 871 Excess prevalence of micronuclei has been observed in a group of people exposed to 137Cs 872 contamination at near background dose rates from a research reactor in Taiwan (Jen et al., 2002). 873 Another population in Taiwan was exposed to prolonged, near background dose-rate 60Co 874 contamination, and again excess prevalence of chromosome translocations and deletions were 875 observed (Hsieh et al., 2002). This population has also been followed up for cancer, and significant 876 excess incidence of leukemia, and a borderline significant excess incidence of breast cancer 877 observed (Hwang et al., 2008), in both cases consistent with, although slightly less than, risks that 878 can be derived by linear extrapolation from the Japanese atomic-bomb survivors. Reduced fertility 879 has also been observed in this cohort, with fertility decreasing according to dose rate, especially of 880 mothers, but not in relation to cumulative dose (Lin et al., 2010); fertility recovered to normal among 881 those who moved out of the 60Co contaminated buildings. 882 883 In contrast, studies of populations living in areas of high natural background radiation have 884 failed to reveal convincing evidence for increases in leukemia, thyroid cancer or other malignancies 885 (Nair et al., 2009; Tao et al., 2012; Wang et al., 1990), yet increased chromosome aberrations are 886 observed in the resident populations (Hayata et al., 2004; Wang et al., 1990). Such data, limited by 887 both sample size and low doses, indicate caution when interpreting the biological significance of 888 chromosome aberrations in circulating lymphocytes as well as the absence of an effect in exposed 889 populations. Nonetheless, the absence of a leukemia response and a lower chromosome-aberration 33 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 890 yield than anticipated from studies of acute exposures (Tucker et al., 1997a) conceivably might be 891 attributable to a decrease in the tumorigenic effectiveness of low-LET radiation with decreasing dose 892 rate, as has been observed for tumorigenic effects on many tissues in laboratory animals (NCRP, 893 1980; Upton, 1990). 894 895 3.1.6 Transgenerational Studies 896 897 To date, no radiation-related genetic effects (i.e., hereditary diseases) have been 898 demonstrated in a human population exposed to ionizing radiation. A number of long-term studies of 899 the children of survivors of the atomic bombings in Hiroshima and Nagasaki have not detected any 900 transmitted genetic effects (transgenerational effects) of radiation exposure (Neel et al., 1980; Satoh 901 et al., 1996). 902 903 Children of cancer survivors have also been studied to learn of possible adverse genetic effects 904 associated with curative radiation treatments of the parents (Boice et al., 2003; Green et al., 2009; 905 Winther and Olsen, 2012). Blood samples taken from survivors of cancer, their spouses or partners 906 and their children have been analyzed for genomic instability, inherited mutations in minisatellite 907 DNA and in mitochondrial DNA, chromosome radiosensitivity and DNA polymorphic variation, and 908 the occurrence of cytogenetic abnormalities. Molecular studies found no evidence for inheritance of 909 minisatellite DNA mutations or mitochondrial DNA changes related to gonadal radiation (Guo et al., 910 2012; Tawn et al., 2011). There was no evidence for genomic instability (Tawn et al., 2005) or 911 consistent evidence for G2 chromosome radiosensitivity (Wilding et al., 2007), although inheritance 912 of chromosome radiosensitivity was evident (Curwen et al., 2010). The molecular analyses 913 supported the epidemiologic findings of no detectable heritable genetic changes in the risk of 914 cytogenetic abnormalities, single gene disorders, birth defects, stillbirths, neonatal deaths and cancer 915 in the children of men exposed to testicular irradiation or women exposed to ovarian irradiation 916 (Signorello et al., 2012; Winther et al., 2012). These epidemiologic and cellular studies of the 917 children of cancer survivors are consistent with the studies of the children of atomic-bomb survivors 918 in Japan (Schull, 2003). 919 34 NCRP SC 1-21 Draft of March 31, 2015 [MR] 920 NOT TO BE DISSEMINATED OR REFERENCED In contrast however, Dubrova and colleagues have described an elevated minisatellite mutation 921 rate in the offspring of workers involved in radiation cleanup after the Chernobyl accident (Dubrova 922 et al., 1996; 1997), as well as populations living along the Techa River (Dubrova et al., 2006). 923 However, these studies have been somewhat controversial (Livshits et al., 2001; Slebos et al., 2004). 924 Possible reasons for the discrepancy between these observations have been considered by Bouffler 925 et al. (2006), Little (2014) and Tawn et al. (2015). 926 927 928 3.1.7 Gene and Radiation Interaction: The Women’s Environmental Cancer and Radiation Epidemiology Study 929 930 The Women’s Environmental Cancer and Radiation Epidemiology (WECARE) Study is an 931 international multicenter, population-based case-control study of breast cancer designed specifically 932 to examine the joint roles of interaction of gene carrier status and radiation exposure in the etiology 933 of breast cancer (Stovall et al., 2008). The underlying hypothesis is that a woman who carries certain 934 genetic mutations will be more susceptible to radiation-associated breast cancer than a woman who 935 is not a carrier. The study is nested within seven population-based cancer registries. 1,508 women 936 with asynchronous contralateral breast cancer were individually matched to 2,200 controls with 937 unilateral breast cancer on date and age at diagnosis of the first breast cancer, race, and registry 938 region. 939 940 Only young women with breast cancer were included because they were more likely to be 941 genetically predisposed to breast cancer and to have had a greater susceptibility to radiation-related 942 cancer. All study subjects were identified, recruited, and interviewed through seven population- 943 based cancer registries. The average age of cases and controls was 45 y and the average time 944 between primaries for cases was 5 y. All women were interviewed using a questionnaire that 945 included known and suspected risk factors for breast cancer and detailed treatment. Tumor 946 characteristic information and biospecimens were also collected. For each patient who received 947 radiation therapy, absorbed dose was estimated for each of the four quadrants and nipple area for the 948 contralateral breast. The average dose to the contralateral breast was 1 Gy (Stovall et al., 2008). 949 35 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 950 To date, the WECARE Study participants have been screened for candidate breast cancer 951 susceptibility genes, genes that lie in the ATM-CHEK2 pathway and across the genome using an 952 agnostic approach in order to determine genetic variation associated with contralateral breast cancer 953 and radiation-associated contralateral breast cancer in particular. Screening 2,100 WECARE Study 954 participants for the ATM gene yielded an elevated risk among carriers who were treated with 955 radiation [RR for carrying missense mutations and receiving >1 Gy (3.3, 95 % CI 1.4 to 8.0)]. The 956 risk was stronger among those who were treated at young ages and who carried mutations predicted 957 to be deleterious [RR for those under age 45 y at exposure (10.4, 95 % CI 2.3 to 47.2) (Stovall et al., 958 2008)]. There was no elevated risk in the absence of radiation. The WECARE Study incorporates 959 epidemiologic methods, genetic data, and individual dosimetry for each participant, making the 960 study a paradigm for future interdisciplinary integrated genetic radiation research. 961 962 3.1.8 Retinoblastoma 963 964 Retinoblastoma is an embryonic childhood cancer of the eye that originates in the cells of the 965 retina (Gallie et al., 1999). It has both a heritable and a nonheritable etiology (Knudson, 1971). 966 Similar to other familial susceptibility syndromes, hereditary retinoblastoma is an autosomal- 967 dominant cancer resulting from deleterious mutations of a single gene of high penetrance, the RB1 968 tumor-suppressor gene (Taylor et al., 2007). Most patients with hereditary retinoblastoma develop 969 cancer in both eyes, but carriers of low-penetrance alleles may develop cancer in one eye only or not 970 at all. Nonhereditary retinoblastoma rises sporadically as a result of biallelic somatic mutation and 971 usually affects only one eye (Little et al., 2012a; Lohmann, 2010). Unlike patients with sporadic 972 (nonhereditary) disease, children with hereditary retinoblastoma are at a notable increased risk for 973 developing a second cancer later in life (Eng et al., 1993; Kleinerman et al., 2007). Radiation therapy 974 further increases the risk of sarcoma and second cancer development (Kleinerman et al., 2005; 975 NCRP, 2011; Wong et al., 1997; Yu et al., 2009). Radiation-related sarcomas occur most frequently 976 around the orbit of the eye that received curative treatments, whereas the nonradiation associated 977 sarcomas occur in other parts of the body. For germline (hereditary) retinoblastoma, every cell in the 978 body contains a deleterious mutation of the retinoblastoma tumor-suppressor gene and studies have 979 attempted to evaluate the possible interaction between high-dose radiation and underlying genetic 980 susceptibilities in causing second primary cancers (Kleinerman, 2009). While there is compelling 36 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 981 evidence for an enhancement of risk of certain types of second cancer following radiation therapy 982 among children with heritable retinoblastoma contrasted with children with the nonheritable type of 983 retinoblastoma (i.e., for a gene and radiation interaction), the data are limited by small numbers and 984 statistical uncertainties (Draper et al., 1986; Wong et al., 1997; Yu et al., 2009). For some cancer 985 sites (e.g., breast) there appears to be little or no excess radiation risk among those with heritable 986 retinoblastoma, in contrast to the much larger risk in those without the heritable disease (Little et al., 987 2014b). Nonetheless, it is interesting that studies of cells from retinoblastoma families involving the 988 -H2AX assay (Kato et al., 2007) and the G2 chromosome-radiosensitivity assay (Wilson et al., 989 2010) indicate elevated radiation sensitivities with respect to cell killing and DNA repair or DNA 990 damage processing functions. Conceivably these cellular studies point to a defect in genome 991 maintenance, including the capacity to process radiation-induced DNA damage, that results in 992 increased germline mutation rates, predisposition to cancer, and increased susceptibility to radiation- 993 related cancer. Identification of such genes or factors involved could provide insights into 994 995 996 mechanisms leading to retinoblastoma, cancer predisposition and radiation carcinogenesis. 997 radiation therapy-related cancers (Travis et al., 2006). Germline mutations in the RB1 tumor- 998 suppressor gene predispose children to a high risk of osteosarcomas, soft-tissue sarcomas, and a 999 number of other cancers. Radiation therapy further enhances the risk of tumors arising in the Retinoblastoma provides an example of how genetic mutations may influence the risk of 1000 radiation field. Mutations in the RB1 gene and perhaps other genes appear important in the cellular 1001 response to DNA damage and may confer an increased risk of radiation-related cancer. It is unclear, 1002 however, the extent to which radiation therapy for hereditary retinoblastoma, a genetic disorder due 1003 to mutations in a gene of high penetrance, can be generalized to genetic conditions of lower 1004 penetrance (or to polygenic circumstances) following much lower absorbed doses at or below 1005 1006 1007 1008 100 mGy. 1009 3.2. Epidemiologic Studies Focused on Noncancer Outcomes There is emerging evidence of radiation-associated excess risk of circulatory disease in a number 1010 of groups exposed at moderate doses (e.g., 500 mGy) (Little et al., 2012b). In particular, radiation- 1011 associated excess risk of mortality and morbidity for a number of circulatory disease endpoints has 1012 been observed in the Japanese atomic-bomb survivor data (Shimizu et al., 2010); Yamada et al. (2004). 37 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1013 There is very rich covariate information available within the AHS subcohort of atomic-bomb 1014 survivors, and the radiation associations for a number of these have been analyzed in a number of 1015 studies. For example, elevated levels of the pro-inflammatory cytokines IL6, CRP, TNF-α and INF-γ, 1016 but also increased levels of the (generally) anti-inflammatory cytokine IL10, have been observed in 1017 the Japanese atomic-bomb survivors (Hayashi et al., 2003; 2005). There was also a dose-related 1018 elevation in the erythrocyte sedimentation rate and in the levels of IgG, IgA and total 1019 immunoglobulins, all markers of systemic inflammation, in this cohort (Hayashi et al., 2005). 1020 Certain T-cell and B-cell population numbers are known to vary with radiation dose among the 1021 Japanese atomic-bomb survivors (Kusunoki et al., 1998), suggesting a role for the immune system. 1022 The atomic-bomb survivors also demonstrate dose-dependent decreases in levels of CD4+ helper T 1023 cells (Hayashi et al., 2003); decreased levels of helper T cells have also been found in blood samples 1024 from Japanese atomic-bomb survivors with myocardial infarction (Kusunoki et al., 1999). However, 1025 none of this covariate data has been analyzed together with circulatory disease outcome data. There 1026 is known to be similarly rich covariate information in the Mayak nuclear worker data, in which there 1027 is also significant radiation-associated excess risk of circulatory disease (Azizova et al., 2010a; 1028 2010b; 2011; 2014; Moseeva et al., 2014). However, this covariate data has not been separately 1029 1030 analyzed, nor is there any analysis in which this has been combined with health outcome data. 1031 The literature on radiation and cataract and other lenticular opacities has been the subject of a 1032 number of recent reviews (Ainsbury et al., 2009; ICRP, 2012; Little, 2013; Shore et al., 2010), 1033 including a recent systematic review (Hammer et al., 2013), all suggesting that there may be 1034 radiation-associated risk at moderate and low doses. However, there appear to be no studies 1035 combining analysis of cataract outcome data with intermediate bioindicator or biomarker covariates. 1036 For all other noncancer disease endpoints, the evidence for radiation etiology is much weaker, 1037 whether in directly exposed populations (Little, 2013) or in the offspring of exposed individuals 1038 (Little et al., 2013). 1039 38 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1040 1041 1042 4. Bioindicators from Radiation Biology Studies 1043 outcomes (and to a lesser extent noncancer outcomes) was introduced in Section 1.2. This included a 1044 description of how bioindicators of effect could be used as the key events for such an approach. In 1045 Section 4, a discussion is presented of the types of bioindicators that can be measured in cellular, 1046 laboratory animal and human systems, with a parallelogram approach for linking these 1047 measurements to adverse health outcomes in humans. In this way, it is feasible to both describe the 1048 shape of the dose response at low doses and low dose rates as well as the risk estimates themselves 1049 for these exposure scenarios. The rapid development of new technologies that permit whole-genome 1050 sequencing in a very short time has and will continue to enhance the ability to identify and measure 1051 many of these types of informative bioindicators. To fully utilize this type of approach to develop a 1052 bioindicator of response, it is likely that a systems biology type approach (Section 5.2.2) will be 1053 1054 1055 1056 1057 needed. 1058 indirectly through production of reactive oxygen species, producing a plethora of lesions, including 1059 many types of base damage such as broken rings, oxidized bases, small and large adducts including 1060 DNA-DNA and DNA-protein crosslinks, single-strand breaks (SSBs), and DSBs, including 1061 relatively reparable simple DSBs, and more difficult to repair complex DNA lesions. Under typical 1062 conditions, ~40 DSBs are created per cell per gray, and many more base lesions and SSBs are 1063 produced than DSBs (Ward, 1998). However, DSBs have the greatest biologic significance since 1064 DSBs correlate with cytotoxicity, chromosome aberration formation, and cellular transformation 1065 1066 1067 (Frankenberg-Schwager, 1990). 1068 2011). Specific lesions are typically repaired by a specific pathway or subpathway, but if the primary 1069 pathway fails, repair may be attempted by another pathway due to redundancies in the process. DNA 1070 damage and repair do not occur in a static environment, but rather in the context of other dynamic 1071 processes including DNA replication and transcription, and in a structurally complex and dynamic The concept of using a key event/adverse outcome pathway approach for estimating cancer 4.1 Deoxyribonucleic Acid Damage and Repair Ionizing radiation causes damage to DNA by direct energy absorption and to a larger extent, There are at least five types of DNA repair pathways, each with subpathways (Shaheen et al., 39 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1072 chromatin environment. DNA repair systems maintain genome stability and confer resistance to 1073 ionizing radiation and other DNA damaging agents, and defects in DNA repair systems typically 1074 predispose to cancer and other diseases (Friedberg et al., 2005). Three sets of pathways act on 1075 single-strand damage, including base-excision repair (nonbulky lesions) (Krokan and Bjoras, 2013), 1076 nucleotide excision repair [bulky, helix distorting lesions (Kamileri et al., 2012)], and mismatch 1077 repair (Hsieh and Yamane, 2008). Classical and alternative nonhomologous end joining, and 1078 homologous recombination, are redundant DSB repair pathways that can restore the chemical 1079 1080 1081 integrity of DNA, but with lesser or greater capacity to accurately restore genetic sequences. 1082 when forks encounter other types of lesions (single-strand breaks, many types of base damage, most 1083 DNA adducts, pyrimidine dimers, and intra- and inter-strand crosslinks) (Allen et al., 2011; 1084 Budzowska and Kanaar, 2009). DSBs are marked by phosphorylated histone H2AX (-H2AX) 1085 (Rogakou et al., 1998), which plays important roles in DNA damage checkpoint signaling and DSB 1086 repair (Chanoux et al., 2008; Downey and Durocher, 2006). -H2AX is a fairly reliable biomarker of 1087 DSBs, detected by fluorescence microscopy using phospho-specific antibodies, hence -H2AX is 1088 often used in kinetic studies to monitor both DSB induction and repair (Lobrich et al., 2010), and as 1089 a radiation biodosimeter (Rothkamm et al., 2013). Proteins involved in nonhomologous end joining 1090 and homologous recombination migrate to DSBs, such as the early DNA damage response protein 1091 53BP1, and the late acting homologous recombination protein RAD51. The resulting protein 1092 aggregates (“repair factories”) can be detected with fluorescence microscopy using cognate 1093 antibodies as subnuclear foci. These foci thus complement -H2AX as additional biomarkers of 1094 DSBs and their repair (Allen et al., 2011). -H2AX foci represent a robust biomarker of DNA 1095 damage by radiation, and induced foci or residual (unrepaired) -H2AX foci show promise as 1096 potential biomarkers of risk for various cancers (Fernandez et al., 2013; He et al., 2013; Matthaios 1097 1098 1099 et al., 2013). DSBs are produced immediately by radiation, or they can arise later during DNA replication 4.2 Cellular Signaling at Low Doses 1100 1101 1102 Cells respond to DNA damage in part through the protein kinase mediated activation of signaling pathways that can culminate in cell-cycle arrest, stimulation of DNA repair, induction of cell death, 40 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1103 or terminal differentiation and senescence. The cellular DNA-damage response includes two major 1104 checkpoint signaling pathways; one responding to DSBs through ATM activation that leads to 1105 CHEK2 activation and p53 stabilization (Postiglione et al., 2010), and one centered on ATM and 1106 Rad3-related activation, which activates CHEK1 in response to single-strand breaks and gaps (i.e., at 1107 replication forks) (Fokas et al., 2014). Damage to cellular components in addition to DNA can also 1108 trigger signal transduction pathways, such as the ceramide pathway that responds to damage to the 1109 cell membrane (Zeidan and Hannun, 2010), redox signaling from mitochondria (Hei et al., 2008), as 1110 well as signaling between directly hit and non-hit bystander cells (Section 4.6.2). 1111 1112 Checkpoint pathways are not “on or off” but show different responses depending on the level of 1113 damage. This is reflected in different gene expression responses at different levels of radiation 1114 exposure and in different physiological outcomes (Ding et al., 2005; Gridley et al., 2009; Lu et al., 1115 2010; Mezentsev and Amundson, 2011). DNA damage response thresholds are genetically regulated 1116 (Peng et al., 2010; Putnam et al., 2009), and thresholds may vary for each pathway (Fernet et al., 1117 2010). With minimal DNA damage, as with low-dose ionizing radiation, some cells may not activate 1118 the DNA repair process (Rothkamm and Lobrich, 2003) while others may activate repair pathways 1119 but not cell-cycle arrest or death pathways. Activation of repair or other damage signaling pathways 1120 by low-dose ionizing radiation probably underlies adaptive responses that enhance survival, prevent 1121 genomic instability, or limit deleterious effects when cells are subsequently exposed to large ionizing 1122 radiation doses (Section 4.6.1). DNA repair efficiency may vary with the level of damage, with more 1123 efficient repair at low ionizing radiation doses than at high doses (Neumaier et al., 2012). At higher 1124 levels of damage, cells arrest at G1, S, and/or G2/M phases to prevent cell-cycle progression with 1125 DNA damage to reduce the chance of mutations, aneuploidy, and mitotic catastrophe, and above 1126 threshold levels of damage (which vary with cell type), cell death and nonproliferation pathways are 1127 activated, including apoptosis, autophagy, necrosis and senescence (Branzei and Foiani, 2009; 1128 Budzowska and Kanaar, 2009; Eriksson and Stigbrand, 2010; Galluzzi and Kroemer, 2008; 1129 1130 1131 1132 1133 Vakifahmetoglu et al., 2008; Vandenabeele et al., 2010). 1134 of radiation dose, dose rate and radiation quality, and are the best-documented biomarker of 4.3 Chromosome Alterations Following exposure to ionizing radiation, chromosomal rearrangements are induced as a function 41 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1135 radiation exposure, even to doses as low as 200 mGy (Lloyd et al., 1992; Pohl-Ruling et al., 1983; 1136 Tucker and Luckinbill, 2011). Chromosomal rearrangements arise as a consequence of the 1137 processing of directly and indirectly induced DNA DSBs and at high dose rates increase in a linear- 1138 quadratic fashion for sparsely ionizing radiation, suggesting that the observed rearrangements are the 1139 result of two or more interacting molecular events. There is a long and well-documented literature on 1140 the dose-response relationships for the induction of chromosomal aberrations evaluated after Giemsa 1141 staining, or by fluorescence in situ hybridization using chromosome-specific molecular probes, and 1142 more recently by combinatorial hybridization techniques (reviewed in Cornforth, 2001). 1143 1144 Using these more modern technologies it appears that the shape of the dose-response profile for 1145 chromosomal rearrangements is influenced by the production of complex rearrangements involving 1146 three or more chromosomes (Loucas et al., 2013), especially following exposure to low dose-rate 1147 radiation exposures (Loucas et al., 2004). 1148 1149 While chromosomal alterations are a well-documented biomarker of exposure and dose, and 1150 remain the gold standard in this area following potential radiation exposure (Tucker, 2008), it should 1151 be noted that rearrangement frequency can vary significantly between tissues (Brooks et al., 2003). 1152 Ongoing studies suggest that chromosomal aberrations might predict human cancer independently of 1153 exposure to carcinogens (Bonassi et al., 2000; Hagmar et al., 1994). However, it remains to be seen 1154 if a molecular, biochemical, or cellular signature of radiation exposure can be correlated with 1155 chromosomal aberration formation (a biomarker of radiation exposure) and be used as a predictor of 1156 health risk(s) associated with irradiation and predict adverse effects, particularly at low radiation 1157 doses. 1158 1159 4.4 Mutations 1160 1161 Mutations are essential features of cancer (Hanahan and Weinberg, 2011), and ionizing 1162 radiation was the first environmental mutagen identified (Muller, 1927). Mutations can serve as 1163 bioindicators of radiation effects as a function of dose, dose rate and radiation quality, and most 1164 mutation studies to date have made use of model systems that assess changes at reporter loci that are 1165 not directly linked to tumor formation. 42 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1166 1167 Most mutation studies to date have utilized model reporter gene systems in which changes in the 1168 DNA sequence of a gene result in the failure to produce a functional protein product, allowing 1169 chemical selection of the mutant cells. Such mutations can reflect small changes in the protein 1170 product that render it unable to perform its normal function, or the failure to produce any of the 1171 protein, but the proteins involved generally are not related to cancer development.. However, 1172 information learned from these studies can be useful in helping to guide our understanding of 1173 mutational mechanisms and the types of mutational events likely to occur at loci that are directly 1174 linked to tumor formation following exposure to ionizing radiation. All of the studies presented in 1175 Section 4.4 focus on the identification of mutations that are thought to be directly induced by the 1176 radiation at short times after exposure. 1177 1178 Tumor suppressor genes generally require mutation of two alleles, often involving a point 1179 mutation and subsequent loss of the second allele to enable tumor formation. Model systems have 1180 been engineered to allow quantification of mutations at such heterozygous autosomal loci, and 1181 analysis of the molecular changes in DNA. In these models, one copy of a target gene, such as 1182 thymidine kinase (TK1, or Tk) or adenine phosphoribosyltransferase (APRT, or Aprt), has been 1183 inactivated and a mutagenic event that results in the loss of production of any functional protein 1184 from the second copy of the gene can be selected using a chemical that will kill only the normal 1185 cells. Mutations can also occur in dominantly acting oncogenes. In this case, mutation of a single 1186 copy of the gene can lead to tumorigenesis, but the mutational mechanisms are the same. 1187 1188 Radiation has been shown to induce mutations by many mechanisms, including all types of 1189 single basepair substitutions (Grosovsky et al., 1988), small insertions and deletions within the gene 1190 itself (intragenic events), deletion of the whole gene, multi-locus deletions, recombination-mediated 1191 events leading to loss of heterozygosity, and loss of whole chromosomes (a form of aneuploidy) 1192 (reviewed in Turker et al., 2009; Wiese et al., 2001). 1193 1194 Studies using single copy genes, such as the X-linked hypoxanthine phosphoribosyltransferase 1195 (HPRT, or Hprt) gene, can detect a subset of the radiation-induced mutational mechanisms that can 1196 occur at an autosomal locus (Liber et al., 1989). Similarly, the S1/MIC1/CD59 locus in artificially 43 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1197 constructed human x hamster hybrid cells has been used to demonstrate that ionizing radiation often 1198 produces chromosomal scale events, including whole chromosome loss, although such events are 1199 often lethal in normal human cells (Kraemer et al., 2001). 1200 1201 There are also data on radiation mutagenesis at low doses and/or low dose rates. In one study, 1202 induction of HPRT mutations in a human lymphoblast cell line by protracted exposure could not be 1203 distinguished from the mutant frequencies measured in samples exposed to the same total doses in a 1204 single acute exposure, suggesting that there was no dose-rate effect (Grosovsky and Little, 1985). 1205 Other studies using the same cell line have found an inverse dose rate effect for TK1 mutation that 1206 was dependent on dose rate (Amundson and Chen, 1996; Brenner et al., 1996). 1207 1208 The Aprt heterozygous mouse model provides a good example of translation of mutagenesis 1209 studies from in vitro to in vivo. Mutations have been characterized in kidney cells irradiated in vitro, 1210 or in cells retrieved from irradiated kidneys in mice that had total body exposures. All of the types of 1211 mutational events that occurred in kidney cells exposed in vitro also occurred in vivo, from small 1212 intragenic events to whole chromosome loss (Ponomareva et al., 2002; Turker et al., 2009; 2013). 1213 The chromosomal milieu of the Aprt locus in the mouse is similar to that for the TK1 locus in human 1214 cells. This is important for cross-species extrapolations of mutational risk, although one class of 1215 radiation-induced mutations (whole chromosome loss) that can be observed in the engineered mouse 1216 model is not detected in the human cells, most likely due to functional hemizygosity of linked loci in 1217 the “outbred” human genome that would result in cell death from such an event in the human cells. 1218 1219 Recent studies on radiation-related acute myeloid leukemia (AML) in mice have examined 1220 mutations at the PU.1 locus on mouse chromosome 2. Deletion of one allele of PU.1 was proposed 1221 as a candidate bioindicator for radiogenic AML in a susceptible mouse strain (Peng et al. 2009). 1222 Further work indicates that point mutation of the second PU.1 allele is not radiation related, and that 1223 mutation of other genes located near PU.1 may be particularly important in radiogenic AML in 1224 different mouse strains (Genik et al., 2014). 1225 1226 Radiation-induced autosomal mutations have also been studied in Dlb-1 heterozygous mice. 1227 Mutations were measured in intestinal stem cells and their progeny (cells which are of particular 44 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1228 interest since mutations in stem cells are thought to be important in the carcinogenic process). Fewer 1229 gamma ray-induced mutations were observed at the Dlb-1 locus in the mouse small Muintestine 1230 when the dose rate was lowered to 0.01 Gy min–1 as compared with acute exposures at 1.8 Gy min–1 1231 (Winton et al., 1989). However, this study was limited to high total doses. 1232 1233 In the early 1950s and 1960s hybrid male mice were irradiated and then mated to unirradiated 1234 females that harbored a series of seven recessive loci and mutations were assessed in these marker 1235 loci in the offspring. Mutant frequency results indicated that low-LET radiation delivered to stem 1236 cell spermatogonia at exposure rates of 0.8 R min–1 and below induced only about one-third as many 1237 mutations in the offspring as those delivered as single acute exposures (Russell and Kelly, 1982; 1238 Russell et al., 1958). While the total exposures in these experiments were not low (86 to 1,000 R), 1239 the exposure rates were as low as 1 mR min–1 and lower in some cases. The data gathered in these 1240 very large-scale studies were re-examined in light of the availability of fine-structure genetic or 1241 molecular analyses of the seven marker loci and their surrounding chromosomal regions (Russell 1242 and Hunsicker, 2012), and indicated that the reduction in mutant frequency at the lower exposure- 1243 rates is associated with a decline in “large lesion” mutations. Since these data reflect radiation effects 1244 on the irradiated male germline, it is more challenging to extrapolate these findings to somatic cells 1245 in adult humans, where most human cancers develop. 1246 1247 New approaches to the analysis of mutations have become available with the advent of next- 1248 generation DNA sequencing, in addition to molecular cytogenetic approaches that can detect copy 1249 number variations with increasing precision. These methods are being considered now for use in the 1250 analysis of irradiated cells in culture as well as in irradiated tissues. Associating radiation-induced 1251 changes with particular loss of heterozygosity or other events that may be causal to tumor formation 1252 will be challenging, given the wide variety of molecular events that lead to radiation-induced 1253 mutations and the large number of driver and passenger mutations found in human tumors (Bozic 1254 et al., 2010; Vogelstein et al., 2013). An example of the use of such an approach comes from 1255 experimental studies of genetic and chemically-induced lung cancers in mice (Westcott et al., 2014). 1256 In this study, the carcinogen-induced tumors display signatures of the initiating chemical 1257 carcinogens. The situation may be more challenging for ionizing radiation-induced events, where 1258 specific radiation-signature events have been more difficult to identify. 45 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1259 1260 4.5 Radiation-Induced Genomic Instability 1261 1262 Genomic instability is characterized by an increased rate of genetic change in the progeny of an 1263 irradiated cell. Instability manifests as alterations in chromosomal rearrangements, micronuclei, 1264 increased gene copy number, minisatellite and microsatellite instabilities in DNA repeat sequences, 1265 transformation, and cell killing and can be induced through both targeted and nontargeted bystander- 1266 like processes (Little, 2000; Morgan et al., 2002). Genome instability is a key feature of cancer cells 1267 and it is now well established that defects in DNA repair and DNA damage response systems 1268 predispose to cancer. Defects in DNA repair or DNA damage response systems can allow more rapid 1269 accumulation of instability that underlies cancer development. However it is still controversial 1270 whether there is a causal association between instability and cancer (Little, 2010). Genomic 1271 instability can be produced from endogenous or induced DNA damage, and as for nontargeted 1272 bystander effects, the mechanisms of instability share features with inflammatory responses 1273 characterized by intercellular signaling, production of chemokines, cytokines, and reactive oxygen 1274 and nitrogen species. 1275 1276 Ionizing radiation causes DNA damage that directly leads to large- or small-scale genetic 1277 changes, ranging from single-base (point) mutations to gene rearrangements, translocations, and 1278 whole chromosome gain or loss (Section 4.4). Studies over the past decade have shown that ionizing 1279 radiation (low- and high-LET) and nonionizing radiation (e.g., ultraviolet light) also induce genome 1280 instability many cell generations after the exposure (Durant et al., 2006; Huang et al., 2004; 2007; 1281 Morgan, 2003a; 2003b). Some of these instability phenotypes are correlated and therefore likely 1282 share a common means of induction or are otherwise related mechanistically (Limoli et al., 1997). 1283 For example, cells that display ionizing radiation-induced chromosome instability typically show 1284 delayed death, probably reflecting the low viability of cells with large-scale karyotypic changes and 1285 aneuploidy as a result of massive gene expression imbalances. In contrast, ionizing radiation- 1286 induced delayed hyper-recombination is not associated with delayed death; therefore chromosome 1287 instability and delayed hyper-recombination are mechanistically distinct processes (Huang et al., 1288 2004). 1289 46 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1290 One commonality among the many instabilities investigated to date is the fact that they can be 1291 induced by both high (1 to 10 Gy) and low (0.01 to 0.1 Gy) ionizing radiation doses. However, in 1292 some instances a threshold dose has been observed and there are also examples of the effect 1293 saturating at a specific radiation dose (Limoli et al., 1999). Like many immediate effects of 1294 radiation, the induction of genome instability is also subject to adaptive responses. As an example, 1295 exposing cells to low doses of ionizing radiation offered some protection for induction of 1296 instabilities by a subsequent higher dose (Huang et al., 2007). However, the adaptive response was 1297 obviously not a complete or even a strong protective mechanism, since even low doses were 1298 sufficient to induce instabilities. 1299 1300 Two other features common to ionizing radiation-induced instabilities is that they occur at high 1301 frequencies (often 10 % or more of cells that survive low to moderate doses) and they typically do 1302 not show a dose response (Little, 2000; Morgan, 2003a). These features suggest that the target for 1303 induced instability is large. It is difficult to imagine instabilities arising from gene inactivation (even 1304 if only one of a large family of genes such as those in the DNA damage response pathway needed to 1305 be inactivated). Instead, the target is likely to be as large as the nucleus or even the whole cell. 1306 1307 There is considerable variation in the manifestation of induced instability between cell lines in 1308 vitro, and organisms in vivo. This variation in response has a genetic component (Ponnaiya et al., 1309 1997) and may reflect the age of the organism at the time of irradiation as well as the cells/organisms 1310 ability to detoxify cellular reactive oxygen/nitrogen species (Spitz et al., 2004). Once initiated 1311 however, there is evidence that instability can be perpetuated over time by dicentric chromosome 1312 formation followed by bridge breakage fusion cycles (Marder and Morgan, 1993) as well as 1313 recombinational events involving interstitial telomere like repeat sequences (Day et al., 1998). There 1314 is also increasing evidence that epigenetic factors (Aypar et al., 2011; Koturbash et al., 2006), 1315 dysfunctional mitochondria (Kim et al., 2006a; 2006b), and inflammatory type reactions (Lorimore 1316 and Wright, 2003; Mukherjee et al., 2012), presumably involving reactive oxygen and nitrogen 1317 species as well as cytokines and chemokines might be involved in driving the unstable phenotype 1318 (Laiakis et al., 2007; Hei et al., 2008). To this end there is convincing evidence for such reactions 1319 being involved in another nontargeted effect associated with ionizing radiation, the bystander effect 1320 (Morgan et al., 2002). Clearly the link between induced instability and bystander effects suggests 47 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1321 common processes and inflammatory type reactions will likely be the subject of future investigation, 1322 as will the clinical implications of induced instability in terms of a marker for increased cancer 1323 risk/early detection, and as a consequence of radiation therapy that may result in the induction of a 1324 therapy-related second malignancy (Goldberg, 2003). 1325 1326 While the precise molecular, cellular, organ, and organismal mechanisms contributing to 1327 radiation-induced genomic instability are not known, the phenotype is well documented in a number 1328 of in vivo model systems (Morgan, 2003b; Watson et al., 2001). This raises a number of questions 1329 regarding this intriguing phenotype. 1330 1331 What, if any, specific types of radiation-induced genomic instability are capable of 1332 transforming a cell from a normal to a cancer cell, or furthering the progression of a 1333 premalignant cell towards a more aggressive cancer phenotype? 1334 1335 1336 What, if any, is the role of induced instability in secondary cancers observed in radiation therapy patients? Could analysis of radiation-induced genomic instability in peripheral blood lymphocytes be 1337 used as a biomarker of an individual’s radiation sensitivity and potential radiation-related 1338 cancer risk? 1339 1340 Recently the integration of radiobiological effects in a two-step clonal expansion model of 1341 carcinogenesis and applications to radioepidemiologic data have been proposed (Jacob et al., 2010). 1342 First, a model version with radiation-induced genomic instability was shown to be a possible 1343 explanation for the age dependence of the radiation-related cancer mortality in the Techa River 1344 Cohort. Second, it was demonstrated that inclusion of a bystander effect with a dose threshold allows 1345 an improved description of the lung cancer mortality risk for the Mayak workers cohort due to 1346 incorporation of plutonium. It is envisaged that a further combination of epidemiologic, biological 1347 and modeling interactions will address these questions in the future (Eidemuller et al., 2011; Kaiser 1348 1349 et al., 2014). 48 NCRP SC 1-21 Draft of March 31, 2015 [MR] 1350 NOT TO BE DISSEMINATED OR REFERENCED 4.6 Modulators of Response 1351 1352 Classical radiation biology was built on the concept of target theory wherein the deleterious 1353 effects of exposure including chromosome aberrations, mutations and carcinogenesis are assumed to 1354 arise from damage to a cellular target (e.g., nuclear DNA) via energy deposition in the cell at risk 1355 (Hall and Giaccia, 2011; Lea, 1946; UNSCEAR, 1993). The deposition of energy after irradiation is 1356 well understood, but the biological processes that modify response to that damage may vary between 1357 individuals and in response to low versus high dose and dose rate. 1358 1359 In recent years, much research has focused on responses to doses of 100 mGy and below. 1360 While some responses appear to follow a continuous dose-response relationship from high to low 1361 doses, some other responses may only occur at either low or high doses, thus complicating the 1362 extrapolation from high dose to low dose. Current radiation risk extrapolations to low doses assume 1363 that: 1364 1365 the primary mode of action is linearly related to dose; and 1366 cancer is clonal in origin (i.e., the individual cell is the unit of risk), although cellular 1367 responses are likely modulated by the tissue microenvironment. 1368 1369 Although the targeted effects of radiation (i.e., responses in hit cells) may be proportional to dose, 1370 other processes, including effects in non-hit cells (e.g., induced genomic instability and bystander 1371 effects) may show nonlinear responses. Some of these low-dose responses are highlighted in 1372 Section 4.6. 1373 1374 4.6.1 Adaptive Responses 1375 1376 Two different adaptive responses have been described. The classical adaptive response was first 1377 described by Olivieri et al. (1984). They demonstrated that when human lymphocytes were exposed 1378 to a very low dose of radiation, a priming dose, followed a short time later with a larger higher dose, 1379 the challenge dose, the frequency of chromosomal aberrations induced by the challenge dose was 1380 less than that from the challenge dose given alone. A second adaptive response suggests that when 49 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1381 cells with a high background frequency of phenotypic alterations (e.g., mutations or transformation) 1382 were exposed to very low doses of ionizing radiation, these doses can actually reduce that frequency 1383 to below background (Azzam et al., 1996; Redpath et al., 2001). 1384 1385 While adaptive responses are well characterized in certain situations in vitro, there is also some 1386 evidence that they can occur in humans occupationally (Barquinero et al., 1995) or clinically 1387 (Monsieurs et al., 2000) exposed to ionizing radiation. As always in a biological system there is 1388 considerable variation both in vitro (Bosi and Olivieri, 1989) and in vivo (Padovani et al., 1995; 1389 Tedeschi et al., 1995). In fact, not only are reduced levels of DNA alterations observed in some 1390 systems, in other individuals no response has been observed, while in others an additive increase in 1391 the manifestations of irradiation has been observed (Tapio and Jacob, 2007; UNSCEAR, 1994; 1392 2012). 1393 1394 Adaptive response(s), if reproducibly induced by low doses of ionizing radiation, have potential 1395 clinical and radiation protection implications. For example, that radiation signals danger to the 1396 immune system through the Toll-like receptor family can cause cells to release damage-associated 1397 molecular patterns to activate canonical inflammatory pathways and/or initiate immunity (McBride 1398 et al., 2004). Through induction of superoxide dismutase and anti-inflammatory responses, low-dose 1399 ionizing radiation could also be used prior to surgery or other therapies that cause inflammation or 1400 ischemia. Howell et al. (2013) indicated that exposure of mice in utero to a dose rate of 10 to 1401 13 mGy d–1 [Chernobyl soils (primary radionuclides being 137Cs and 90Sr) embedded into bioplastic] 1402 for 10 d had no deleterious effects on litter size, bone marrow stem cells, and white blood cell 1403 counts, yet this exposure had significant protective effects when pups were later exposed to an acute 1404 dose of 2.4 Gy (calibrated 137Cs source). Alternatively, if low dose radiation exposures can induce an 1405 adaptive response then a CT scan for treatment planning purposes may result in the subsequent 1406 fractionated radiation dose being less effective in tumor cell kill, thus limiting the impact of radiation 1407 therapy. These outcomes are likely to vary between individuals and a future challenge is to 1408 determine which individuals will manifest one or other of these alternatives. 1409 50 NCRP SC 1-21 Draft of March 31, 2015 [MR] 1410 NOT TO BE DISSEMINATED OR REFERENCED 4.6.2 Bystander Effects 1411 1412 Bystander effects occur in nonirradiated cells that receive a signal from an irradiated cell. Such 1413 signals may be secreted and/or shed by irradiated cells or communicated via molecules transported 1414 between cells by cell-to-cell gap junction communication. Not all cells are able to produce bystander 1415 signals or respond to them. The bystander effect has been the subject of a number of recent reviews 1416 (Blyth and Sykes, 2011; Butterworth et al., 2013; Ilnytskyy and Kovalchuk, 2011; Mancuso et al., 1417 2012). 1418 1419 The key question in the context of this Commentary is whether bystander effects can be 1420 beneficial or detrimental to the nonirradiated cell, tissue, or organism. On the cellular level there is 1421 evidence of both potentially protective effects, such as apoptosis (Belyakov et al., 2005) and 1422 differentiation (Belyakov et al., 2006), and detrimental effects, such as DNA damage (Dickey et al., 1423 2009; Nagasawa and Little, 1992), chromosomal instability (Dickey et al., 2009), mutation (Zhang 1424 et al., 2009; Zhou et al., 2000), and transformation (Sawant et al., 2001). More recently, bystander 1425 responses including mutagenesis (Chai et al., 2013a; 2013b) and carcinogenesis (Mancuso et al., 1426 2008; 2011; 2013) have been convincingly demonstrated in animals in vivo. Neither the net 1427 protective or detrimental effect of the different processes affected, nor their relevance to human risk 1428 are yet fully understood. 1429 1430 It is sometimes thought that the implications of bystander effects for radiation protection and 1431 ultimately for interpreting epidemiologic studies are not likely to be significant – in that they are 1432 likely already “built into” organ risks and consequent tissue weighting factors. However, at low 1433 doses and/or dose rates this is probably incorrect. The bystander effect would not be observed (e.g., 1434 in the LSS data) because most cells are traversed by at least 100 electron tracks (= 0.1 Gy). But this 1435 is not the case extrapolating to an occupational setting, for example, where, with low-LET doses of 1436 1 mGy y–1, most cells would receive only one electron track per year. By definition, bystander 1437 effects imply that the target for radiation effects in low-dose exposures will be greater than the target 1438 volume actually irradiated. 1439 51 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1440 A number of bystander effect models have been fitted to experimental (Brenner et al., 2001; 1441 Little et al., 2005; Nikjoo and Khvostunov, 2003) and epidemiologic (Brenner and Sachs, 2003; 1442 Little, 2004; Little and Wakeford, 2001) data. The available data suggest that despite compelling 1443 experimental findings there is not strong evidence of the involvement of the bystander effect as a 1444 modifier of radiation-related cancer risk in humans (Little, 2004; Little and Wakeford, 2001; Little 1445 et al., 2009). However, as bystander responses could alter the shape of the dose-response curve at 1446 low doses, an understanding of this area could add confidence to biological models and 1447 extrapolations. 1448 1449 4.6.3 Genetic Susceptibility and Interactions with Radiation 1450 1451 Over the past decade or so, there has been an extensive increase in knowledge of the genetic 1452 basis of diseases, especially cancers of many different types (Weinberg, 2013). For example, for so- 1453 called high penetrance genes, excess spontaneous cancers are expressed in a large proportion of 1454 carriers. In addition, there are gene-gene and gene-environment interactions that can lead to 1455 variations in the likelihood of cancer across the population. How such information might convert 1456 into inter-individual differences in susceptibility to radiation-related cancer has been quite 1457 extensively discussed by ICRP (1998), NA/NRC (2006), and UNSCEAR (2001; 2008a). However, it 1458 remains unclear as to the magnitude of the effect of any such sensitivity even at the individual level, 1459 let alone a population level. 1460 1461 Sankaranarayanan and Chakraborty (2001) conducted an extensive analysis of potential impacts 1462 at the individual and population levels. They used a Mendelian one-locus, two allele autosomal 1463 dominant model for predicting the impact of cancer predisposition and increased radiosensitivity on 1464 the risk of radiation-related cancers in the population and in relatives of affected individuals using 1465 breast cancer due to BRCA-1 mutations. The general conclusions from their study were that, when 1466 the proportion of cancers due to the susceptible genotype is small (<10 %), the attributable risks are 1467 small. These risks only become large when cancer susceptibility is very large (>10-fold). On the 1468 other hand, when the proportion of cancers resulting from the susceptible genotypes is large (10 %), 1469 there can be significant increases in attributable risk for relatively small increases in cancer 1470 susceptibility (>10-fold) and radiosensitivity (>100-fold) in the susceptible populations. This means 52 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1471 that the increase in cancer risk to a heterogeneous population of susceptible and nonsusceptible 1472 genotypes will generally be quite small if the proportion of susceptible individuals is small and the 1473 radiation sensitivity relatively small. On the other hand, cancer risks assessed on the basis of an 1474 individual (in radiation therapy situations, for example) can be greatly influenced by genotype. For 1475 the purposes of radiation protection, dose limits for the public will not be significantly influenced by 1476 genetic radiosensitivity whereas they certainly could be in some occupational settings and for 1477 individual assessments for defined medical exposures. 1478 1479 There is evidence to suggest that efficiency in DNA DSB-repair modulates radiation toxicity risk 1480 because patients with genetic syndromes involving DNA DSB-repair factors, such as ataxia 1481 telangiectasia, have well recognized acute and late sensitivities to radiation. However, apart from 1482 these rare genetic syndromes, the genetic determinants of radiation toxicity are largely unknown. 1483 However, progress is being made in demonstrating experimentally that there are complex 1484 interactions that underlie the expression of cancer-predisposing genes of lower penetrance 1485 (NA/NRC, 2006). This knowledge highlights the difficulty that will be encountered in trying to 1486 incorporate facets of genetic radiosensitivity into the radiation risk assessment process, especially at 1487 low doses and doses rates, and determinants of clinical endpoints. 1488 1489 4.6.4 Interactions with Environmental and Lifestyle Factors 1490 1491 Lifetime risk estimates are reported for radiation exposures essentially considering the radiation 1492 exposure as the single stressor. Modulating factors have been identified to some extent and a few 1493 have been incorporated into the risk paradigm (e.g., age at exposure, sex, smoking for radon 1494 exposures) (Section 2.2). A major issue is to be able to identify those cancers associated with 1495 radiation exposure among the high frequency of background cancers, so that the impact of any 1496 modulating factor on the radiation component can be ascertained. This is currently an intractable 1497 problem since radiation-specific disease signatures are not known. The issue has been addressed by 1498 UNSCEAR (2000) and by Wakeford (2012). However, there is no simple approach to the problem. 1499 As a starting point it is necessary to be able to describe the nature of exposures to which individuals 1500 or populations are subject. Because the U.S. Environmental Protection Agency’s guidelines for 1501 estimation of risks to environmental stressors (chemicals and others) requires a consideration of 53 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1502 aggregate or cumulative risk, methods have been proposed and are being developed for this purpose 1503 (EPA, 2003). A novel approach that takes advantage of current technical advances in exposure 1504 science, including biomonitoring, is the concept of a so-called exposome [see an overview at NIOSH 1505 (2014) and reviews by Rappaport (2011) and Vrijheid (2014)]. The exposome is broadly defined as a 1506 measure of the total exposure of an individual in their lifetime and the relationship of this exposure 1507 to health outcomes. In this context, exposure begins before birth and includes both environmental 1508 and occupational exposures. As noted by NIOSH (2014), “Understanding how exposures from our 1509 environment, diet, lifestyle etc. interact with our own unique characteristics such as genetics, 1510 physiology, and epigenetics impact our health” is how the exposome will be articulated. This issue is 1511 very pertinent to this Commentary for which ionizing radiation is just one of the environmental/ 1512 occupational exposures and considerations of risk from radiation will need to be placed in the 1513 purview of total exposure and total risk. Exposomics is the name given to the study of the exposome 1514 and utilizes internal and external exposure methods. 1515 1516 As discussed by Rappaport (2011), it is appropriate for the application of an exposome-based 1517 approach for exposure assessment, to regard the “environment” as the body’s internal chemical (and 1518 physical) environment and to define “exposures” as levels of biologically active chemical and 1519 physical agents in and impacting this environment. He further proposes that to assess the exposome, 1520 the more applicable approach might be a top down one based on biomonitoring, rather than a 1521 bottom-up one that relies on analysis of exposures from external sources (e.g., air water, food). This 1522 general approach is extended by Wild et al. (2013) to provide a view of how an understanding of the 1523 complete exposure profiles of individuals in large populations can be linked to a detailed analysis of 1524 their omics profiles. As noted such approaches are in their early stages of development but already 1525 indicate the reasonable likelihood of linking total exposure measures to biological effects and 1526 subsequently adverse health outcomes. In the context of this Commentary, the “omics” component is 1527 intended to indicate that molecular methods based on omics techniques (e.g., genomics, 1528 transcriptomics, proteomics, and metabolomics) can be used in the selection of informative 1529 biomarkers and bioindicators of external exposure and internal dose. The more that is known about 1530 the etiology of diseases such as cancer, the more readily such informative biological surrogates can 1531 be identified. 1532 54 NCRP SC 1-21 Draft of March 31, 2015 [MR] 1533 NOT TO BE DISSEMINATED OR REFERENCED 5. Recommendations for Closing the Gaps towards an Integrated Approach 1534 1535 The current state of knowledge on the effects of low-dose radiation is incomplete and uncertain 1536 with respect to understanding the shape of the dose-response relationship and the level of risk at low 1537 doses. These deficiencies have long-term and profound consequences with regard to radiation 1538 protection guidance, compensation programs, and environmental contamination issues. There is a 1539 need to identify novel approaches to reduce these uncertainties. Research is needed to identify, 1540 evaluate, and utilize informative bioindicators of the health effects of interest (i.e., cancer and 1541 noncancer effects). Ideally, a bioindicator would be a quantitative predictor of the adverse health 1542 effect and measureable over the dose range of interest, particularly including low doses (i.e., 1543 <100 mGy). Such information would extend the knowledge of the dose-response relationship at 1544 doses below those that can be observed through epidemiologic studies. 1545 1546 To be able to utilize an approach for incorporating radiation biology data into the risk assessment 1547 process for low-dose and low dose-rate exposures, it is necessary to establish a framework for 1548 research planning and data collection. Such a framework has been presented in this Commentary and 1549 is basically one that involves the use of adverse outcome pathways and key events defining such 1550 pathways. Research should address this need in a prospective manner rather than in a retrospective 1551 one. Lastly, to ensure progress in multi-disciplinary radiation research and continuity of low-dose 1552 radiation research in general, it is essential that low-dose facilities are maintained and broadened and 1553 educational training programs are expanded to meet the growing needs of cross-disciplinary 1554 radiation science in the next generation of radiation researchers. 1555 1556 5.1 Targeted Research to Identify Adverse Outcome Pathways and Bioindicators of Response 1557 1558 To utilize such an approach outlined above, it is essential to identify adverse outcome pathways 1559 for radiation-related adverse health outcomes and the key events that characterize these. The types of 1560 bioindicators that will be informative in this regard are discussed above in Section 4. It remains very 1561 difficult to develop the required information for human tumors because of the lack of ability to 1562 accurately identify radiation-related tumors. Thus, it is necessary, in part, to use animal models of 1563 human tumors together with cellular systems to support the evaluation of the potential predictive 55 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1564 value of any prospective bioindicator. This general approach is depicted in the parallelogram 1565 presented in Figure 1.3. It has been proposed that adverse outcome pathways are likely to be 1566 conserved across species in support of integrated approaches to testing for chemical risks (Tollefsen 1567 et al., 2014) 1568 1569 It is initially necessary to establish the robustness of specific bioindicators and other intermediate 1570 endpoints for predicting the adverse outcome under study. Thus, it is required that any biological 1571 endpoint utilized for extrapolation purposes be predictive of the apical endpoint for which risk is 1572 being estimated. Such predictions can describe the shape of the dose-response curve or an estimate 1573 of the risk itself, depending on the nature of the specific biological endpoint. Of course, it is 1574 pertinent to establish, whenever possible, if there are additional dose-dependent modifications 1575 beyond the position of a particular bioindicator along the AOP. This can, in part, be overcome by 1576 using a suite of key-event bioindicators for steps along the AOP. As discussed in Sections 1 and 4, 1577 biomarkers are considered to be surrogates for key events in the formation of the apical endpoint, 1578 whereas bioindicators are defined as being key events themselves in an adverse outcome pathway for 1579 a particular adverse health outcome. For a quantitative assessment of risk, it is necessary to make use 1580 of bioindicators at dose levels below those at which the apical endpoint itself can be assessed with 1581 any degree of accuracy. In the case of cancer, it is also feasible to assess pre-neoplastic lesions as 1582 predictors of cancer outcome in some human studies. These are likely to be the most accurate 1583 predictors of a cancer outcome, given their proximity to the malignant cancer itself. 1584 1585 There are basically two classes of predictive biomarkers or bioindicators: those that can be 1586 predictive of the adverse health outcome (cancer or noncancer) and those that are predictive 1587 specifically of radiation-related cancer or noncancer outcomes. Knowledge of the cancer process has 1588 increased dramatically leading to the description of the Hallmarks of Cancer by Hanahan and 1589 Weinberg (2000; 2011) and the concept of gene alterations in cancer cells being classified as 1590 “drivers” and “passengers” (Pleasance et al., 2010). The use of this type of information forms the 1591 basis for the U.S. Environmental Protection Agency’s Guidelines for Carcinogen Risk Assessment 1592 (EPA, 2005). The approach is predicated on the fact that for the majority of chemicals (and exposure 1593 scenarios) there are very few epidemiologic data sets available other than those for occupational 1594 exposures. Thus, data from other sources (laboratory animal and in vitro cell studies) are used to 56 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1595 support the epidemiology. The specific approach relies upon the assessment of key events in the 1596 cancer process to be used as bioindicators in a qualitative manner today, but is anticipated to be more 1597 quantitative in the near future. A similar use of key events has been proposed for noncancer 1598 endpoints using toxicity data (Julien et al., 2009; Seed et al., 2005). It would seem feasible for the 1599 general principles developed for these diverse set of exposures and disease endpoints to be applied 1600 for the enhancement of the radiation risk assessment process. The aim, in this case, would be to 1601 design research that specifically measured key events for a particular disease outcome — in the first 1602 instance, likely to be cancer given the wealth of knowledge on the underlying molecular mechanisms 1603 of cancer development. 1604 1605 At this time, it is not readily feasible to assess the robustness of surrogate markers or key events 1606 in the radiation (or chemical) risk assessment process because too little reliable information has been 1607 developed. There is some developing information that will prove useful with enhancement. For 1608 example, Jarabek et al. (2009) describe the use of specific DNA adducts as bioindicators for use in 1609 the cancer risk assessment process, and chromosome alterations have been used to predict cancer 1610 outcomes at the group (not the individual) level (Bonassi et al., 2008). The real problem is the lack 1611 of bioindicators that are specific for radiation exposure. The advent of ultra-high-throughput DNA 1612 sequencing and the identification of a limited set of genes that can be mutated in cancer cells [about 1613 120 according to Greenman et al. (2007)] could lead to the identification of radiation-specific 1614 signature alterations. 1615 1616 5.2 Biologically-Based Modeling 1617 1618 The limitations of the direct use of epidemiologic data for estimating cancer and noncancer risks 1619 at low radiation doses were discussed in (Sections 2.1, 2.2 and 2.3). In cases where such direct use 1620 has been presented (e.g., Kendall et al. 2013; Mathews et al., 2013; Pearce et al., 2012), possible 1621 confounders [in particular in relation to the brain cancer findings in the study of Pearce et al. (2012)] 1622 and modest power [in the case of the study of Kendall et al. (2013)] make interpretation somewhat 1623 problematic. The present Commentary is predicated on the potential use of biological data to support 1624 the current risk assessment approach. The specific application would be to enhance the extrapolation 1625 from available high/medium dose epidemiologic data to dose levels and dose rates that are necessary 57 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1626 for radiation protection purposes (e.g., <100 mGy). In this regard, it has been proposed that some 1627 form of biologically-based dose-response modeling would meet this need (e.g., Conolly et al., 2004; 1628 Curtis et al., 2002; Little et al., 2008a; NCRP, 2012; Shuryak et al., 2010). However, here also, 1629 modeling on such a biological basis is uncertain outside the range of the available data that are used 1630 as input parameters to a biologically-based model. 1631 1632 5.2.1 Biologically-Based Dose-Response Approach 1633 1634 For risk assessment for environmental chemicals, because of the paucity of epidemiologic data, it 1635 is recommended that a biologically-based dose-response (BBDR) approach be used for predicting 1636 adverse health outcomes under low-dose chronic exposure scenarios (EPA, 2005). However, the 1637 approach has been used very sparingly in the regulatory process itself, largely because of the lack of 1638 availability of the appropriate data sets to provide input parameters to a BBDR model. The need for 1639 expanded use of BBDR models has been concisely expressed in Section 5.7 of NCRP (2012): 1640 1641 “The challenge of developing a biologically-based computational model to minimize 1642 uncertainty in dose-response modeling can be summarized as: understanding a 1643 sufficient amount of the relevant biology; acquiring enough data to parameterize the 1644 1645 1646 model; and developing the computational model.” Of course, this simple statement hides a degree of complexity that must be addressed. For 1647 example, how much of the detailed biology do we need to know and when are there sufficient data to 1648 sustain model development? This situation can be significantly alleviated if research is directed 1649 towards the needs identified by BBDR models. Research targeted to the risk assessment process is 1650 needed. 1651 1652 The initial development of BBDR as applied to carcinogenesis is described by Moolgavkar and 1653 Knudson (1981) and Moolgavkar and Venzon (1979) in their two-stage cancer model [usually 1654 referred to as the Moolgavkar-Venzon-Knudson model]. This simplified model has been expanded 1655 upon in subsequent years, as reviewed in Little (2010). The basic principle is that carcinogenesis can 1656 be considered as a two-step process. These two critical steps in carcinogenesis are considered to be 58 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1657 specific, irreversible, and inherited (at the cell level). The first step is assumed to give a small growth 1658 advantage through partial alteration of growth control, and the second is assumed to lead to total 1659 abrogation of growth control. Other assumptions include: that cancer arises from a single cell; 1660 transformation of stem cells are independent events; and the time from transformation of a single 1661 cell to a tumor is constant. Thus, what is developed is essentially a “working” model rather than one 1662 that is truly representative of the cancer process as described, for example, by Hanahan and 1663 Weinberg (2000; 2011). 1664 1665 The two-stage carcinogenesis model has been applied to a number of situations, for example, by 1666 Heidenreich et al. (2004) and Krewski et al. (2003) for radiation and by Conolly et al. (2004) for 1667 formaldehyde exposures. There remain some concerns that the use of this two-stage model does not 1668 make use of the considerable body of data that has been generated over the past 20 y on the 1669 understanding of cancer development and, in the present context, of radiation-related effects and 1670 radiation carcinogenesis. Multi-stage generalizations of the two-stage model have been developed 1671 (Little, 1995), and applied to model ionizing radiation exposure (Little, 1996; Little et al., 2002). 1672 Further generalizations of the model have been developed to incorporate genomic instability, and 1673 used for modeling population colon cancer data (Little and Li, 2007; Little and Wright, 2003; Little 1674 et al., 2008b). However, it is equally fair to note that no viable alternative BBDR has been proposed 1675 and applied to radiation risk assessment, with the possible exception of the multistage model 1676 developed by Shuryak et al. (2010) for the analysis of cancer risk patterns as a function of age-at- 1677 exposure in Japanese atomic-bomb survivors. The uncertainties inevitably associated with BBDR 1678 approaches are discussed in detail in NCRP (2012). 1679 1680 In addition to a simplified model approach (such as with the two-stage carcinogenesis model), it 1681 is feasible to use more realistic models of carcinogenesis, that is, models that establish if specific 1682 processes such as genomic instability or adaptive responses are predictive of the cancer response in 1683 specific studies (e.g., Eidemuller et al., 2011). Third, and perhaps the most informative type of 1684 BBDR model, are ones that are developed to examine the role of specific measured biological 1685 variables in the genesis of radiogenic cancer in a particular study. Examples of this informative 1686 approach have been reported by Heidenreich and Rosemann (2012) and Heidenreich et al. (2013). 59 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1687 Further development of this latter type of model using key-event bioindicators as input parameters is 1688 encouraged. 1689 1690 5.2.2 Systems Biology 1691 1692 Systems biology, defined here as the quantitative study of biological networks and pathways as 1693 integrated systems rather than as a set of isolated parts, is a young but rapidly evolving field that 1694 takes a data-intensive approach to biologically-based modeling. Systems radiation biology 1695 emphasizes the interactions of cell signaling pathways and networks, the distribution of effects and 1696 responses throughout these networks, as well as the redundancy between and within such networks 1697 for maintenance of cellular homeostasis and subsequent tissue function. This approach uses ‘omics- 1698 level data (e.g., genomics, transcriptomics, proteomics and metabolomics) to reverse engineer the 1699 biological circuitry of a cell, tissue, or organism with the goal of being able to predict the response to 1700 a perturbation, such as the mutation of a gene that effectively removes a node from the network, or 1701 an extrinsic stress, such as radiation exposure. Ultimately, systems biology also attempts to consider 1702 tissue responses in the context of the whole irradiated system rather than as the response of 1703 individual cells. 1704 1705 While much is known about the DNA damage induced by the deposition of energy from 1706 exposure to ionizing radiation and the subsequent cellular responses, there is a significant gap in our 1707 understanding of how these might lead to detrimental health effects years after exposure. Current 1708 radiation modeling and risk evaluation tend to be limited to simplistic models that do not reflect the 1709 inherent biological diversity of humans or the mechanisms that underlie radiation risks (Barcellos- 1710 Hoff, 2008). For instance, the interactions between irradiated cells within a tissue and the 1711 modulating effects of the tissue microenvironment (Barcellos-Hoff, 2005) are not accounted for in 1712 many models. Linking mechanisms of cellular and molecular responses to radiation exposure to 1713 macroscopic processes at the tissue, organ, and whole organism levels would improve our ability to 1714 predict cancer risk in response to irradiation. This is our multiscale systems challenge: to understand 1715 the sequence of events occurring in tissues following irradiation and why in some instances exposure 1716 can lead to cancer or other phenotypic alterations, and in other instances no long-term phenotypic 1717 changes are observed. 60 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1718 1719 Recent advances in high-throughput ‘omics technologies enable interrogation of induced 1720 radiation effects at the genomic [reviewed in Amundson (2008)], proteomic (Lin et al., 2009; 1721 Menard, et al. 2006; Park et al., 2006), and metabolomic (Lanz et al., 2009; Tyburski et al., 2008; 1722 2009; Zhang, et al., 2014) levels. Radiation systems biology attempts to integrate the resulting 1723 ‘omics data to enable modeling of radiation dose and dose-rate effects in a complex tissue as a 1724 function of time, and relate these to observable functional physiological and /or physical changes in 1725 that tissue after irradiation. 1726 1727 It is clear that vast amounts of data can, and will, be generated. The usefulness of these data will 1728 depend on the development of computational modeling approaches to organize an abundance of 1729 complex biological data in a predictive, multicellular framework. The challenge is to understand the 1730 flow of information between cell types in a given tissue and why different tissues vary in their 1731 response to radiation. This demands approaches that can elucidate the connectivity among and 1732 between signaling networks rather than focusing on reducing analysis to individual signaling 1733 components. 1734 1735 Network construction of radiation responses will require quality, dose-dependent measurements 1736 over a protracted time frame (Eschrich et al., 2009). Multiple doses, dose rates and time points will 1737 need to be interrogated at the global level in order to address the question at hand. The impact of 1738 post-transcriptional and post-translational modifications as well as potential epigenetic modifications 1739 that influence cell signaling will have to be evaluated and incorporated into the model. In this way 1740 the combined behavior of interdependent processes that dictate cell responses and the fundamental 1741 properties of the networks involved, such as redundancy, modularity, robustness, and feedback 1742 1743 1744 1745 control can be assessed. The systems biology approach for radiation risk assessment will require: 1746 multi-level ‘omics data from well-designed experiments; 1747 knowledge of the pathways involved; 1748 developing the computational modeling approaches to organize complex biological data in a 1749 predictive, multi-cellular framework that can be integrated with radiation risk models; 61 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1750 a long-term commitment from federal funding agencies; and 1751 dedicated cross-disciplinary teams of investigators to link studies at all levels to relevant 1752 1753 1754 1755 1756 health outcomes. 5.3 Registries of Medical-Imaging Exposures Linked to Outcomes and Biological Materials The use of ionizing radiation for medical imaging has grown significantly in recent years. In the 1757 United States, exposure to ionizing radiation from medical-imaging procedures3 in 2006 was 1758 approximately six times greater than in 1980 (NCRP, 2009d), with an average effective dose to a 1759 member of the U.S. population of ~3 mSv, comparable to the annual effective dose from natural 1760 background radiation in 2006; the dose from natural background was the same as in 1980. This 1761 increased dose from medical-imaging procedures was attributed to the greatly increased usage in 1762 2006 of higher-dose procedures particularly computed tomography (CT) and nuclear medicine 1763 compared to 1980 (NCRP, 2009d). Brenner and Hall (2007) reported that almost 82 million CT 1764 procedures were performed annually in the United States in 2011, up from 46 million in 2000 and 1765 13 million in 1990. Mettler et al. (2009), reported that cardiac diagnostic nuclear procedures 1766 increased from 1 % of the total number of diagnostic nuclear medicine examinations performed in 1767 1973 to 57 % in 2005). Further, Brenner and Hricak (2010) state that an increase in emerging 1768 imaging modalities such as positron-emission tomography CT, single-photon emission CT, and 1769 potentially CT for screening of high-risk asymptomatic patients (e.g., smokers screened for early 1770 1771 1772 lung cancer detection) are likely. 1773 patient demographics, as available in other countries (e.g., Nordic countries) could be useful for 1774 tracking long-term sequelae of these procedures, including the potential for carcinogenic risk 1775 associated with extensive medical imaging using these technologies. An ideal situation would also 1776 include the collection and storage of biospecimens. In addition, there may be other potentially 1777 informative radiation-exposed groups, such as astronauts, where follow-up and collection of samples 1778 would be valuable. However, such a comprehensive endeavor may not be economically feasible as Therefore registries of medical-imaging procedures, including indication for examination and 3 Medical-imaging procedures included computed tomography, converntional radiography and fluoroscopy, interventional fluoroscopy, and nuclear medicine. 62 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1779 the cost of establishing and maintaining the registry, harmonizing the input and follow-up 1780 1781 1782 1783 1784 1785 1786 procedures and the complex issues of sample collection and storage may be extensive. 1787 information and strong quantitative data to inform risk assessments for human exposure. This 1788 research requires the availability of facilities that can provide accurate dose delivery at the relevant 1789 dose-rates. The necessary facilities have variable requirements. Some must be able to accommodate 1790 cell culture systems, including tissue-mimetic models, and these require ancillary equipment to 1791 provide detailed dosimetry and to maintain defined oxygen tensions. Some animal work will be 1792 possible on a small scale using these facilities, but larger-scale, dedicated facilities with the 1793 capability to conduct long-term exposures at low dose rates are critical for some aspects of this 1794 work. Specialized facilities are also needed for studies with low-energy sources, charged-particle 1795 microbeams, and inhaled sources, and to investigate long-term low dose-rate effects. Support for the 1796 maintenance and development of this diverse group of facilities is essential to the conduct of a robust 1797 1798 1799 1800 1801 research program at low doses and low dose rates. 5.4.2 Interdisciplinary Training 1802 critical elements that are required to support this highly interdisciplinary field (Boice, 2014b; NCRP, 1803 2015). Individuals trained across disciplines are essential to meet the health needs of the nation as 1804 we evaluate radiation risk. Current training programs are often narrowly focused and rarely include 1805 the breadth of coursework and practical training needed to develop robust research programs. 1806 Integrated training programs with representation of the disciplines of radiation biology, molecular 1807 biology, pathology, radiation dosimetry, health physics, epidemiology, and statistics should be 1808 developed and supported to ensure the availability of a cadre of scientists who can advance our 1809 understanding of radiation health effects at low doses. 5.4 Facilities and Interdisciplinary Training 5.4.1 Facilities Basic and applied research studies at low doses and low dose rates can provide mechanistic The science of radiation protection will continue to develop only if individuals are trained in 1810 63 NCRP SC 1-21 Draft of March 31, 2015 [MR] 1811 NOT TO BE DISSEMINATED OR REFERENCED Glossary 1812 1813 absorbed dose (D): The quotient of dε by dm, where dε is the mean energy imparted to matter of mass dm 1814 (i.e., D = dε /dm). The unit for D is joule per kilogram (J kg–1) with the special name gray (Gy) (see also 1815 organ dose). 1816 1817 1818 1819 1820 1821 ascertainment bias: Arises when there is variation in ascertainment of disease status, perhaps correlated with exposure variables. adaptive response: In radiobiology, the theory that a low dose of ionizing radiation can reduce the effect of a higher dose of ionizing radiation when the higher dose is administered after a short time delay. Adult Health Study (AHS): A subcohort of the Life Span Study, which provides biennial medical follow-up for a subcohort of about 20,000 members of the Life Span Study (see Life Span Study). 1822 as low as reasonably achievable (ALARA): A principle of radiation protection philosophy that requires that 1823 exposures to ionizing radiation should be kept as low as reasonably achievable, economic and social 1824 factors being taken into consideration. The protection from radiation exposure is ALARA when the 1825 expenditure of further resources would be unwarranted by the reduction in exposure that would be 1826 achieved. 1827 background radiation: The amount of radiation to which a member of the population is exposed from 1828 natural sources, such as terrestrial radiation from naturally-occurring radionuclides in the soil, cosmic 1829 radiation originating in outer space, and naturally-occurring radionuclides deposited in the human body. 1830 The natural background radiation received by an individual depends on geographic location and living 1831 habits. In the United States, the average effective dose from background radiation is ~1 mSv y–1, 1832 excluding indoor radon which amounts to ~2 mSv y–1 on average. 1833 Berkson error: The Berkson error model assumes that true dose is equal to the observed dose plus an error 1834 introduced by individual peculiarity, which is defined as a random variable that has a mean of zero and is 1835 independent of observed dose. 1836 bias (epidemiology): Any process at any stage in the conduct of the study that tends to produce results or 1837 conclusions that differ systematically from the truth (see follow-up bias, ascertainment bias, recall bias, 1838 and confounding factor). 1839 bioindicator: A cellular alteration that is on the pathway to the disease endpoint itself, such as a specific 1840 mutation in a target cell that is associated with tumor formation. Thus, a bioindicator can be perceived as 1841 informing on the shape of dose-response curve for the disease outcome or on cancer frequency itself, and 1842 therefore, is equivalent to a key event. 1843 1844 biomarker: A biological phenotype (e.g., chromosome alteration, DNA adduct, gene expression change, specific metabolite) that can be used to indicate a response at the cell or tissue level to an exposure. In this 64 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1845 regard, it is generally a measure of the potential for development of an adverse outcome such as cancer 1846 (e.g., a predictor of exposure level). 1847 bystander effect: In radiobiology, the term is used to describe an effect on cells in which the energy had not 1848 been directly deposited. In most instances, the cells so affected were neighbors of the cells directly 1849 impacted by the radiation. 1850 1851 cancer: A general term for more than 100 diseases characterized by abnormal and uncontrolled growth of cells. 1852 carcinogen: An agent that is associated with an increased risk of cancer. 1853 carcinogenesis: Induction of cancer by radiation or any other agent (a somatic effect). 1854 case-control study: An epidemiologic study in which people with disease and a similarly composed control 1855 1856 group are compared in terms of exposures to a putative causative agent. Chernobyl recovery workers: The civil and military personnel cleanup workers who were called upon to 1857 deal with consequences of the 1986 Chernobyl nuclear disaster on the site of the event in the 1858 former Soviet Union. 1859 Classical measurement error: The Classical measurement error model assumes that observed dose equals 1860 true dose plus measurement error, where measurement error is a random variable that is independent of 1861 the true dose and has a mean of zero. 1862 cohort study: An epidemiologic study in which groups of people (the cohort) are identified with respect to 1863 the presence or absence of exposure to a disease-causing agent, and in which the outcomes of disease 1864 rates are compared; also called a follow-up study. 1865 confounding factor: In statistics, an extraneous variable in a statistical model that correlates (directly or 1866 inversely) with both the dependent variable and the independent variable (e.g., a factor that is correlated 1867 both with the disease under study and with an exposure of interest). 1868 1869 1870 1871 1872 chromosome aberration: A missing, extra, or irregular portion of chromosomal DNA. It can be from an atypical number of chromosomes or a structural abnormality in one or more chromosomes. cytogenetic: Relating to the branch of genetics that is concerned with the study of the structure and function of the cell, especially the chromosomes. deoxyribonucleic acid (DNA): Genetic material of cells; a complex molecule of high molecular weight 1873 consisting of deoxyribose, phosphoric acid, and four bases which are arranged as two long chains that 1874 twist around each other to form a double helix joined by hydrogen bonds between the complementary 1875 components. 1876 1877 DNA damage: An alteration in the chemical structure of DNA, such as a break in a strand of DNA, a base missing from the backbone of DNA, or a chemically changed base. 65 NCRP SC 1-21 Draft of March 31, 2015 [MR] 1878 1879 1880 NOT TO BE DISSEMINATED OR REFERENCED dose: A general term used when the context is not specific to a particular dose quantity. When the context is specific, the name for the quantity is used (e.g., absorbed dose). dose and dose-rate effectiveness factor (DDREF): A judged factor that generalizes the usually lower 1881 biological effectiveness (per unit of dose) of radiation exposures at low doses and low dose rates as 1882 compared with exposures at high doses and high dose rates. 1883 dose limit: A limit on radiation dose that is applied for exposure to individuals or groups of individuals in 1884 order to prevent the occurrence of radiation-induced tissue reactions or to limit the probability of 1885 radiation-related stochastic effects to an acceptable level (see tissue reaction and stochastic effect). 1886 dose rate: Dose delivered per unit time. Can refer to any dose quantity (e.g., organ dose). 1887 dose reconstruction: Retrospective assessment of dose to identifiable or representative individuals or 1888 populations by any means, especially absorbed dose to specific organs or tissues. 1889 dose response: The way in which an effect, or response, depends on dose. 1890 effective dose: The sum over specified tissues of the products of the equivalent dose in a tissue or organ and 1891 the tissue weighting factor for that tissue or organ. Equivalent dose is the product of the radiation 1892 weighting factor and the mean absorbed dose in a tissue or organ (organ dose).The SI unit for effective 1893 dose is joule per kilogram (J kg–1), with the special name sievert (Sv). A similar quantity used by the 1894 Nuclear Regulatory Commission is named total effective dose equivalent. 1895 1896 electron: Subatomic charged particle. Negatively charged particles are parts of stable atoms. Both negatively and positively charged electrons may be expelled from the radioactive atom when it disintegrates. 1897 exposome: (see exposomics). 1898 exposomics: The name given to the study of the exposome. The exposome is broadly defined as a measure of 1899 the total exposure of an individual in a lifetime and the relationship of this exposure to health outcomes. 1900 The “omics” component is intended to indicate that molecular methods based on omics techniques (e.g., 1901 genomics, transcriptomics, proteomics, and metabolomics) can be used in the selection of informative 1902 biomarkers and bioindicators of the exposure. 1903 excess absolute risk (EAR): The difference in absolute risk between exposed and control populations. 1904 excess relative risk (ERR): The ratio of the excess risk of a specified disease to the probability of the same 1905 effect in the unexposed population (i.e., the relative risk minus one). The relative is ratio of the incidence 1906 rate of a given disease in those exposed to the incidence rate of that disease in those not exposed. 1907 exposure: In this Commentary, exposure is used in a general sense meaning to be irradiated. 1908 follow-up bias: Arises when there is a lack of follow-up information, for example if persons have, unknown 1909 to the investigator, migrated outside of the study area, so that their health status cannot be reported. 1910 gamma rays: Short-wavelength electromagnetic radiation of nuclear origin (approximate range of energy: 1911 10 keV to 9 MeV). 66 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1912 genomic instability (delayed genetic effects): An increased rate of acquiring genetic change. 1913 genotoxin: Any substance capable of causing damage to cellular DNA and thus 1914 producing mutations or cancer. 1915 gray (Gy): The International System (SI) unit of absorbed dose of radiation, 1 Gy = 1 J kg–1. 1916 germline mutation: Any detectable and heritable variation in the lineage of germ cells. Mutations in these 1917 cells are transmitted to offspring, 1918 incidence: The rate of occurrence of a disease, usually expressed in number of cases per million. 1919 ionizing radiation: Any electromagnetic or particulate radiation capable of producing ions, directly or 1920 1921 1922 1923 1924 indirectly, in its passage through matter. key event: An empirically observable precursor step that is itself a necessary element of the mode of action or is a biologically-based marker for such an element (see also bioindicator). Life Span Study (LSS): Study of the Japanese atomic-bomb survivors; the sample consists of 93,741 persons (in city) of whom 86,572 survivors have a defined dose assigned to them. 1925 lifetime risk: The probability during one’s lifetime of expressing a given health outcome. 1926 linear energy transfer (LET): Average amount of energy lost per unit of particle track length and expressed 1927 in keV μm–1. 1928 low-LET: Radiation having a low linear energy transfer; for example, electrons, x rays, and gamma rays. 1929 high-LET: Radiation having a high linear energy transfer; for example, alpha particles and interaction 1930 products of fast neutrons. 1931 linear-nonthreshold model: In radiation protection, a model that assumes that the long term, biological 1932 damage caused by ionizing radiation (essentially the cancer risk) is directly proportional to the dose. 1933 linkage study: A study that looks for patterns of inheritance of genetic markers in large families in which a 1934 1935 particular condition is common. mode of action: For both cancer and noncancer endpoints, a sequence of key events and processes starting 1936 with interaction of an agent with a cell, proceeding through operational and anatomical changes, and 1937 resulting in formation of an adverse outcome (see key event). 1938 Monte Carlo simulation: Computation of a probability distribution of an output of a model on the basis of 1939 repeated calculations using random sampling of input variables from specified probability distributions. 1940 natural background radiation (see background radiation): 1941 neutrons: Particles with a mass similar to that of a proton, but with no electrical charge. Because they are 1942 1943 1944 electrically neutral, they cannot be accelerated in an electrical field. noncancer: Health effects other than cancer (e.g., cataracts, cardiovascular disease) that occur in the exposed individual. 67 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 1945 nonionizing radiation: Electromagnetic radiation that includes the ultraviolet, visible, infrared, microwave, 1946 radiofrequency, and extremely-low-frequency portions of the electromagnetic spectrum. Unlike ionizing 1947 radiation, nonionizing radiation is unable to ionize atoms in its interactions with matter. 1948 1949 1950 1951 1952 1953 1954 occupational exposure: The exposure received by an individual in a restricted area, or in the course of employment in which the individual’s duties necessarily involve exposure to radiation. ’omics: Refers to molecular methods for measuring all of a certain molecular specie in a cell (e.g., genomics, transcriptomics, proteomics, and metabolomics). organ dose: The mean absorbed dose in an organ or tissue, obtained by integrating or averaging absorbed doses at points in the organ or tissue. parallelogram approach: As initially applied to the estimation of heritable risk: the principle that genetic 1955 damage in human germ cells can be estimated by measuring a common end point in humans and animals, 1956 such as mutations or chromosomal aberrations in lymphocytes, and a corresponding genetic end point in 1957 germ cells of animals, the desired target tissue generally inaccessible in man. 1958 partial body: Exposure to only part of a body, as opposed to exposure of the whole body. 1959 polygene: A group of nonallelic genes that together influence a phenotypic trait. 1960 quality factor: The factor by which absorbed dose at a point is modified in order to express the effectiveness 1961 of a given type of ionizing radiation in inducing stochastic effects. 1962 radiation: (1) The energy propagated as waves; radiation or radiant energy, when unqualified, usually refers 1963 to electromagnetic radiation; commonly classified by frequency (e.g., infrared, visible, ultraviolet, x rays, 1964 and gamma rays), and (2) corpuscular emission, such as alpha and beta particles. 1965 radiation weighting factor: A factor used to allow for differences in the biological effectiveness between 1966 different radiations when calculating equivalent dose. These factors are independent of the tissue or organ 1967 irradiated. Equivalent dose is the product of the radiation weighting factor and the mean absorbed dose in 1968 a tissue or organ (organ dose). 1969 1970 radiosensitivity: The relative susceptibility of cells, tissues, organs or organisms to the harmful effect of ionizing radiation. 1971 recall bias: Arises when information, for example on exposure, is collected retrospectively, and patients, or 1972 their relatives, are subject to differential recall of this information, depending on their disease status. 1973 relative biological effectiveness: A factor used to compare the biological effectiveness of absorbed doses 1974 from different types of ionizing radiation, determined experimentally. Relative biological effectiveness is 1975 the ratio of the absorbed dose of a reference radiation (often taken as 250 kVp x rays) to the absorbed 1976 dose of the radiation in question required to produce an identical biological effect in a particular 1977 experimental organism or tissue. 1978 risk: The probability of a specified effect or response occurring. 68 NCRP SC 1-21 Draft of March 31, 2015 [MR] 1979 1980 NOT TO BE DISSEMINATED OR REFERENCED risk estimate: The number of cases (or deaths) that are projected to occur in a specified exposed population per unit dose for a defined exposure regime and expression period. 1981 sievert (Sv): The special name for the unit of effective dose. 1 Sv = 1 J kg–1. 1982 somatic mutation: A mutation occurring in a somatic cell as opposed to a germ cell. 1983 stochastic effect: An effect for which the probability of occurrence, rather than its severity, is a function of 1984 1985 1986 1987 1988 1989 radiation dose without threshold (e.g., cancer). systems biology: The quantitative study of biological networks and pathways as integrated systems rather than as a set of isolated parts. tissue reaction: Injury in population of cells, characterized by a threshold dose and an increase in the severity of the reaction as the dose is increased further. tissue weighting factor: A factor representing the ratio of risk of stochastic effects attributable to 1990 irradiation of a given organ or tissue to the total risk when the whole body is irradiated 1991 uniformly. The factor is assumed to be independent of the type of radiation or energy of the 1992 radiation. 1993 total effective dose equivalent: (see effective dose). 1994 uncertainty: Lack of sureness or confidence in predictions of models or results of measurements. 1995 Uncertainties may be categorized as those due to stochastic variation, or as those due to lack of 1996 knowledge founded on an incomplete characterization, understanding or measurement of a system. 1997 69 NCRP SC 1-21 Draft of March 31, 2015 [MR] 1998 NOT TO BE DISSEMINATED OR REFERENCED Symbols, Abbreviations and Acronyms 1999 2000 AHS Adult Health Study (Japanese atomic-bomb survivors) 2001 ALARA as low as reasonably achievable 2002 AML acute myeloid leukemia 2003 ATM ataxia-telangiectasia mutated 2004 BBDR biologically-based dose-response 2005 BEIR Committee on the Biological Effects of Ionizing Radiation 2006 CT computed tomography 2007 DDREF dose and dose-rate effectiveness factor 2008 DNA deoxyribonucleic acid 2009 DSBs double-strand breaks (DNA) 2010 EAR excess absolute risk 2011 ERR excess relative risk 2012 GWAS genome-wide association study(ies) 2013 LET linear energy transfer 2014 LSS Life Span Study (Japanese atomic-bomb survivors) 2015 SNP single nucleotide polymorphism 2016 SSBs single-strand breaks (DNA) 2017 TERT, hTERT telomerase reverse transcriptase (hTERT in humans) 2018 WARP Where are the Radiation Professionals? 2019 WECARE Women’s Environmental Cancer and Radiation Epidemiology (Study) 2020 70 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2021 NOT TO BE DISSEMINATED OR REFERENCED References 2022 2023 ADELEYE, Y., ANDERSEN, M., CLEWELL, R., DAVIES, M., DENT, M., EDWARDS, S., FOWLER, P., 2024 MALCOMBER, S., NICOL, B., SCOTT, A., SCOTT, S., SUN, B., WESTMORELAND, C., WHITE, A., 2025 ZHANG, O. and CARMICHAEL, P.L. (2014). “Implementing Toxicity Testing in the 21st Century 2026 (TT21C): Making safety decisions using toxicity pathways and progress in a prototype risk assessment,” 2027 Toxicology pii: S0300-483X(14)00032-8. doi: 10.1016/j.tox.2014.02.007 (Epub ahead of print). 2028 2029 2030 AINSBURY, E.A., BOUFFLER, S.D., DORR, W., GRAW, J., MUIRHEAD, C.R., EDWARDS, A.A. and COOPER, J. (2009). “Radiation cataractogenesis: A review of recent studies,” Radiat. Res. 172(1), 1–9. AINSBURY, E.A., BAKHANOVA, E., BARQUINERO, J.F., BRAI, M., CHUMAK, V., CORRECHER, V., 2031 DARROUDI, F., FATTIBENE, P., GRUEL, G., GUCLU, I., HORN, S., JAWORSKA, A., KULKA, U., 2032 LINDHOLM, C., LLOYD, D., LONGO, A., MARRALE, M., MONTEIRO GIL, O., OESTREICHER, 2033 U., PAJIC, J., RAKIC, B., ROMM, H., TROMPIER, F., VERONESE, I., VOISIN, P., VRAL, A., 2034 WHITEHOUSE, C.A., WIESER, A., WODA, C., WOJCIK, A. and ROTHKAMM, K. (2011). “Review 2035 of retrospective dosimetry techniques for external ionising radiation exposures,” Radiat. Prot. Dosim. 2036 147(4), 573–592. 2037 2038 2039 2040 2041 2042 2043 2044 ALLEN, C., ASHLEY, A.K., HROMAS, R. and NICKOLOFF, J.A. (2011). “More forks on the road to replication stress recovery,” J. Mol. Cell. Biol. 3(1), 4–12. AMUNDSON, S.A. (2008). “Functional genomics in radiation biology: A gateway to cellular systems-level studies,” Radiat. Environ. Biophys. 47(1), 25–31. AMUNDSON, S.A. and CHEN, D.J. (1996). “Inverse dose-rate effect for mutation induction by gamma-rays in human lymphoblasts,” Int. J. Radiat. Biol. 69(5), 555–563. AYPAR, U., MORGAN, W.F. and BAULCH, J.E. (2011). “Radiation-induced genomic instability: Are epigenetic mechanisms the missing link?,” Int. J. Radiat. Biol. 87(2), 179–191. 2045 AZIZOVA, T.V., MUIRHEAD, C.R., DRUZHININA, M.B., GRIGORYEVA, E.S., VLASENKO, E.V., 2046 SUMINA, M.V., O’HAGAN, J.A., ZHANG, W., HAYLOCK, R.G. and HUNTER, N. (2010a). 2047 “Cardiovascular diseases in the cohort of workers first employed at Mayak PA in 1948–1958,” Radiat. 2048 Res. 174(2), 155–168. 2049 AZIZOVA, T.V., MUIRHEAD, C.R., DRUZHININA, M.B., GRIGORYEVA, E.S., VLASENKO, E.V., 2050 SUMINA, M.V., O’HAGAN, J.A., ZHANG, W., HAYLOCK, R.G. and HUNTER, N. (2010b). 2051 “Cerebrovascular diseases in the cohort of workers first employed at Mayak PA in 1948–1958,” Radiat. 2052 Res. 174(6), 851–864. 2053 2054 AZIZOVA, T.V., MUIRHEAD, C.R., MOSEEVA, M.B., GRIGORYEVA, E.S., SUMINA, M.V., O'HAGAN, J., ZHANG, W., HAYLOCK, R.J. and HUNTER, N. (2011). “Cerebrovascular diseases in 71 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2055 nuclear workers first employed at the Mayak PA in 1948–1972,” Radiat. Environ. Biophys. 50(4), 539– 2056 552. 2057 AZIZOVA, T.V., HAYLOCK, R.G., MOSEEVA, M.B., BANNIKOVA, M.V. and GRIGORYEVA, E.S. 2058 (2014). “Cerebrovascular diseases incidence and mortality in an extended Mayak Worker Cohort 2059 1948–1982,” Radiat. Res. 182(5), 529–544. 2060 AZZAM, E.I., DE TOLEDO, S.M., RAAPHORST, G.P. and MITCHEL, R.E. (1996). “Low-dose ionizing 2061 radiation decreases the frequency of neoplastic transformation to a level below the spontaneous rate in 2062 C3H 10T1/2 cells,” Radiat. Res. 146(4), 369–373. 2063 BARBER, R., PLUMB, M.A., BOULTON, E., ROUX, I. and DUBROVA, Y.E. (2002). “Elevated mutation 2064 rates in the germ line of first- and second-generation offspring of irradiated male mice,” Proc. Natl. Acad. 2065 Sci. USA 99(10), 6877–6882. 2066 2067 2068 2069 2070 2071 2072 BARCELLOS-HOFF, M.H. (2005). “Integrative radiation carcinogenesis: Interactions between cell and tissue responses to DNA damage,” Semin. Cancer Biol. 15(2), 138–148. BARCELLOS-HOFF, M.H. and COSTES, S.V. (2006). “A systems biology approach to multicellular and multi-generational radiation responses,” Mutat. Res. 597(1–2), 32–38. BARCELLOS-HOFF, M.H. (2008). “Cancer as an emergent phenomenon in systems radiation biology,” Radiat. Environ. Biophys 47(1), 33–38. BARQUINERO, J.F., BARRIOS, L., CABALLIN, M.R., MIRO, R., RIBAS, M., SUBIAS, A. and 2073 EGOZCUE, J. (1995). “Occupational exposure to radiation induces an adaptive response in human 2074 lymphocytes,” Int. J. Radiat. Biol. 67(2), 187–191. 2075 BELYAKOV, O.V., MITCHELL, S.A., PARIKH, D., RANDERS-PEHRSON, G., MARINO, S.A., 2076 AMUNDSON, S.A., GEARD, C.R. and BRENNER, D.J. (2005) “Biological effects in unirradiated 2077 human tissue induced by radiation damage up to 1 mm away,” Proc. Natl. Acad. Sci. USA, 102(40), 2078 14203–14208. 2079 BELYAKOV, O.V., FOLKARD, M., MOTHERSILL, C., PRISE, K.M. and MICHAEL, B.D. (2006). 2080 “Bystander-induced differentiation: A major response to targeted irradiation of a urothelial explant 2081 model,” Mutat. Res. 597(1–2), 43–49. 2082 BHATTI, P., DOODY, M.M., PRESTON, D.L., KAMPA, D., RON, E., WEINSTOCK, R.W., SIMON, S., 2083 EDWARDS, A.A. and SIGURDSON, A.J. (2008). “Increased frequency of chromosome translocations 2084 associated with diagnostic x-ray examinations,” Radiat. Res. 170(2), 149–155. 2085 BISSELL, M.J., WARNER, H.R., BERGET, S.M., FRY, R.J.M., HANAWALT, P.C., KASTAN, M., 2086 KORNBERG, A., LUTZE-MANN, L., SOUZA, K.A., ULLRICH, R., VIJG, J. and CHATTERJEE, A. 2087 (1997). Modeling Human Risk: Cell and Molecular Biology in Context, DE-97007347, LBNL-40278 2088 (National Technical Information Service, Springfield, Virginia). 72 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2089 2090 2091 2092 2093 2094 2095 2096 2097 2098 2099 NOT TO BE DISSEMINATED OR REFERENCED BITHELL, J.F. and STEWART, A.M. (1975). “Pre-natal irradiation and childhood malignancy: A review of British data from the Oxford Survey,” Br. J. Cancer 31(3), 271–287. BLYTH, B.J. and SYKES, P.J. (2011). “Radiation-induced bystander effects: What are they, and how relevant are they to human radiation exposures?” Radiat. Res. 176(2), 139–157. BOICE, J.D., JR. (2012). “A study of one million U.S. radiation workers and veterans: A new NCRP initiative (DOE Grant Awarded September 2012),” Health Phys. News XL(11), 7–10. BOICE, J.D., JR. (2014a). “Invited editorial: The importance of radiation worker studies,” J. Radiol. Prot. 34(2014), E7–E12. BOICE, J.D., JR. (2014b). “WARP—where are the radiation professionals?” Health Phys. News XLLII(4), 22. BOICE, J.D., JR., BLETTNER, M., KLEINERMAN, R.A., STOVALL, M., MOLONEY, W.C., 2100 ENGHOLM, G., AUSTIN, D.F., BOSCH, A., COOKFAIR, D.L. and KREMENTZ, E.T. (1987). 2101 “Radiation dose and leukemia risk in patients treated for cancer of the cervix,” J. Natl. Cancer Inst. 79(6), 2102 1295–1311. 2103 BOICE, J.D., JR., TAWN, E.J., WINTHER, J.F., DONALDSON, S.S., GREEN, D.M., MERTENS, A.C., 2104 MULVIHILL, J.J., OLSEN, J.H., ROBISON, L.L. and STOVALL, M. (2003). “Genetic effects of 2105 radiation therapy for childhood cancer,” Health Phys. 85(1), 65–80. 2106 BONASSI, S., HAGMAR, L., STROMBERG, U., MONTAGUD, A.H., TINNERBERG, H., FORNI, A., 2107 HEIKKILA, P., WANDERS, S., WILHARDT, P., HANSTEEN, I.L., KNUDSEN, L.E. and NORPPA, 2108 H. (2000). “Chromosomal aberrations in lymphocytes predict human cancer independently of exposure to 2109 carcinogens,” Cancer Res. 60(6), 1619–1625. 2110 BONASSI, S., NORPPA, H., CEPPI, M., STROMBERG, U., VERMEULEN, R., ZNAOR, A., CEBULSKA- 2111 WASILEWSKA, A., FABIANOVA, E., FUCIC, A., GUNDY, S., HANSTEEN, I.L., KNUDSEN, L.E., 2112 LAZUTKA, J., ROSSNER, P., SRAM, R.J. and BOFFETTA, P. (2008). “Chromosomal aberration 2113 frequency in lymphocytes predicts the risk of cancer: Results from a pooled cohort study of 22 358 2114 subjects in 11 countries,” Carcinogenesis 29(6), 1178–1183. 2115 BOOBIS, A.R., COHEN, S.M., DELLARCO, V., MCGREGOR, D., MEEK, M.E., VICKERS, C., 2116 WILLCOCKS, D. and FARLAND, W. (2006). “IPCS framework for analyzing the relevance of a cancer 2117 mode of action for humans,” Crit. Rev. Toxicol. 36(10), 781–792. 2118 2119 2120 2121 BOSI, A. and OLIVIERI, G. (1989). “Variability of the adaptive response to ionizing radiations in humans,” Mutat. Res. 211(1), 13–17. BOSS, M., BRISTOW, R. and DEWHIRST, M. (2014). “Linking the history of radiation biology to the hallmarks of cancer,” Radiat. Res. 181(6), 561–577. 73 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2122 NOT TO BE DISSEMINATED OR REFERENCED BOUFFLER, S.D., BRIDGES, B.A., COOPER, D.N., DUBROVA, Y., MCMILLAN, T.J., THACKER, J., 2123 WRIGHT, E.G. and WATERS, R. (2006). “Assessing radiation-associated mutational risk to the 2124 germline: Repetitive DNA sequences as mutational targets and biomarkers,” Radiat. Res. 165(3), 249– 2125 268. 2126 BOUVILLE, A., TOOHEY, R.E., BOICE, J.D., JR., BECK, H.L., DAUER, L.T., ECKERMAN, K.F., 2127 HAGEMEYER, D., LEGGETT, R.W., MUMMA, M., NAPIER, B., PRYOR, K.H., ROSENSTEIN, M., 2128 SCHAUER, D.A., SHERBINI, S., STRAM, D.O., THOMPSON, J.L., TILL, J.E., YODER, C. and 2129 ZEITLIN, C. (2015). “Dose reconstruction for the million worker study: Status and guidelines,” Health 2130 Phys. 108(2), 206–220. 2131 BOZIC, I., ANTAL, T., OHTSUKI, H., CARTER, H., KIM, D., CHEN, S., KARCHIN, R., KINZLER, 2132 K.W., VOGELSTEIN, B. and NOWAK, M.A. (2010). “Accumulation of driver and passenger mutations 2133 during tumor progression,” Proc. Natl. Acad. Sci. (USA) 107(43), 18545–18550. 2134 2135 2136 2137 2138 2139 2140 2141 2142 2143 2144 BRANZEI, D. and FOIANI, M. (2009). “The checkpoint response to replication stress,” DNA Repair (Amst.) 8(9), 1038–1046. BRENNER, D.J. (1994). “The significance of dose rate in assessing the hazards of domestic radon exposure,” Health Phys. 67(1), 76–79. BRENNER, D.J. and HALL, E.J. (2007). “Computed tomography—an increasing source of radiation exposure,” N. Engl. J. Med. 357(22), 2277–2284. BRENNER, D.J. and HRICAK, H. (2010). “Commentary: Radiation exposure from medical imaging: Time to regulate?” JAMA 304(2), 208–209. BRENNER, D.J. and SACHS, R.K. (2003). “Domestic radon risks may be dominated by bystander effects— but the risks are unlikely to be greater than we thought,” Health Phys. 85(1), 103–108. BRENNER, D.J., HAHNFELDT, P., AMUNDSON, S.A. and SACHS, R.K. (1996). “Interpretation of 2145 inverse dose-rate effects for mutagenesis by sparsely ionizing radiation,” Int. J. Radiat. Biol. 70(4), 447– 2146 458. 2147 2148 2149 2150 2151 2152 BRENNER, D.J., LITTLE, J.B. and SACHS, R.K. (2001). “The bystander effect in radiation oncogenesis: II. A quantitative model,” Radiat. Res. 155(3), 402–408. BREWEN, J.G., PRESTON, R.J., JONES, K.P. and GOSSLEE, D.G. (1973). “Genetic hazards of ionizing radiations: cytogenetic extrapolations from mouse to man,” Mutat. Res. 17(2), 245–254. BROOKS, A.L., LEI, X.C. and RITHIDECH, K. (2003). “Changes in biomarkers from space radiation may reflect dose not risk,” Adv. Space Res. 31(6), 1505–1512. 2153 BUCKTON, K.E. (1983). “Chromosome aberrations in patients treated with x-irradiation for ankylosing 2154 spondylitis,” pages 491 to 511 in Radiation-Induced Chromosome Damage in Man, Ishihara, T. and 2155 Sasaki, M.S. Eds. (AR Liss, New York). 74 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2156 2157 2158 NOT TO BE DISSEMINATED OR REFERENCED BUDZOWSKA, M. and KANAAR, R. (2009). “Mechanisms of dealing with DNA damage-induced replication problems,” Cell Biochem. Biophys. 53(1), 17–31. BURAK, L.E., KODAMA, Y., NAKANO, M., OHTAKI, K., ITOH, M., OKLADNIKOVA, N.D., 2159 VASILENKO, E.K., COLOGNE, J.B. and NAKAMURA, N. (2001). “FISH examination of lymphocytes 2160 from Mayak workers for assessment of translocation induction rate under chronic radiation exposures,” 2161 Int. J. Radiat. Biol. 77(8), 901–908. 2162 BUTTERWORTH, K.T., MCMAHON, S.J., HOUNSELL, A.R., O'SULLIVAN, J.M. and PRISE K.M. 2163 (2013) “Bystander signalling: Exploring clinical relevance through new approaches and new models,” 2164 Clin. Oncol. (R. Coll. Radiol.), 25(10), 586–592. 2165 CARDIS, E., VRIJHEID, M., BLETTNER, M., GILBERT, E., HAKAMA, M., HILL, C., HOWE, G., 2166 KALDOR, J., MUIRHEAD, C.R., SCHUBAUER-BERIGAN, M., YOSHIMURA, T., BERMANN, F., 2167 COWPER, G., FIX, J., HACKER, C., HEINMILLER, B., MARSHALL, M., THIERRY-CHEF, I., 2168 UTTERBACK, D., AHN, Y.O., AMOROS, E., ASHMORE, P., AUVINEN, A., BAE, J.M., BERNAR, 2169 J., BIAU, A., COMBALOT, E., DEBOODT, P., DIEZ SACRISTAN, A., EKLOF, M., ENGELS, H., 2170 ENGHOLM, G., GULIS, G., HABIB, R.R., HOLAN, K., HYVONEN, H., KEREKES, A., 2171 KURTINAITIS, J., MALKER, H., MARTUZZI, M., MASTAUSKAS, A., MONNET, A., MOSER, M., 2172 PEARCE, M.S., RICHARDSON, D.B., RODRIGUEZ-ARTALEJO, F., ROGEL, A., TARDY, H., 2173 TELLE-LAMBERTON, M., TURAI, I., USEL, M. and VERESS, K. (2007). “The 15-Country 2174 Collaborative Study of Cancer Risk among Radiation Workers in the Nuclear Industry: Estimates of 2175 radiation-related cancer risks,” Radiat. Res. 167(4), 396–416. 2176 2177 2178 2179 CARLS, N. and SCHIESTL, R.H. (1999). “Effect of ionizing radiation on transgenerational appearance of p(un) reversions in mice,” Carcinogenesis 20(12), 2351–2354. CARNES, B.A., GRAHN, D. and HOEL, D. (2003). “Mortality of atomic bomb survivors predicted from laboratory animals,” Radiat. Res. 160, 159–167. 2180 CARROLL, R.J., RUPPERT, D., STEFANSKI, L.A. and CRAINICEANU, C.M. (2006). Measurement Error 2181 in Nonlinear Models. A Modern Perspective, 2nd ed. (Chapman and Hall/CRC, Boca Raton, Florida). 2182 CHAI, Y., CALAF, G.M., ZHOU, H., GHANDHI, S.A., ELLISTON, C.D., WEN, G., NOHMI, T., 2183 AMUNDSON, S.A. and HEI, T.K. (2013a). “Radiation induced COX-2 expression and mutagenesis at 2184 non-targeted lung tissues of gpt delta transgenic mice,” Br. J. Cancer, 108(1), 91–98. 2185 CHAI, Y., LAM, R.K., CALAF, G.M., ZHOU, H., AMUNDSON, S. and HEI, T.K. (2013b). “Radiation- 2186 induced non-targeted response in vivo: Role of the TGFβ-TGFBR1-COX-2 signalling pathway,” Br. J. 2187 Cancer, 108(5), 1106–1112. 75 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2188 NOT TO BE DISSEMINATED OR REFERENCED CHANOUX, R.A., YIN, B., URTISHAK, K.A., ASARE, A., BASSING, C.H. and BROWN, E.J. (2008). 2189 “ATR and H2AX cooperate in maintaining genome stability under replication stress,” J. Biol. Chem. 2190 284(9), 5994–6003. 2191 CONOLLY, R.B., KIMBELL, J.S., JANSZEN, D., SCHLOSSER, P.M., KALISAK, D., PRESTON, J. and 2192 MILLER, F.J. (2004). “Human respiratory tract cancer risks of inhaled formaldehyde: Dose-response 2193 predictions derived from biologically-motivated computational modeling of a combined rodent and 2194 human dataset,” Toxicol. Sci. 82(1), 279–296. 2195 2196 2197 CORNFORTH, M.N. (2001). “Analyzing radiation-induced complex chromosome rearrangements by combinatorial painting,” Radiat Res. 155(5), 643–659. CUCINOTTA, F.A., KIM, M.H.Y. and CHAPPELL, L.J. (2013a). Space Radiation Cancer Risk Projections 2198 and Uncertainties – 2012, NASA/TP-2013-217375, http://www.csc.caltech.edu/references/ 2199 SpaceRadiationCancerRiskProjectionsandUncertainties.pdf (accessed November 8, 2013) (National 2200 Aeronautics and Space Administration, Washington). 2201 CURTIS, S.B., LUEBECK, E.G., HAZELTON, W.D. and MOOLGAVKAR, S.H. (2002). “A new 2202 perspective of carcinogenesis from protracted high-LET radiation arises from the two-stage clonal 2203 expansion model,” Adv. Space. Res. 30(4), 937–944. 2204 CURWEN, G.B., CADWELL, K.K., WINTHER, J.F., TAWN, E.J., REES, G.S., OLSEN, J.H., 2205 RECHNITZER, C., SCHROEDER, H., GULDBERG, P., CORDELL, H.J. and BOICE, J.D., JR. (2010). 2206 “The heritability of G2 chromosomal radiosensitivity and its association with cancer in Danish cancer 2207 survivors and their offspring,” Int. J. Radiat. Biol. 86(11), 986–995. 2208 DAUER, L.T., BROOKS, A.L., HOEL, D.G., MORGAN, W.F., STRAM, D. and TRAN, P. (2010). “Review 2209 and evaluation of updated research on the health effects associated with low-dose ionising radiation,” 2210 Radiat. Prot. Dosim. 140(2), 103–136. 2211 DAY, J.P., LIMOLI, C.L. and MORGAN, W.F. (1998). “Recombination involving interstitial telomere 2212 repeat-like sequences promotes chromosomal instability in Chinese hamster cells,” Carcinogenesis 19(2), 2213 259–265. 2214 DICKEY, J.S., BAIRD, B.J., REDON, C.E., SOKOLOV, M.V., SEDELNIKOVA, O.A. and BONNER, 2215 W.M. (2009). “Intercellular communication of cellular stress monitored by γ-H2AX induction,” 2216 Carcinogenesis 30(10), 1686–1695. 2217 DING, L.H., SHINGYOJI, M., CHEN, F., HWANG, J.J., BURMA, S., LEE, C., CHENG, J.F. and CHEN, 2218 D.J. (2005). “Gene expression profiles of normal human fibroblasts after exposure to ionizing radiation: 2219 A comparative study of low and high doses,” Radiat. Res. 164(1), 17–26. 2220 2221 DOWNEY, M. and DUROCHER, D. (2006). “H2AX as a checkpoint maintenance signal,” Cell Cycle 5(13), 1376–1381. 76 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2222 2223 2224 NOT TO BE DISSEMINATED OR REFERENCED DRAPER, G.J., SANDERS, B.M. and KINGSTON, J.E. (1986). “Second primary neoplasms in patients with retinoblastoma,” Br. J. Cancer 53(5), 661–671. DUBROVA, Y.E., GRANT, G., CHUMAK, A.A., STEZHKA, V.A. and KARAKASIAN, A.N. (1996). 2225 “Elevated minisatellite mutation rate in the post-Chernobyl families from Ukraine,” Am. J. Hum. Genet. 2226 71(4), 801–809. 2227 DUBROVA, Y.E., NESTEROV, V.N., KROUCHINSKY, N.G., OSTAPENKO, V.A., VERGNAUD, G., 2228 GIRAUDEAU, F., BUARD, J. and JEFFREYS, A.J. (1997). “Further evidence for elevated human 2229 minisatellite mutation rate in Belarus eight years after the Chernobyl accident,” Mutat. Res. 381(2), 267– 2230 278. 2231 2232 2233 DUBROVA, Y.E., PLOSHCHANSKAYA, O.G., KOZIONOVA, O.S. and AKLEYEV, A.V. (2006). “Minisatellite germline mutation rate in the Techa River population,” Mutat. Res. 602(1–2), 74-82. DURANT, S.T., PAFFETT, K.S., SHRIVASTAV, M., TIMMINS, G.S., MORGAN, W.F. and NICKOLOFF, 2234 J.A. (2006). “UV radiation induces delayed hyperrecombination associated with hypermutation in human 2235 cells,” Mol. Cell. Biol. 26(16), 6047–6055. 2236 EIDEMULLER, M., HOLMBERG, E., JACOB, P., LUNDELL, M. and KARLSSON, P. (2011). “Breast 2237 cancer risk after radiation treatment at infancy: Potential consequences of radiation-induced genomic 2238 instability,” Radiat. Prot. Dosim. 143(2–4), 375–379. 2239 ENG, C., LI, F.P., ABRAMSON, D.H., ELLSWORTH, R.M., WONG, F.L., GOLDMAN, M.B., SEDDON, 2240 J., TARBELL, N. and BOICE, J.D., JR. (1993). “Mortality from second tumors among long-term 2241 survivors of retinoblastoma,” J. Natl. Cancer Inst. 85(14), 1121–1128. 2242 EPA (2003). U.S. Environmental Protection Agency. Framework for Cumulative Risk Assessment, 2243 EPA/630/P-02/001F, http://www.epa.gov/raf/publications/pdfs/frmwrk_cum_risk_assmnt.pdf (accessed 2244 January 16, 2015) (U.S. Environmental Protection Agency, Washington). 2245 EPA (2005). U.S. Environmental Protection Agency. Guidelines for Carcinogen Risk Assessment, 2246 EPA/630/P-03/001F, http://www.epa.gov/raf/publications/pdfs/CANCER_GUIDELINES_FINAL_3-25- 2247 05.PDF (accessed January 14, 2015) (U.S. Environmental Protection Agency, Washington). 2248 2249 ERIKSSON, D. and STIGBRAND, T. (2010). “Radiation-induced cell death mechanisms,” Tumour Biol 31(4), 363–372. 2250 ESCHRICH, S., ZHANG, H., ZHAO, H., BOULWARE, D., LEE, J.H., BLOOM, G. and TORRES-ROCA, 2251 J.F. (2009). “Systems biology modeling of the radiation sensitivity network: A biomarker discovery 2252 platform,” Int. J. Radiat. Oncol. Biol. Phys. 75(2), 497–505. 2253 2254 EVANS, H.J., BUCKTON, K.E., HAMILTON, G.E. and CAROTHERS, A. (1979). “Radiation-induced chromosome aberrations in nuclear-dockyard workers,” Nature 277(5697), 531–534. 77 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2255 NOT TO BE DISSEMINATED OR REFERENCED FERNANDEZ, M.I., GONG, Y., YE, Y., LIN, J., CHANG, D.W., KAMAT, A.M. and WU, X. (2013). 2256 “γ-H2AX level in peripheral blood lymphocytes as a risk predictor for bladder cancer,” Carcinogenesis 2257 34(11), 2543–2547. 2258 FERNET, M., MOULLAN, N., LAUGE, A., STOPPA-LYONNET, D. and HALL, J. (2004). “Cellular 2259 responses to ionising radiation of AT heterozygotes: Differences between missense and truncating 2260 mutation carriers,” Br. J. Cancer 90(4), 866–873. 2261 FERNET, M., MEGNIN-CHANET, F., HALL, J. and FAVAUDON, V. (2010). “Control of the G2/M 2262 checkpoints after exposure to low doses of ionising radiation: Implications for hyper-radiosensitivity,” 2263 DNA Repair (Amst.) 9(1), 48–57. 2264 FOKAS, E., PREVO, R., HAMMOND, E.M., BRUNNER, T.B., MCKENNA, W.G. and MUSCHEL, R.J. 2265 (2014). “Targeting ATR in DNA damage response and cancer therapeutics,” Cancer Treat. Rev. 40(1), 2266 109–117. 2267 2268 2269 2270 2271 2272 2273 2274 FRANKENBERG-SCHWAGER, M. (1990). “Induction, repair and biological relevance of radiation-induced DNA lesions in eukaryotic cells,” Radiat. Environ. Biophys. 29(4), 273–292. FRIEDBERG, E.C., WALKER, G.C., SIEDE, W., WOOD, R.D., SCHULTZ, R.A. and ELLENBERGER, T. (2005). DNA Repair and Mutagenesis, 2nd ed. (ASM Press. Washington). GALLIE, B.L., CAMPBELL, C., DEVLIN, H., DUCKETT, A. and SQUIRE, J.A. (1999). “Developmental basis of retinal-specific induction of cancer by RB mutation,” Cancer Res. 59(7 Suppl.), 1731s–1735s. GALLUZZI, L. and KROEMER, G. (2008). “Necroptosis: A specialized pathway of programmed necrosis,” Cell 135(7), 1161–1163. 2275 GENIK, P.C., VYAZUNOVA, I., STEFFEN, L.S., BACHER, J.W., BIELEFELDT-OHMANN, H., 2276 MCKERCHER, S., ULLRICH, R.L., FALLGREN, C.M., WEIL, M.M. and RAY, F.A. (2014). 2277 “Leukemogenesis in heterozygous PU.1 knockout mice,” Radiat. Res. 182(3), 310–315. 2278 GENOVESE, G., KAHLER, A.K., HANDSAKER, R.E., LINDBERG, J., ROSE, S.A., BAKHOUM, S.F., 2279 CHAMBERT, K., MICK, E., NEALE, B.M., FROMER, M., PURCELL, S.M., SVANTESSON, O., 2280 LANDEN, M., HOGLUND, M., LEHMANN, S., GABRIEL, S.B., MORAN, J.L., LANDER, E.S., 2281 SULLIVAN, P.F., SKLAR, P., GRONBERG, H., HULTMAN, C.M. and MCCARROLL, S.A. (2014). 2282 “Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence,” N. Engl. J. Med. 2283 371(26), 2477–2487. 2284 2285 2286 GOLDBERG, Z. (2003). “Clinical implications of radiation-induced genomic instability,” Oncogene 22(45), 7011–7017. GRAHN, D. (1960). “Genetic control of physiological processes: The genetics of radiation toxicity in 2287 animals,” pages 181 to 190 in A Symposium on Radioisotopes in the Biosphere, Caldecott, R.S. and 2288 Snyder, L.A., Eds. (University of Minnesota, Minneapolis). 78 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2289 NOT TO BE DISSEMINATED OR REFERENCED GRAHN, D, and SACHER, GA. (1968). “Fractionation and protraction factors and the late effects of 2290 radiation in small mammals,” pages 2-1 to 2-27 in The Proceedings of a Symposium on Dose Rate in 2291 Mammalian Radiation Biology, Brown, D.G., Cragle, R.G. and Noonan, T.R., Eds., U.S. Atomic Energy 2292 Commission CONF-680410 (National Technical Information Service, Springfield, Virginia). 2293 GRANT, E.J., SUGIYAMA, H., OZASA, A.V.K. and RERF-NCI Cancer Risk Analysis Group (2014). 2294 “Abstract: Solid cancer incidence among atomic bomb survivors: Overview of dosimetry, follow-up and 2295 risk updates,” Conference on Radiation and Health, http://www.ameetingbydesign.com/ambddrop/ 2296 files/basic-html/page39.html (accessed January 16, 2015) (Radiation Effects Research Foundation, 2297 Hiroshima). 2298 GREEN, D.M., SKLAR, C.A., BOICE, J.D., JR., MULVIHILL, J.J., WHITTON, J.A., STOVALL, M. and 2299 YASUI, Y. (2009). “Ovarian failure and reproductive outcomes after childhood cancer treatment: Results 2300 from the Childhood Cancer Survivor Study,” J. Clin. Oncol. 27(14), 2374–2381. 2301 GREENMAN, C., STEPHENS, P., SMITH, R., DALGLIESH, G.L., HUNTER, C., BIGNELL, G., DAVIES, 2302 H., TEAGUE, J., BUTLER, A., STEVENS, C., EDKINS, S., O’MEARA, S., VASTRIK, I., SCHMIDT, 2303 E.E., AVIS, T., BARTHORPE, S., BHAMRA, G., BUCK, G., CHOUDHURY, B., CLEMENTS, J., 2304 COLE, J., DICKS, E., FORBES, S., GRAY, K., HALLIDAY, K., HARRISON, R., HILLS, K., 2305 HINTON, J., JENKINSON, A., JONES, D., MENZIES, A., MIRONENKO, T., PERRY, J., RAINE, K., 2306 RICHARDSON, D., SHEPHERD, R., SMALL, A., TOFTS, C., VARIAN, J., WEBB, T., WEST, S., 2307 WIDAA, S., YATES, A., CAHILL, D.P., LOUIS, D.N., GOLDSTRAW, P., NICHOLSON, A.G., 2308 BRASSEUR, F., LOOIJENGA, L., WEBER, B.L., CHIEW, Y.E., DEFAZIO, A., GREAVES, M.F., 2309 GREEN, A.R., CAMPBELL, P., BIRNEY, E., EASTON, D.F., CHENEVIX-TRENCH, G., TAN, M.H., 2310 KHOO, S.K., TEH, B.T., YUEN, S.T., LEUNG, S.Y., WOOSTER, R., FUTREAL, P.A. and 2311 STRATTON, M.R. (2007). “Patterns of somatic mutation in human cancer genomes,” Nature 446(7132), 2312 153–158. 2313 GRIDLEY, D.S., RIZVI, A., LUO-OWEN, X., MAKINDE, A.Y. and PECAUT, M.J. (2009). “Low dose, 2314 low dose rate photon radiation modifies leukocyte distribution and gene expression in CD4(+) T cells,” J. 2315 Radiat. Res. (Tokyo) 50(2), 139–150. 2316 2317 2318 GROSOVSKY, A.J. and LITTLE, J.B. (1985). “Evidence for linear response for the induction of mutations in human cells by x-ray exposures below 10 rads,” Proc. Natl. Acad. Sci. (USA) 82(7), 2092–2095. GROSOVSKY, A.J., DE BOER, J.G., DE JONG, P.J., DROBETSKY, E.A. and GLICKMAN, B.W. (1988). 2319 “Base substitutions, frameshifts, and small deletions constitute ionizing radiation-induced point mutations 2320 in mammalian cells,” Proc. Natl. Acad. Sci USA 85(1), 185–188. 2321 2322 GUO, Y., CAI, Q., SAMUELS, D.C., YE, F., LONG, J., LI, C.I., WINTHER, J.F., TAWN, E.J., STOVALL, M., LAHTEENMAKI, P., MALILA, N., LEVY, S., SHAFFER, C., SHYR, Y., SHU, X.O. and BOICE, 79 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2323 J.D., JR. (2012). “The use of next generation sequencing technology to study the effect of radiation 2324 therapy on mitochondrial DNA mutation,” Mutat. Res. 744(2), 154–160. 2325 HAGMAR, L., BROGGER, A., HANSTEEN, I.L., HEIM, S., HOGSTEDT, B., KNUDSEN, L., LAMBERT, 2326 B., LINNAINMAA, K., MITELMAN, F., NORDENSON, I., REUTERWALL, C., SALOMAN, S., 2327 SKERFVING, S. and SORSA, M. (1994). “Cancer risk in humans predicted by increased levels of 2328 chromosomal aberrations in lymphocytes: Nordic study group on the health risk of chromosome damage,” 2329 Cancer Res. 54(11), 2919–2922. 2330 HAGMAR, L., STROMBERG, U., TINNERBERG, H. and MIKOCZY, Z. (2004). “Epidemiological 2331 evaluation of cytogenetic biomarkers as potential surrogate end-points for cancer,” IARC Sci. Publ. (157), 2332 207–215. 2333 2334 2335 HALL, E.J. and GIACCIA, A.J. (2011). Radiobiology for the Radiologist, 7th ed. (Lippincott Williams and Wilkins, Philadelphia). HAMMER, G.P., SCHEIDEMANN-WESP, U., SAMKANGE-ZEEB, F., WICKE, H., NERIISHI, K. and 2336 BLETTNER, M. (2013). “Occupational exposure to low doses of ionizing radiation and cataract 2337 development: A systematic literature review and perspectives on future studies,” Radiat. Environ. 2338 Biophys. 52(3), 303–319. 2339 HANAHAN, D. and WEINBERG, R.A. (2000). “The hallmarks of cancer,” Cell 100(1), 57–70. 2340 HANAHAN, D. and WEINBERG, R.A. (2011). “Hallmarks of cancer: The next generation,” Cell 144(5), 2341 2342 2343 2344 646–674. HARVEY, E.B., BOICE, J.D., JR., HONEYMAN, M. and FLANNERY, J.T. (1985). “Prenatal x-ray exposure and childhood cancer in twins,” N. Engl. J. Med. 312(9), 541–545. HAYASHI, T., KUSUNOKI, Y., HAKODA, M., MORISHITA, Y., KUBO, Y., MAKI, M., KASAGI, F., 2345 KODAMA, K., MACPHEE, D.G. and KYOIZUMI, S. (2003). “Radiation dose-dependent increases in 2346 inflammatory response markers in A-bomb survivors,” Int. J. Radiat. Biol. 79(2), 129–136. 2347 HAYASHI, T., MORISHITA, Y., KUBO, Y., KUSUNOKI, Y., HAYASHI, I., KASAGI, F., HAKODA, M., 2348 KYOIZUMI, S. and NAKACHI, K. (2005). “Long-term effects of radiation dose on inflammatory 2349 markers in atomic bomb survivors,” Am. J. Med. 118(1), 83–86. 2350 HAYATA, I., WANG, C., ZHANG, W., CHEN, D., MINAMIHISAMATSU, M., MORISHIMA, H., WEI, L. 2351 and SUGAHARA, T. (2004). “Effect of high-level natural radiation on chromosomes of residents in 2352 southern China,” Cytogenet. Genome Res. 104(1–4), 237–239. 2353 HE, Y., GONG, Y., LIN, J., CHANG, D.W., GU, J., ROTH, J.A. and WU, X. (2013). “Ionizing radiation- 2354 induced γ-H2AX activity in whole blood culture and the risk of lung cancer,” Cancer Epidemiol. 2355 Biomarkers Prev. 22(3), 443–451. 80 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2356 HEI, T.K., ZHOU, H., IVANOV, V.N., HONG, M., LIEBERMAN, H.B., BRENNER, D.J., AMUNDSON, 2357 S.A. and GEARD, C.R. (2008). “Mechanism of radiation-induced bystander effects: A unifying model,” 2358 J. Pharm. Pharmacol. 60(8), 943–950. 2359 HEIDENREICH, W.F. and ROSEMANN, M. (2012). “Genetic background and 227Thorium as risk factors in 2360 biologically based models for induction of bone cancer in mice,” Radiat. Environ. Biophys. 51(2),179– 2361 185. 2362 HEIDENREICH, W.F., TOMASEK, L., ROGEL, A., LAURIER, D. and TIRMARCHE, M. (2004). “Studies 2363 of radon-exposed miner cohorts using a biologically based model: Comparison of current Czech and 2364 French data with historic data from China and Colorado,” Radiat. Environ. Biophys. 43(4), 247–256. 2365 2366 HEIDENREICH, W.F., SARAN, A., ATKINSON, M. and PAZZAGLIA, S. (2013). “A mechanistic model for medulloblastoma induction in mice,” Radiat. Res. 179(5), 610–614. 2367 HOWE, G.R. and MCLAUGHLIN, J. (1996). “Breast cancer mortality between 1950 and 1987 after exposure 2368 to fractionated moderate-dose-rate ionizing radiation in the Canadian Fluoroscopy Cohort Study and a 2369 comparison with breast cancer mortality in the Atomic Bomb Survivors Study,” Radiat. Res. 145(6), 694– 2370 707. 2371 2372 2373 2374 HOWELL, E.K., GASCHAK, S.P., GRIFFITH, K.D. and RODGERS, B.E. (2013). “Radioadaptive response following in utero low-dose irradiation,” Radiat. Res. 179(1), 29–37. HSIEH, P. and YAMANE, K. (2008). “DNA mismatch repair: Molecular mechanism, cancer, and ageing,” Mech. Ageing Dev. 129(7–8), 391–407. 2375 HSIEH, W.A., NI, C., HWANG, J.J., FANG, J.S., LIN, S.P., LIN, Y.A., HUANG, T.W. and CHANG, W.P. 2376 (2002). “Evaluation of the frequencies of chromosomal aberrations in a population exposed to prolonged 2377 low dose-rate 60Co gamma-irradiation,” Int. J. Radiat. Biol. 78(7), 625–633. 2378 HSU, W.L., PRESTON, D.L., SODA M, SUGIYAMA, H., FUNAMOTO, S., KODAMA, K., KIMURA, A., 2379 KAMADA, N., DOHY, H., TOMONAGA, M., IWANAGA, M., MIYAZAKI, Y., CULLINGS, H.M., 2380 SUYAMA, A., OZASA, K., SHORE, R.E. and MABUCHI, K. (2013). “The incidence of leukemia, 2381 lymphoma and multiple myeloma among atomic bomb survivors: 1950–2001,” Radiat. Res. 179(3), 361– 2382 382. 2383 HUANG, L., GRIMM, S., SMITH, L.E., KIM, P.M., NICKOLOFF, J.A., GOLOUBEVA, O.G. and 2384 MORGAN, W.F. (2004). “Ionizing radiation induces delayed hyperrecombination in mammalian cells,” 2385 Mol. Cell. Biol. 24(11), 5060–5068. 2386 HUANG, L., KIM, P.M., NICKOLOFF, J.A. and MORGAN, W.F. (2007). “Targeted and non-targeted 2387 effects of low-dose ionizing radiation on delayed genomic instability in human cells,” Cancer Res. 67(3), 2388 1099–1104. 81 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2389 NOT TO BE DISSEMINATED OR REFERENCED HWANG, S.L., HWANG, J.S., YANG, Y.T., HSIEH, W.A., CHANG, T.C., GUO, H.R., TSAI, M.H., 2390 TANG, J.L., LIN, I.F. and CHANG, W.P. (2008). “Estimates of relative risks for cancers in a population 2391 after prolonged low-dose-rate radiation exposure: A follow-up assessment from 1983 to 2005,” Radiat. 2392 Res. 170(2), 143–148. 2393 ICRP (1991). International Commission on Radiological Protection. 1990 Recommendations of the 2394 International Commission on Radiological Protection. ICRP Publication 60, Ann. ICRP 21(1–3) (Sage 2395 Publications, Thousand Oaks, California). 2396 2397 2398 ICRP (1998). International Commission on Radiological Protection. Genetic Susceptibility to Cancer, ICRP Publication 79, Ann. ICRP 28(1–2) (Sage Publications, Thousand Oaks, California). ICRP (2007). International Commission on Radiological Protection. The 2007 Recommendations of the 2399 International Commission on Radiological Protection, ICRP Publication 103, Ann. ICRP 37(2–4) (Sage 2400 Publications, Thousand Oaks, California). 2401 ICRP (2012). International Commission on Radiological Protection. ICRP Statement on Tissue Reactions and 2402 Early and Late Effects of Radiation in Normal Tissues and Organs—Threshold Doses for Tissue 2403 Reactions in a Radiation Protection Context, ICRP Publication 118, Ann. ICRP 41(1/2) (Sage 2404 Publications, Thousand Oaks, California). 2405 2406 2407 ILNYTSKYY, Y. and KOVALCHUK, O. (2011). “Non-targeted radiation effects-an epigenetic connection,” Mutat. Res. 714(1–2), 113–125. IVANOV, V.K., TSYB, A.F., KHAIT, S.E., KASHCHEEV, V.V., CHEKIN, S.Y., MAKSIOUTOV, M.A. 2408 and TUMANOV, K.A. (2012). “Leukemia incidence in the Russian cohort of Chernobyl emergency 2409 workers,” Radiat. Environ. Biophys. 51(2), 143–149. 2410 JACOB, P., MECKBACH, R., KAISER, J.C. and SOKOLNIKOV, M. (2010). “Possible expressions of 2411 radiation-induced genomic instability, bystander effects or low-dose hypersensitivity in cancer 2412 epidemiology,” Mutat. Res. 687(1–2), 34–39. 2413 2414 2415 JABLON, S. and KATO, H. (1970). “Childhood cancer in relation to prenatal exposure to atomic-bomb radiation,” Lancet 2(7681), 1000–1003. JARABEK, A.M., POTTENGER, L.H., ANDREWS, L.S., CASCIANO, D., EMBRY, M.R., KIM, J.H., 2416 PRESTON, R.J., REDDY, M.V., SCHOENY, R., SHUKER, D., SKARE, J., SWENBERG, J., 2417 WILLIAMS, G.M. and ZEIGER, E. (2009). “Creating context for the use of DNA adduct data in cancer 2418 risk assessment: I. Data organization,” Crit. Rev. Toxicol. 39(8), 659–678. 2419 JEN, M.H., HWANG, J.J., YANG, J.Y., NABYVANETS, Y.B., HSIEH, W.A., TSAI, M.H., GUO, S.D. and 2420 CHANG, W.P. (2002). “Micronuclei and nuclear anomalies in urinary exfoliated cells of subjects in 2421 radionuclide-contaminated regions,” Mutat. Res. 520(1–2), 39–46. 82 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2422 NOT TO BE DISSEMINATED OR REFERENCED JONES, I.M., GALICK, H., KATO, P., LANGLOIS, R.G., MENDELSOHN, M.L., MURPHY, G.A., 2423 PLESHANOV, P., RAMSEY, M.J., THOMAS, C.B., TUCKER, J.D., TUREVA, L., VOROBTSOVA, I. 2424 and NELSON, D.O. (2002). “Three somatic genetic biomarkers and covariates in radiation-exposed 2425 Russian cleanup workers of the Chernobyl nuclear reactor 6-13 years after exposure,” Radiat. Res. 2426 158(4), 424–442. 2427 JULIEN, E., BOOBIS, A.R. and OLIN, S.S. (2009). “The key events dose-response framework: A cross- 2428 disciplinary mode-of-action based approach to examining dose-response and thresholds,” Crit. Rev. Food 2429 Sci. Nutr. 49(8), 682–689. 2430 2431 2432 2433 2434 KAISER, J.C., MECKBACH, R. and JACOB, P. (2014). “Genomic instability and radiation risk in molecular pathways to colon cancer,” PLoS One 9(10):e111024. KAMILERI, I., KARAKASILIOTI, I. and GARINIS, G.A. (2012). “Nucleotide excision repair: New tricks with old bricks,” Trends Genet. 28(11), 566–573. KATO, T.A., WILSON, P.F., NAGASAWA, H., FITZEK, M.M., WEIL, M.M., LITTLE, J.B. and 2435 BEDFORD, J.S. (2007). “A defect in DNA double strand break processing in cells from unaffected 2436 parents of retinoblastoma patients and other apparently normal humans,” DNA Repair (Amst). 6(6), 818– 2437 829. 2438 KATO, T.A., WILSON, P.F., NAGASAW, H., PENG, Y., WEIL, M.M., LITTLE, J.B. and BEDFORD, J.S. 2439 (2009). “Variations in radiosensitivity among individuals: A potential impact on risk assessment?” Health 2440 Phys. 97(5), 470–480. 2441 KENDALL, G.M, LITTLE, M.P., WAKEFORD, R., BUNCH, K.J., MILES, J.C., VINCENT, T.J., MEARA, 2442 J.R. and MURPHY, M.F. (2013). “A record-based case-control study of natural background radiation and 2443 the incidence of childhood leukaemia and other cancers in Great Britain during 1980–2006,” Leukemia 2444 27(1), 3–9. 2445 KIM, G.J., CHANDRASEKARAN, K. and MORGAN, W.F. (2006a). “Mitochondrial dysfunction, 2446 persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: A 2447 review,” Mutagenesis 21(6), 361–367. 2448 2449 2450 2451 2452 KIM, G.J., FISKUM, G.M. and MORGAN, W.F. (2006b). “A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability,” Cancer Res. 66(21), 10377–10383. KLEINERMAN, R.A. (2009). “Radiation-sensitive genetically susceptible pediatric sub-populations,” Pediatr. Radiol. 39(Suppl. 1), S27–S31. KLEINERMAN, R.A., LITTLEFIELD, L.G., TARONE, R.E., MACHADO, S.G., BLETTNER, M., 2453 PETERS, L.J. and BOICE, J.D., JR. (1989). “Chromosome aberrations in peripheral lymphocytes and 2454 radiation dose to active bone marrow in patients treated for cancer of the cervix,” Radiat. Res. 119(1), 2455 176–190. 83 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2456 NOT TO BE DISSEMINATED OR REFERENCED KLEINERMAN, R.A., LITTLEFIELD, L.G., TARONE, R.E., SAYER, A.M., HILDRETH, N.G., 2457 POTTERN, L.M., MACHADO, S.G. and BOICE, J.D., JR. (1990). “Chromosome aberrations in relation 2458 to radiation dose following partial-body exposures in three populations,” Radiat. Res. 123(1), 93–101. 2459 KLEINERMAN, R.A., LITTLEFIELD, L.G., TARONE, R.E., SAYER, A.M., COOKFAIR, D.L., 2460 WACTAWSKI-WENDE, J., INSKIP, P.D., BLOCK, A., RAMESH, K.H. and BOICE, J.D., JR. (1994). 2461 “Chromosome aberrations in lymphocytes from women irradiated for benign and malignant 2462 gynecological disease,” Radiat. Res. 139(1), 40–46. 2463 KLEINERMAN, R.A., TUCKER, M.A., TARONE, R.E., ABRAMSON, D.H., SEDDON, J.M., STOVALL, 2464 M., LI, F.P. and FRAUMENI, J.F., JR. (2005). “Risk of new cancers after radiation therapy in long-term 2465 survivors of retinoblastoma: An extended follow-up,” J. Clin. Oncol. 23(10), 2272–2279. 2466 KLEINERMAN, R.A, ROMANYUKHA, A.A., SCHAUER, D.A. and TUCKER, J.D. (2006). “Retrospective 2467 assessment of radiation exposure using biological dosimetry: Chromosome painting, electron 2468 paramagnetic resonance and the glycophorin a mutation assay,” Radiat. Res. 166(1 Pt 2), 287–302. 2469 KLEINERMAN, R.A., TUCKER, M.A., ABRAMSON, D.H., SEDDON, J.M., TARONE, R.E. and 2470 FRAUMENI, J.F., JR. (2007). “Risk of soft tissue sarcomas by individual subtype in survivors of 2471 hereditary retinoblastoma,” J. Natl. Cancer Inst. 99(1), 24–31. 2472 2473 2474 KNUDSON, A.G., JR. (1971). “Mutation and cancer: Statistical study of retinoblastoma,” Proc. Natl. Acad. Sci. USA 68(4), 820–823. KODAMA, Y., PAWEL, D., NAKAMURA, N., PRESTON, D., HONDA, T., ITOH, M., NAKANO, M., 2475 OHTAKI, K., FUNAMOTO, S. and AWA, A.A. (2001). “Stable chromosome aberrations in atomic 2476 bomb survivors: Results from 25 years of investigation,” Radiat. Res. 156(4), 337–346. 2477 KOTURBASH, I., BAKER, M., LOREE, J., KUTANZI, K., HUDSON, D., POGRIBNY, I., 2478 SEDELNIKOVA, O., BONNER, W. and KOVALCHUK, O. (2006). “Epigenetic dysregulation underlies 2479 radiation-induced transgenerational genome instability in vivo,” Int. J. Radiat. Oncol. Biol. Phys. 66(2), 2480 327–330. 2481 KRAEMER, S.M., VANNAIS, D.B., KRONENBERG, A., UENO, A. and WALDREN, C.A. (2001). 2482 “Gamma-ray mutagenesis studies in a new human-hamster hybrid, A(L)CD59(+/-), which has two human 2483 chromosomes 11 but is hemizygous for the CD59 gene,” Radiat. Res. 156(1), 10–19. 2484 KRESTININA, L.Y., DAVIS, F.G., SCHONFELD, S., PRESTON, D.L., DEGTEVA, M., EPIFANOVA, S. 2485 and AKLEYEV, A.V. (2013). “Leukaemia incidence in the Techa River Cohort: 1953–2007,” Br. J. 2486 Cancer 109(11), 2886–2893. 2487 KREWSKI, D., ZIELINSKI, J.M., HAZELTON, W.D., GARNER, M.J. and MOOLGAVKAR, S.H. (2003). 2488 “The use of biologically based cancer risk models in radiation epidemiology,” Radiat. Prot. Dosim. 2489 104(4), 367–376. 84 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2490 2491 2492 NOT TO BE DISSEMINATED OR REFERENCED KROKAN, H.E. and BJORAS, M. (2013). “Base excision repair,” Cold Spring Harb. Perspect. Biol. 5(4), a012583. KUSUNOKI, Y., KYOIZUMI, S., HIRAI, Y., SUZUKI, T., NAKASHIMA, E., KODAMA, K. and 2493 SEYAMA, T. (1998). “Flow cytometry measurements of subsets of T, B and NK cells in peripheral blood 2494 lymphocytes of atomic bomb survivors,” Radiat. Res. 150(2), 227–236. 2495 KUSUNOKI, Y., KYOIZUMI, S., YAMAOKA, M., KASAGI, F., KODAMA, K. and SEYAMA, T. (1999). 2496 “Decreased proportion of CD4 T cells in the blood of atomic bomb survivors with myocardial infarction,” 2497 Radiat. Res. 152(5), 539–543. 2498 LANZ, C., PATTERSON, A.D., SLAVIK, J., KRAUSZ, K.W., LEDERMANN, M., GONZALEZ, F.J. and 2499 IDLE, J.R. (2009). “Radiation metabolomics. 3. Biomarker discovery in the urine of gamma-irradiated 2500 rats using a simplified metabolomics protocol of gas chromatography-mass spectrometry combined with 2501 random forests machine learning algorithm,” Radiat. Res. 172(2), 198–212. 2502 LAIAKIS, E.C., BAULCH, J.E. and MORGAN, W.F. (2007). “Cytokine and chemokine responses after 2503 exposure to ionizing radiation: Implications for the astronauts,” Adv. Space Res. 39(6), 1019–1025. 2504 LEA, D.E. (1946). Actions of Radiations on Living Cells (Cambridge University Press, Cambridge). 2505 LEE DO, Y., BOWEN, B.P., NGUYEN, D.H., PARSA, S., HUANG, Y., MAO, J.H. and NORTHEN, T.R. 2506 (2012). “Low-dose ionizing radiation-induced blood plasma metabolic response in a diverse genetic 2507 mouse population,” Radiat. Res. 178(6), 551–555. 2508 LIBER, H., YANDELL, D.W. and LITTLE, J.B. (1989). “A comparison of mutation induction at the tk and 2509 hprt loci in human lymphoblastoid cells; quantitative differences are due to an additional class of 2510 mutations at the autosomal tk locus,” Mutat. Res. 216(1), 9–17. 2511 LIMOLI, C.L., KAPLAN, M.I., CORCORAN, J., MEYERS, M., BOOTHMAN, D.A. and MORGAN, W.F. 2512 (1997). “Chromosomal instability and its relationship to other end points of genomic instability,” Cancer 2513 Res. 57(24), 5557–5563. 2514 LIMOLI, C.L., CORCORAN, J.J., MILLIGAN, J.R., WARD, J.F. and MORGAN, W.F. (1999). “Critical 2515 target and dose and dose-rate responses for the induction of chromosomal instability by ionizing 2516 radiation,” Radiat. Res. 151(6), 677–685. 2517 LIN, R.X., ZHAO, H.B., LI, C.R., SUN, Y.N., QIAN, X.H. and WANG, S.Q. (2009). “Proteomic analysis of 2518 ionizing radiation-induced proteins at the subcellular level,” J. Proteome Res. 8(1), 390–399. 2519 LIN, C.M., CHANG, W.P., DOYLE, P., WANG, J.D., LEE, L.T., LEE, C.L. and CHEN, P.C. (2010). 2520 “Prolonged time to pregnancy in residents exposed to ionising radiation in cobalt-60-contaminated 2521 buildings,” Occup. Environ. Med. 67(3), 187–195. 85 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2522 NOT TO BE DISSEMINATED OR REFERENCED LITTLE, M.P. (1995). “Are two mutations sufficient to cause cancer? Some generalizations of the two- 2523 mutation model of carcinogenesis of Moolgavkar, Venzon, and Knudson, and of the multistage model of 2524 Armitage and Doll,” Biometrics 51(4), 1278–1291. 2525 2526 LITTLE, M.P. (1996). “Generalisations of the two-mutation and classical multi-stage models of carcinogenesis fitted to the Japanese atomic bomb survivor data,” J. Radiol. Prot. 16(1), 7–24. 2527 LITTLE, J.B. (2000). “Radiation carcinogenesis,” Carcinogenesis 21(3), 397–404. 2528 LITTLE, M.P. (2004). “The bystander effect model of Brenner and Sachs fitted to lung cancer data in 11 2529 cohorts of underground miners, and equivalence of fit of a linear relative risk model with adjustment for 2530 attained age and age at exposure,” J. Radiol. Prot. 24(3), 243–255. 2531 2532 2533 2534 2535 2536 2537 LITTLE, M.P. (2010). “Cancer models, genomic instability and somatic cellular Darwinian evolution,” Biol. Direct 5, 19. LITTLE, M.P. (2013). “A review of non-cancer effects, especially circulatory and ocular diseases,” Radiat. Environ. Biophys. 52(4), 435–449. LITTLE, M.P. (2014). “Germline minisatellite mutations in the offspring of irradiated parents,” J. Radiat. Prot. 35(1), E1–E4. LITTLE, M.P. and BOICE, J.D., JR. (1999). “Comparison of breast cancer incidence in the Massachusetts 2538 Tuberculosis Fluoroscopy Cohort and in the Japanese atomic bomb survivors,” Radiat. Res. 151(2), 218– 2539 224. 2540 2541 2542 2543 2544 2545 LITTLE, M.P. and LI, G. (2007). “Stochastic modelling of colon cancer: Is there a role for genomic instability?” Carcinogenesis 28(2), 479–487. LITTLE, M.P. and WAKEFORD, R. (2001). “The bystander effect in C3H 10T1/2 cells and radon-induced lung cancer,” Radiat. Res. 156(6), 695–699. LITTLE, M.P. and WRIGHT, E.G. (2003). “A stochastic carcinogenesis model incorporating genomic instability fitted to colon cancer data,” Math. Biosci. 183(2), 111–134. 2546 LITTLE, M.P., MUIRHEAD, C.R. and CHARLES, M.W. (1999a). “Describing time and age variations in the 2547 risk of radiation-induced solid tumour incidence in the Japanese atomic bomb survivors using generalized 2548 relative and absolute risk models,” Stat. Med. 18(1), 17–33. 2549 LITTLE, M.P., WEISS, H.A., BOICE, J.D. JR., DARBY, S.C., DAY, N.E. and MUIRHEAD, C.R. (1999b). 2550 “Risks of leukemia in Japanese atomic bomb survivors, in women treated for cervical cancer, and in 2551 patients treated for ankylosing spondylitis,” Radiat. Res. 152(3), 280–292. 2552 LITTLE, M.P., HAYLOCK, R.G.E. and MUIRHEAD, C.R. (2002). “Modelling lung tumour risk in radon- 2553 exposed uranium miners using generalizations of the two-mutation model of Moolgavkar, Venzon and 2554 Knudson,” Int. J. Radiat. Biol. 78(1), 49–68. 86 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2555 NOT TO BE DISSEMINATED OR REFERENCED LITTLE, M.P., FILIPE, J.A., PRISE, K.M., FOLKARD, M. and BELYAKOV, O.V. (2005). “A model for 2556 radiation-induced bystander effects, with allowance for spatial position and the effects of cell turnover,” 2557 J. Theor. Biol. 232(3), 329–338. 2558 LITTLE, M.P., HOEL, D.G., MOLITOR, J., BOICE, J.D., WAKEFORD, R. and MUIRHEAD, C.R. (2008a). 2559 “New models for evaluation of radiation-induced lifetime cancer risk and its uncertainty employed in the 2560 UNSCEAR 2006 report,” Radiat. Res. 169(6), 660–676. 2561 LITTLE, M.P., VINEIS, P. and LI, G. (2008b). “A stochastic carcinogenesis model incorporating multiple 2562 types of genomic instability fitted to colon cancer data,” J. Theoret. Biol. 254(2), 229-238 [Erratum 2563 255(2), 268]. 2564 LITTLE, M.P., WAKEFORD, R., TAWN, E.J., BOUFFLER, S.D. and BERRINGTON DE GONZALEZ, A. 2565 (2009). “Risks associated with low doses and low dose rates of ionizing radiation: Why linearity may be 2566 (almost) the best we can do,” Radiology 251(1), 6–12. 2567 LITTLE, M.P., KLEINERMAN, R.A., STILLER, C.A., LI, G., KROLL, M.E. and MURPHY, M.F. (2012a). 2568 “Analysis of retinoblastoma age incidence data using a fully stochastic cancer model,” Int. J. Cancer 2569 130(3), 631–640. 2570 LITTLE, M.P., AZIZOVA, T.V., BAZYKA, D., BOUFFLER, S.D., CARDIS, E., CHEKIN, S., CHUMAK, 2571 V.V., CUCINOTTA, F.A., DE VATHAIRE, F., HALL, P., HARRISON, J.D., HILDEBRANDT, G., 2572 IVANOV, V., KASHCHEEV, V.V., KLYMENKO, S.V., KREUZER, M., LAURENT, O., OZASA, K., 2573 SCHNEIDER, T., TAPIO, S., TAYLOR, A.M., TZOULAKI, I., VANDOOLAEGHE, W.L., 2574 WAKEFORD, R., ZABLOTSKA, L.B., ZHANG, W. and LIPSHULTZ, S.E. (2012b). “Systematic 2575 review and meta-analysis of circulatory disease from exposure to low-level ionizing radiation and 2576 estimates of potential population mortality risks,” Environ. Health Perspect. 120(11), 1503–1511. 2577 LITTLE, M.P., GOODHEAD, D.T., BRIDGES, B.A. and BOUFFLER, S.D. (2013). “Evidence relevant to 2578 untargeted and transgenerational effects in the offspring of irradiated parents,” Mutat. Res. Rev. 753(1), 2579 50–67. 2580 LITTLE, M.P., KWON, D., DOI, K., SIMON, S.L., PRESTON, D.L., DOODY, M.M., LEE, T., MILLER, 2581 J.S., KAMPA, D.M., BHATTI, P., TUCKER, J.D., LINET, M.S. and SIGURDSON, A.J. (2014a). 2582 “Association of chromosome translocation rate with low dose occupational radiation exposures in U.S. 2583 radiologic technologists,” Radiat. Res. 182(1), 1–17. 2584 LITTLE, M.P., SCHAEFFER, M.L., REULEN, R.C., ABRAMSON, D.H., STOVALL, M., WEATHERS, 2585 R., DE VATHAIRE, F., DIALLO, I., SEDDON, J.M., HAWKINS, M.M., TUCKER, M.A. and 2586 KLEINERMAN, R.A. (2014b). “Breast cancer risk after radiation therapy for heritable and non-heritable 2587 retinoblastoma: A US–UK study,” Br. J. Cancer 110(10), 2623–2632. 87 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2588 LITTLEFIELD, L.G., KLEINERMAN, R.A., SAYER, A.M., TARONE, R. and BOICE, J.D., JR. (1991). 2589 “Chromosome aberrations in lymphocytes—biomonitors of radiation exposure,” Prog. Clin. Biol. Res. 2590 372, 387–397. 2591 LITTLEFIELD, L.G., TRAVIS, L.B., SAYER, A.M., VOELZ, G.L., JENSEN, R.H. and BOICE, J.D., JR. 2592 (1997). “Cumulative genetic damage in hematopoietic stem cells in a patient with a 40-year exposure to 2593 alpha particles emitted by thorium dioxide,” Radiat. Res. 148(2), 135–144. 2594 LITTLEFIELD, L.G., MCFEE, A.F., SALOMAA, S.I., TUCKER, J.D., INSKIP, P.D., SAYER, A.M., 2595 LINDHOLM, C., MAKINEN, S., MUSTONEN, R., SORENSEN, K., TEKKEL, M., VEIDEBAUM, T., 2596 AUVINEN, A. and BOICE, J.D., JR. (1998). “Do recorded doses overestimate true doses received by 2597 Chernobyl cleanup workers? Results of cytogenetic analyses of Estonian workers by fluorescence in situ 2598 hybridization,” Radiat. Res. 150(2), 237–249. 2599 LIU, J.J., FREEDMAN, D.M., LITTLE, M.P., DOODY, M.M., ALEXANDER, B.H., KITAHARA, C.M., 2600 LEE, T., RAJARAMAN, P., MILLER, J.S., KAMPA, D.M., SIMON S.L., PRESTON, D.L. and LINET, 2601 M.S. (2014). “Work history and mortality risks in 90,268 US radiological technologists,” Occup. Environ. 2602 Med. 71(12), 819–835. 2603 LIVSHITS, L.A., MALYARCHUK, S.G., KRAVCHENKO, S.A., MATSUKA, G.H., LUKYANOVA, E.M., 2604 ANTIPKIN, Y.G., ARABSKAYA, L.P., PETIT, E., GIRAUDEAU, F., GOURMELON, P., 2605 VERGNAUD, G. and LE GUEN, B. (2001). “Children of Chernobyl cleanup workers do not show 2606 elevated rates of mutations in minisatellite alleles,” Radiat. Res. 155(1), 74–80. 2607 LLOYD, D.C., EDWARDS, A.A., LEONARD, A., DEKNUDT, G.L., VERSCHAEVE, L., NATARAJAN, 2608 A.T., DARROUDI, F., OBE, G., PALITTI, F. and TANZARELLA, C. (1992). “Chromosomal 2609 aberrations in human lymphocytes induced in vitro by very low doses of x-rays,” Int. J. Radiat. Biol. 2610 61(3), 335–343. 2611 LOBRICH, M., SHIBATA, A., BEUCHER, A., FISHER, A., ENSMINGER, M., GOODARZI, A.A., 2612 BARTON, O. and JEGGO, P.A. (2010). “H2AX foci analysis for monitoring DNA double-strand break 2613 repair: Strengths, limitations and optimization,” Cell Cycle 9(4), 662–669. 2614 LOHMANN, D. (2010). “Retinoblastoma,” Adv. Exp. Med. Biol. 685, 220–227. 2615 LORIMORE, S.A. and WRIGHT, E.G. (2003). “Radiation-induced genomic instability and bystander effects: 2616 related inflammatory-type responses to radiation-induced stress and injury? A review,” Int. J. Radiat. 2617 Biol. 79(1), 15–25. 2618 LOUCAS, B.D., EBERLE, R., BAILEY, S.M. and CORNFORTH, M.N. (2004). “Influence of dose rate on 2619 the induction of simple and complex chromosome exchanges by gamma rays,” Radiat. Res. 162(4), 339– 2620 349. 88 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2621 NOT TO BE DISSEMINATED OR REFERENCED LOUCAS, B.D., DURANTE, M., BAILEY, S.M. and CORNFORTH, M.N. (2013). “Chromosome damage in 2622 human cells by rays, particles and heavy ions: Track interactions in basic dose-response 2623 relationships,” Radiat. Res. 179(1), 9–20. 2624 LU, T.P., LAI, L.C., LIN, B.I., CHEN, L.H., HSIAO, T.H., LIBER, H.L., COOK, J.A., MITCHELL, J.B., 2625 TSAI, M.H. and CHUANG, E.Y. (2010). “Distinct signaling pathways after higher or lower doses of 2626 radiation in three closely related human lymphoblast cell lines,” Int. J. Radiat. Oncol. Biol. Phys 76(1), 2627 212–219. 2628 LUBIN, J.H., BOICE, J.D., JR., EDLING, C., HORNUNG, R.W., HOWE, G., KUNZ, E., KUSIAK, R.A., 2629 MORRISON, H.I., RADFORD, E.P., SAMET, J.M., TIRMARCHE, M., WOODARD, A. and YAO, 2630 S.X. (1995). “Radon-exposed underground miners and inverse dose-rate (protraction enhancement) 2631 effects,” Health Phys. 69(4), 494–500. 2632 2633 2634 LUEBECK, E.G, CURTIUS, K., JEON, J. and HAZELTON, W.D. (2013). “Impact of tumor progression on cancer incidence curves,” Cancer Res. 73(3), 1086–1096. MANCUSO, M., PASQUALI, E., LEONARDI, S., TANORI, M., REBESSI, S., DI MAJO, V., 2635 PAZZAGLIA, S., PIA TONI, M., PIMPINELLA, M., COVELLI, V. and SARAN, A. (2008). 2636 “Oncogenic bystander radiation effects in Patched heterozygous mouse cerebellum,” Proc. Natl. Acad. 2637 Sci. USA 105(34), 12445–12450. 2638 MANCUSO, M., PASQUALI, E., LEONARDI, S., REBESSI, S., TANORI, M., GIARDULLO, P., BORRA, 2639 F., PAZZAGLIA, S., NAUS, C.C., DI MAJO, V. and SARAN, A. (2011). “Role of connexin43 and ATP 2640 in long-range bystander radiation damage and oncogenesis in vivo,” Oncogene 30(45), 4601–4608. 2641 MANCUSO, M., PASQUALI, E., GIARDULLO, P., LEONARDI, S., TANORI, M., DI MAJO, V., 2642 PAZZAGLIA, S. and SARAN, A. (2012). “The radiation bystander effect and its potential implications 2643 for human health,” Curr. Mol. Med. 12(5), 613–624. 2644 MANCUSO, M., GIARDULLO, P., LEONARDI, S., PASQUALI, E., CASCIATI, A., DE STEFANO, I., 2645 TANORI, M., PAZZAGLIA, S. and SARAN, A. (2013). “Dose and spatial effects in long-distance 2646 radiation signaling in vivo: Implications for abscopal tumorigenesis,” Int. J. Radiat. Oncol. Biol. Phys. 2647 85(3), 813–819. 2648 MAO, J.H., WU, D., DELROSARIO, R., CASTELLANOS, A., BALMAIN, A. and PEREZ-LOSADA, J. 2649 (2008). “Atm heterozygosity does not increase tumor susceptibility to ionizing radiation alone or in a p53 2650 heterozygous background,” Oncogene 27(51), 6596–6600. 2651 2652 MARDER, B.A. and MORGAN, W.F. (1993). “Delayed chromosomal instability induced by DNA damage,” Mol. Cell Biol. 13(11), 6667–6677. 2653 MATHEWS, J.D., FORSYTHE, A.V., BRADY, Z., BUTLER, M.W., GOERGEN, S.K., BYRNES, G.B., 2654 GILES, G.G., WALLACE, A.B., ANDERSON, P.R., GUIVER, T.A., MCGALE, P., CAIN, T.M., 89 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2655 DOWTY, J.G., BICKERSTAFFE, A.C. and DARBY, S.C. (2013). “Cancer risk in 680 000 people 2656 exposed to computed tomography scans in childhood or adolescence: Data linkage study of 11 million 2657 Australians,” BMJ 346, f2360. 2658 2659 MATTHAIOS, D., HOUNTIS, P., KARAKITSOS, P., BOUROS, D. and KAKOLYRIS, S. (2013). “H2AX a promising biomarker for lung cancer: A review,” Cancer Invest. 31(9), 582–599. 2660 MCBRIDE, W.H., CHIANG, C.S., OLSON, J.L., WANG, C.C., HONG, J.H., PAJONK, F., DOUGHERTY, 2661 G.J., IWAMOTO, K.S., PERVAN, M. and LIA, Y.P. (2004). “A sense of danger from radiation,” Radiat. 2662 Res. 162(1), 1–19. 2663 MENARD, C., JOHANN, D., LOWENTHAL, M., MUANZA, T., SPROULL, M., ROSS, S., GULLEY, J., 2664 PETRICOIN, E., COLEMAN, C.N., WHITELEY, G., LIOTTA, L. and CAMPHAUSEN, K. (2006). 2665 “Discovering clinical biomarkers of ionizing radiation exposure with serum proteomic analysis,” Cancer 2666 Res. 66(3), 1844–1850. 2667 METTLER, F.A. JR., BHARGAVAN, M., FAULKNER, K., GILLEY, D.B., GRAY, J.E., IBBOTT, G.S., 2668 LIPOTI, J.A., MAHESH, M., MCCROHAN, J.L., STABIN, M.G., THOMADSEN, B.R. and 2669 YOSHIZUMI, T.T. (2009). “Radiologic and nuclear medicine studies in the United States and worldwide: 2670 Frequency, radiation dose, and comparison with other radiation sources—1950–2007. Radiology 253(2), 2671 520–531. 2672 2673 2674 MEZENTSEV, A. and AMUNDSON, S.A. (2011). “Global gene expression responses to low- or high-dose radiation in a human three-dimensional tissue model,” Radiat. Res. 175(6), 677–688. MONSIEURS, M.A., THIERENS, H.M., VRAL, A.M., VAN DE WIELE, C., DE RIDDER, L.I. and 2675 DIERCKX, R.A. (2000). “Adaptive response in patients treated with 131I,” J. Nucl. Med. 41(1), 17–22. 2676 MONSON, R.R. and MACMAHON, B. (1984). “Prenatal x-ray exposure and cancer in children,” pages 97 to 2677 105 in Radiation Carcinogenesis: Epidemiology and Biological Significance, Boice, J.D. Jr. and 2678 Fraumeni, J.F., Jr., Eds. (Raven Press, New York). 2679 2680 2681 2682 2683 2684 2685 MOOLGAVKAR, S.H. and KNUDSON, A.G., JR. (1981). “Mutation and cancer: A model for human carcinogenesis,” J. Natl. Cancer Inst. 66(6), 1037–1052. MOOLGAVKAR, S.H. and LUEBECK, E.G. (1992). “Multistage carcinogenesis: Population-based model for colon cancer,” J. Natl. Cancer Inst. 84(8), 610–618. MOOLGAVKAR, S.H. and VENZON, D.J. (1979). “Two-event models for carcinogenesis: Incidence curves for childhood and adult tumors,” Math. Biosci. 47(1–2), 55–77. MOORE, D.H., II and TUCKER, J.D. (1999). “Biological dosimetry of Chernobyl cleanup workers: 2686 Inclusion of data on age and smoking provides improved radiation dose estimates,” Radiat. Res. 152(6), 2687 655–664. 90 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2688 2689 2690 NOT TO BE DISSEMINATED OR REFERENCED MORGAN, W.F. (2003a). “Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiationinduced genomic instability and bystander effects in vitro,” Radiat. Res. 159(5), 567–580. MORGAN, W.F. (2003b). “Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation- 2691 induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational 2692 effects,” Radiat. Res. 159(5), 581–596. 2693 2694 2695 2696 MORGAN, W.F. and BAIR, W.J. (2013). “Issues in low dose radiation biology: The controversy continues. A perspective,” Radiat. Res. 179(5), 501–510. MORGAN, W.F., HARTMANN, A., LIMOLI, C.L., NAGAR, S. and PONNAIYA, B. (2002). “Bystander effects in radiation-induced genomic instability,” Mutat. Res. 504(1–2), 91–100. 2697 MOSEEVA, M.B., AZIZOVA, T.V., GRIGORYEVA, E.S. and HAYLOCK, R. (2014). “Risks of circulatory 2698 diseases among Mayak PA workers with radiation doses estimated using the improved Mayak Worker 2699 Dosimetry System 2008,” Radiat. Environ. Biophys. 53(2), 469–477. 2700 2701 2702 MUIRHEAD, C.R. and DARBY, S.C. (1987). “Modelling the relative and absolute risks of radiation-induced cancers,” J. R. Statist. Soc. A 150(2), 83–118. MUIRHEAD, C.R., COX, R., STATHER, J.W., MACGIBBON, B.H., EDWARDS, A.A. and HAYLOCK, 2703 R.G.E. (1993). “Estimates of late radiation risks to the UK population,” pages 15 to 157 in Documents of 2704 the NRPB 4(4) (Public Health England, London). 2705 MUIRHEAD, C.R., O’HAGAN, J.A., HAYLOCK, R.G., PHILLIPSON, M.A., WILLCOCK, T., 2706 BERRIDGE, G.L. and ZHANG, W. (2009). “Mortality and cancer incidence following occupational 2707 radiation exposure: Third analysis of the National Registry for Radiation Workers,” Br. J. Cancer 100(1), 2708 206–212. 2709 MUKHERJEE, D., COATES, P.J., LORIMORE, S.A. and WRIGHT, E.G. (2012). “The in vivo expression of 2710 radiation-induced chromosomal instability has an inflammatory mechanism,” Radiat. Res. 177(1), 18–24. 2711 MULLER, H.J. (1927). “Artificial transmutation of the gene,” Science 66(1699), 84–87. 2712 NAIR, R.R., RAJAN, B., AKIBA, S., JAYALEKSHMI, P., NAIR, M.K., GANGADHARAN, P., KOGA, T., 2713 MORISHIMA, H., NAKAMURA, S. and SUGAHARA, T. (2009). “Background radiation and cancer 2714 incidence in Kerala, India-Karanagappally cohort study,” Health Phys. 96(1), 55–66. 2715 NAKANO, M., KODAMA, Y., OHTAKI, K., NAKASHIMA, E., NIWA, O., TOYOSHIMA, M. and 2716 NAKAMURA, N. (2007). “Chromosome aberrations do not persist in the lymphocytes or bone marrow 2717 cells of mice irradiated in utero or soon after birth,” Radiat. Res. 167(6), 693–702. 2718 NAKANO, M. NISHIMURA, M., HAMASAKI, K., MISHIMA, S., YOSHIDA, M., NAKATA, A., 2719 SHIMADA, Y., NODA, A., NAKAMURA, N. and KODAMA, Y. (2014). “Fetal irradiation of rats 2720 induces persistent translocations in mammary epithelial cells similar to the level after adult irradiation, 2721 but not in hematolymphoid cells,” Radiat. Res. 181(2) 172–176. 91 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2722 2723 2724 NOT TO BE DISSEMINATED OR REFERENCED NA/NRC (2006). National Academies/National Research Council. Health Risks from Exposure to Low Levels of Ionizing Radiation, BEIR VII, Phase 2 (National Academies Press, Washington). NA/NRC (2012). National Academies/National Research Council. Technical Evaluation of the NASA Model 2725 for Cancer Risk to Astronauts Due to Space Radiation (National Academies Press, Washington). 2726 NAGASAWA, H. and LITTLE, J.B. (1992). “Induction of sister chromatid exchanges by extremely low 2727 2728 doses of alpha-particles,” Cancer Res. 52(22), 6394–6396. NCRP (1980). National Council on Radiation Protection and Measurements. Influence of Dose and Its 2729 Distribution in Time on Dose-Response Relationships for Low-LET Radiations, NCRP Report No. 64 2730 (National Council on Radiation Protection and Measurements. Bethesda, Maryland). 2731 NCRP (2001). National Council on Radiation Protection and Measurements. Evaluation of the Linear- 2732 Nonthreshold Dose-Response Model for Ionizing Radiation, NCRP Report No. 136 (National Council on 2733 Radiation Protection and Measurements. Bethesda, Maryland). 2734 NCRP (2005). National Council on Radiation Protection and Measurements. Extrapolation of Radiation- 2735 Induced Cancer Risks from Nonhuman Experimental Systems to Humans, NCRP Report No. 150 2736 (National Council on Radiation Protection and Measurements. Bethesda, Maryland). 2737 NCRP (2007). National Council on Radiation Protection and Measurements. Uncertainties in the 2738 Measurement and Dosimetry of External Radiation, NCRP Report No. 158 (National Council on 2739 Radiation Protection and Measurements. Bethesda, Maryland). 2740 NCRP (2009a). National Council on Radiation Protection and Measurements. Low Dose and Low Dose-Rate 2741 Radiation Effects and Models, Proceedings of the Forty-Fourth Annual Meeting, Health Phys. 97(5), 2742 373–541. 2743 NCRP (2009b). National Council on Radiation Protection and Measurements. Uncertainties in Internal 2744 Radiation Dose Assessment, NCRP Report No. 164 (National Council on Radiation Protection and 2745 Measurements. Bethesda, Maryland). 2746 NCRP (2009c). National Council on Radiation Protection and Measurements). Radiation Dose 2747 Reconstruction: Principles and Practices, NCRP Report No. 163 (National Council on Radiation 2748 Protection and Measurements. Bethesda, Maryland). 2749 NCRP (2009d). National Council on Radiation Protection and Measurements. Ionizing Radiation Exposure of 2750 the Populations of the United States, NCRP Report No.160 (National Council on Radiation Protection 2751 and Measurements. Bethesda, Maryland). 2752 NCRP (2011). National Council on Radiation Protection and Measurements. Second Primary Cancers and 2753 Cardiovascular Disease After Radiation Therapy, NCRP Report No. 170 (National Council of Radiation 2754 Protection and Measurements, Bethesda, Maryland). 92 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2755 NOT TO BE DISSEMINATED OR REFERENCED NCRP (2012). National Council on Radiation Protection and Measurements. Uncertainties in the Estimation 2756 of Radiation Risks and Probability of Disease Causation, NCRP Report No. 171 (National Council on 2757 Radiation Protection and Measurements, Bethesda, Maryland). 2758 NCRP (2015). National Council on Radiation Protection and Measurements. Where Are the Radiation 2759 Professionals (WARP)?, Synopsis of NCRP Statement No. 12, http://ncrponline.org/Publications/ 2760 Statements/WARP_Synopsis_Statement12.pdf (accessed March 22, 2015) (National Council on 2761 Radiation Protection and Measurements, Bethesda, Maryland). 2762 NEEL, J.V., SATOH, C., HAMILTON, H.B., OTAKE, M., GORIKI, K., KAGEOKA, T., FUJITA, M., 2763 NERIISHI, S. and ASAKAWA, J. (1980). “Search for mutations affecting protein structure in children of 2764 atomic bomb survivors: Preliminary report,” Proc. Natl. Acad. Sci. USA 77(7), 4221–4225. 2765 NEUMAIER, T., SWENSON, J., PHAM, C., POLYZOS, A., LO, A.T., YANG, P., DYBALL, J., 2766 ASAITHAMBY, A., CHEN, D.J., BISSELL, M.J., THALHAMMER, S. and COSTES, S.V. (2012). 2767 “Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells,” Proc. 2768 Natl. Acad. Sci. USA 109(2), 443–448. 2769 2770 NIKJOO, H. and KHVOSTUNOV, I.K. (2003). “Biophysical model of the radiation-induced bystander effect,” Int. J. Radiat. Biol. 79(1), 43–52. 2771 NIOSH (2014). National Institute for Occupational Safety and Health. Exposome and Exposomics: Overview, 2772 http://www.cdc.gov/niosh/topics/exposome/ (accessed 2014) (Centers for Disease Control and 2773 Prevention, Atlanta, Georgia). 2774 OHTAKI, K., KODAMA, Y., NAKANO, M., ITOH, M., AWA, A.A., COLOGNE, J. and NAKAMURA, N. 2775 (2004). “Human fetuses do not register chromosome damage inflicted by radiation exposure in lymphoid 2776 precursor cells except for a small but significant effect at low doses,” Radiat. Res. 161(4), 373–379. 2777 OKAYASU, R., SUETOMI, K., YU, Y., SILVER, A., BEDFORD, J.S., COX, R. and ULLRICH, R.L. 2778 (2000). “A deficiency in DNA repair and DNA-PKcs expression in the radiosensitive BALB/c mouse,” 2779 Cancer Res. 60(16), 4342–4345. 2780 2781 2782 OLIVIERI, G., BODYCOTE, J. and WOLFF, S. (1984). “Adaptive response of human lymphocytes to low concentrations of radioactive thymidine,” Science 223(4636), 594–597. OZASA, K., SHIMIZU, Y., SUYAMA, A., KASAGI, F., SODA, M., GRANT, E.J., SAKATA, R., 2783 SUGIYAMA, H. and KODAMA, K. (2012). “Studies of the mortality of atomic bomb survivors, Report 2784 14, 1950–2003: An overview of cancer and noncancer diseases,” Radiat. Res. 177(3), 229–243. 2785 PADOVANI, L., APPOLLONI, M., ANZIDEI, P., TEDESCHI, B., CAPOROSSI, D., VERNOLE, P. and 2786 MAURO, F. (1995). “Do human lymphocytes exposed to the fallout of the Chernobyl accident exhibit an 2787 adaptive response? 1. Challenge with ionizing radiation,” Mutat. Res. 332(1–2), 33–38. 93 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2788 NOT TO BE DISSEMINATED OR REFERENCED PARK, E.C., YOON, J.B., SEONG, J.S., CHOI, K.S., KONG, E.S., KIM, Y.J., PARK, Y.M. and PARK, 2789 E.M. (2006). “Effect of ionizing radiation on rat tissue: Proteomic and biochemical analysis,” Prep. 2790 Biochem. Biotechnol. 36(1), 19–35. 2791 PEARCE, M.S., SALOTTI, J.A., LITTLE, M.P., MCHUGH, K., LEE, C., KIM, K.P., HOWE, N.L., 2792 RONCKERS, C.M., RAJARAMAN, P., CRAFT, A.W., PARKER, L. and BERRINGTON DE 2793 GONZALEZ, A. (2012). “Radiation exposure from CT scans in childhood and subsequent risk of 2794 leukaemia and brain tumours: A retrospective cohort study,” Lancet 380(9840), 499–505. 2795 PENG, Y., BROWN, N., FINNON, R., WARNER, C.L., LIU, X., GENIK, P.C., CALLAN, M.A., RAY, 2796 F.A., BORAK, T.B., BADIE, C., BOUFFLER, S.D., ULLRICH, R.L., BEDFORD, J.S. and WEIL, M.M. 2797 (2009). “Radiation leukemogenesis in mice: Loss of PU.1 on chromosome 2 in CBA and C57BL/6 mice 2798 after irradiation with 1 GeV/nucleon 56Fe ions, x rays or gamma rays. Part I. Experimental observations,” 2799 Radiat. Res. 171(4), 474–483. 2800 PENG, A., LEWELLYN, A.L., SCHIEMANN, W.P. and MALLER, J.L. (2010). “Repo-man controls a 2801 protein phosphatase 1-dependent threshold for DNA damage checkpoint activation,” Curr. Biol. 20(5), 2802 387–396. 2803 PLEASANCE, E.D., STEPHENS, P.J., O’MEARA, S., MCBRIDE, D.J., MEYNERT, A., JONES, D., LIN, 2804 M.L., BEARE, D., LAU, K.W., GREENMAN, C., VARELA, I., NIK-ZAINAL, S., DAVIES, H.R., 2805 ORDONEZ, G.R., MUDIE, L.J., LATIMER, C., EDKINS, S., STEBBINGS, L., CHEN, L., JIA, M., 2806 LEROY, C., MARSHALL, J., MENZIES, A., BUTLER, A., TEAGUE, J.W., MANGION, J., SUN, 2807 Y.A., MCLAUGHLIN, S.F., PECKHAM, H.E., TSUNG, E.F., COSTA, G.L., LEE, C.C., MINNA, J.D., 2808 GAZDAR, A., BIRNEY, E., RHODES, M.D., MCKERNAN, K.J., STRATTON, M.R., FUTREAL, P.A. 2809 and CAMPBELL, P.J. (2010). “A small-cell lung cancer genome with complex signatures of tobacco 2810 exposure,” Nature 463(7278), 184–190. 2811 POHL-RULING, J., FISCHER, P., HAAS, O., OBE, G., NATARAJAN, A.T., VAN BUUL, P.P.W., 2812 BUCKTON, K.E., BIANCHI, N.O., LARRAMENDY, M., KUCEROVA, M., POLIKOVA, Z., 2813 LEONARD, A., FABRY, L., PALITTI, F., SHARMA, T., BINDER, W., MUKHERJEE, R.N. and 2814 MUKHERJEE, U. (1983). “Effect of low-dose acute x-irradiation on the frequencies of chromosomal 2815 aberrationsin human peripheral lymphocytes in vitro,” Mutat. Res. 110(1), 71–82. 2816 PONNAIYA, B., CORNFORTH, M.N. and ULLRICH, R.L. (1997). “Radiation-induced chromosomal 2817 instability in BALB/c and C57BL/6 mice: The difference is as clear as black and white,” Radiat Res. 2818 147(2), 121–125. 94 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2819 PONOMAREVA, O.N., ROSE, J.A., LASAREV, M., RASEY, J. and TURKER, M.S. (2002). “Tissue- 2820 specific deletion and discontinuous loss of heterozygosity are signatures for the mutagenic effects of 2821 ionizing radiation in solid tissues,” Cancer Res. 62(5), 1518–1523. 2822 POSTIGLIONE, I., CHIAVIELLO, A. and PALUMBO, G. (2010). “Twilight effects of low doses of ionizing 2823 radiation on cellular systems: A bird's eye view on current concepts and research,” Med. Oncol. 27(2), 2824 495–509. 2825 2826 2827 2828 PRESTON, R.J. (2015). “Integrating basic radiobiological science and epidemiological studies: Why and how,” Health Phys. 108(2), 125–130. PRESTON, R.J. and WILLIAMS, G.M. (2005). “DNA-reactive carcinogens: Mode of action and human cancer hazard,” Crit. Rev. Toxicol. 35(8–9), 673–683. 2829 PRESTON, D.L., MATTSSON, A., HOLMBERG, E., SHORE, R., HILDRETH, N.G. and BOICE, J.D., JR. 2830 (2002). “Radiation effects on breast cancer risk: A pooled analysis of eight cohorts,” Radiat. Res. 158(2), 2831 220–235. 2832 PRESTON, D., RON, E., TOKUOKA, S., FUNAMOTO, S., NISHI, N., SODA, M., MABUCHI, K. and 2833 KODAMA, K. (2007). “Solid cancer incidence in atomic bomb survivors: 1958–1998,” Radiat. Res. 2834 168(1), 1–64. 2835 PRESTON, D.L., CULLINGS, H., SUYAMA, A., FUNAMOTO, S., NISHI, N., SODA, M., MABUCHI, K., 2836 KODAMA, K., KASAGI, F. and SHORE, R.E. (2008). “Solid cancer incidence in atomic bomb survivors 2837 exposed in utero or as young children,” J. Natl. Cancer Inst. 100(6), 428–436. 2838 PUTNAM, C.D., JAEHNIG, E.J. and KOLODNER, R.D. (2009). “Perspectives on the DNA damage and 2839 replication checkpoint responses in Saccharomyces cerevisiae,” DNA Repair (Amst.) 8(9), 974–982. 2840 RAHU, K., AUVINEN, A., HAKULINEN, T., TEKKEL, M., INSKIP, P.D., BROMET, E.J., BOICE, J.D., 2841 JR. and RAHU, M. (2013). “Chernobyl cleanup workers from Estonia: Follow-up for cancer incidence 2842 and mortality,” J. Radiol. Protect. 33(2), 395–411. 2843 2844 2845 RAPPAPORT, S.M. (2011). “Implications of the exposome for exposure science,” J. Expo. Sci. Environ. Epidemiol. 21(1), 5–9. REDPATH, J. L., LIANG, D., TAYLOR, T. H., CHRISTIE, C. and ELMORE, E. (2001). “The shape of the 2846 dose-response curve for radiation-induced neoplastic transformation in vitro: Evidence for an adaptive 2847 response against neoplastic transformation at low doses of low-LET radiation,” Radiat. Res. 156(6), 700– 2848 707. 2849 ROGAKOU, E.P., PILCH, D.R., ORR, A.H., IVANOVA, V.S. and BONNER, W.M. (1998). “DNA double- 2850 stranded breaks induce histone H2AX phosphorylation on serine 139,” J. Biol. Chem. 1273(10), 5858– 2851 5868. 95 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2852 RON, E., LUBIN, J.H., SHORE, R.E., MABUCHI, K., MODAN, B., POTTERN, L.M., SCHNEIDER, A.B., 2853 TUCKER, M.A. and BOICE, J.D., JR. (1995). “Thyroid-cancer after exposure to external radiation: A 2854 pooled analysis of seven studies,” Radiat. Res. 141(3), 259–277. 2855 2856 2857 ROTHKAMM, K. and LOBRICH, M. (2003). “Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses,” Proc. Natl. Acad. Sci. USA, 100(9), 5057–5062. ROTHKAMM, K., BARNARD, S., AINSBURY, E.A., AL-HAFIDH, J., BARQUINERO, J.F., 2858 LINDHOLM, C., MOQUET, J., PERALA, M., ROCH-LEFEVRE, S., SCHERTHAN, H., THIERENS, 2859 H., VRAL, A. and VANDERSICKEL, V. (2013). “Manual versus automated γ-H2AX foci analysis 2860 across five European laboratories: Can this assay be used for rapid biodosimetry in a large scale radiation 2861 accident?,” Mutat. Res. 756(1–2), 170–173. 2862 RUSSELL, L.B. and HUNSICKER, P.R. (2012). “The effect of dose rate on the frequency of specific-locus 2863 mutations induced in mouse spermatogonia is restricted to larger lesions; a retrospective analysis of 2864 historical data,” Radiat. Res. 177(5), 555–564. 2865 2866 2867 2868 RUSSELL, W.L. and KELLY, E.M. (1982). “Mutation frequencies in male mice and the estimation of genetic hazards of radiation in men,” Proc. Natl. Acad. Sci. USA 79(2), 542–544. RUSSELL, W.L., RUSSELL, L.B. and KELLY, E.M. (1958). “Radiation dose rate and mutation frequency,” Science 128(3338), 1546–15450. 2869 SACKETT, D.L. (1979). “Bias in analytic research,” J. Chronic Dis. 32(1–2), 51–63. 2870 SAKATA, R., KLEINERMAN, R.A., MABUCHI, K., STOVALL, M., SMITH, S.A., WEATHERS, R., 2871 WACTAWSKI-WENDE, J., COOKFAIR, D.L., BOICE, J.D., JR. and INSKIP, P.D. (2012). “Cancer 2872 mortality following radiation therapy for benign gynecologic disorders,” Radiat. Res. 178(4), 266–279. 2873 SANKARANARAYANAN, K. and CHAKRABORTY, R. (2001). “Impact of cancer predisposition and 2874 radiosensitivity on the population risk of radiation-induced cancers,” Radiat. Res. 156(5 Pt 2), 648–656. 2875 SATOH, C., TAKAHASHI, N., ASAKAWA, J., KODAIRA, M., KUICK, R., HANASH, S.M. and NEEL, 2876 J.V. (1996). “Genetic analysis of children of atomic bomb survivors,” Environ. Health Persp. 104(Suppl. 2877 3), 511–519. 2878 SAWANT, S.G., RANDERS-PEHRSON, G., GEARD, C.R., BRENNER, D.J. and HALL, E.J. (2001). “The 2879 bystander effect in radiation oncogenesis: I. Transformation in C3H 10T1/2 cells in vitro can be initiated 2880 in the unirradiated neighbors of irradiated cells,” Radiat. Res. 155(3), 397–401. 2881 2882 2883 SCHULL, W.J. (2003). “The children of atomic bomb survivors: A synopsis,” J. Radiol. Prot. 23(4), 369– 384. SEED, J., CARNEY, E.W., CORLEY, R.A., CROFTON, K.M., DESESSO, J.M., FOSTER, P.M., 2884 KAVLOCK, R., KIMMEL, G., KLAUNIG, J., MEEK, M.E., PRESTON, R.J., SLIKKER, W., JR., 2885 TABACOVA, S., WILLIAMS, G.M., WILTSE, J., ZOELLER, R.T., FENNER-CRISP, P. and PATTON, 96 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2886 D.E. (2005). “Overview: Using mode of action and life stage information to evaluate the human relevance 2887 of animal toxicity data,” Crit. Rev. Toxicol. 35(8–9), 664–672. 2888 SELMANSBERGER, M., FEUCHTINGER, A., ZURNADZHY, L., MICHNA, A., KAISER, J.C., ABEND, 2889 M., BRENNER, A., BOGDANOVA, T., WALCH, A., UNGER, K., ZITZELSBERGER, H. and HESS, J. 2890 (2014). “CLIP2 as radiation biomarker in papillary thyroid carcinoma,” Oncogene. doi: 2891 10.1038/onc.2014.311. 2892 SEVANKAEV, A.V., LLOYD, D.C., EDWARDS, A.A., KHVOSTUNOV, I.K., MIKHAILOVA, G.F., 2893 GOLUB, E.V., SHEPEL, N.N., NADEJINA, N.M., GALSTIAN, I.A., NUGIS, V.YU., BARRIOS L., 2894 CABALLIN, M.R. and BARQUINERO, J.F. (2005). “A cytogenetic follow-up of some highly irradiated 2895 victims of the Chernobyl accident,” Radiat. Prot. Dosim. 113(2), 152–161. 2896 2897 2898 SHAHEEN, M., ALLEN, C., NICKOLOFF, J.A. and HROMAS, R. (2011). “Synthetic lethality: Exploiting the addiction of cancer to DNA repair,” Blood 117(23), 6074–6082. SHILNIKOVA, N.S., PRESTON, D.L., RON, E., GILBERT, E.S., VASSILENKO, E.K., ROMANOV, S.A., 2899 KUZNETSOVA, I.S., SOKOLNIKOV, M.E., OKATENKO, P.V., KRESLOV, V.V. and 2900 KOSHURNIKOVA, N.A. (2003). “Cancer mortality risk among workers at the Mayak nuclear complex,” 2901 Radiat. Res. 159(6), 787–798. 2902 SHIMIZU, Y., KODAMA, K., NISHI, N., KASAGI, F., SUYAMA, A., SODA, M., GRANT, E.J., 2903 SUGIYAMA, H., SAKATA, R., MORIWAKI, H., HAYASHI, M., KONDA, M. and SHORE, R.E. 2904 (2010). “Radiation exposure and circulatory disease risk: Hiroshima and Nagasaki atomic bomb survivor 2905 data, 1950–2003,” BMJ 340, b5349. 2906 2907 2908 2909 SHORE, R.E., NERIISHI, K. and NAKASHIMA, E. (2010). “Epidemiologic studies of cataract risk at lowto-moderate radiation doses: (Not) seeing is believing,” Radiat. Res. 174(6), 889–894. SHURYAK, I., SACHS, R.K. and BRENNER, D.J. (2010). “Cancer risks after radiation exposure in middle age,” J. Natl. Cancer Inst. 102(21), 1628–1636. 2910 SIGNORELLO, L.B., MULVIHILL, J.J., GREEN, D.M., MUNRO, H.M., STOVALL, M., WEATHERS, 2911 R.E., MERTENS, A.C., WHITTON, J.A., ROBISON, L.L. and BOICE, J.D., JR. (2012). “Congenital 2912 anomalies in the children of cancer survivors: A report from the Childhood Cancer Survivor Study,” 2913 J. Clin. Oncol. 30(3), 239–245. 2914 SLEBOS, R.J.C., LITTLE, R.E., UMBACH, D.M., ANTIPKIN, Y., ZADAOROZHNAJA, T.D., MENDEL, 2915 N.A., SOMMER, C.A., CONWAY, K., PARRISH, E., GULINO, S. and TAYLOR, J.A. (2004). 2916 “Mutation research/genetic toxicology and environmental mutagenesis,” Mutat. Res. 559(1–2), 143–151. 2917 SNIJDERS, A.M., MARCHETTI, F., BHATNAGAR, S., DURU, N., HAN, J., HU, Z., MAO, J.H., GRAY, 2918 J.W. and WYROBEK, A.J. (2012). “Genetic differences in transcript responses to low-dose ionizing 2919 radiation identify tissue functions associated with breast cancer susceptibility,” PLoS One 7(10), e45394. 97 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2920 2921 2922 NOT TO BE DISSEMINATED OR REFERENCED SOBELS, F.H. (1993). “Approaches to assessing genetic risks from exposure to chemicals,” Environ. Health Perspect. 101(Supp 3), 327–332. SOWA, M.B., CHRISLER, W.B., ZENS, K.D., ASHJIAN, E.J. and OPRESKO, L.K. (2010). “Three- 2923 dimensional culture conditions lead to decreased radiation induced cytotoxicity in human mammary 2924 epithelial cells,” Mutat. Res. 687(1–2), 78–83. 2925 SPIESS, H. and MAYS, C.W. (1973). “Protraction effect on bone sarcoma-induction of 224Ra in children and 2926 adults,” pages 437–450 in Radionuclide Carcinogenesis, AEC Symposium Series 29, CONF-720505, 2927 Sanders, C. L., Busch, R. H., Ballou, J. E. and Mahlum, D.D., Eds., (National Technical Information 2928 Service, Springfield, Virginia). 2929 SPITZ, D.R., AZZAM, E I., LI, J.J. and GIUS, D. (2004). “Metabolic oxidation/reduction reactions and 2930 cellularresponses to ionizing radiation: A unifying concept in stress response biology,” Cancer Metastasis 2931 Rev. 23(3–4), 311–322. 2932 2933 STEWART, A., WEBB, J., GILES, D. and HEWITT, D. (1956). “Malignant disease in childhood and diagnostic irradiation in utero,” Lancet 268(6940), 447. 2934 STOVALL, M. SMITH, S.A., LANGHOLZ, B.M., BOICE, J.D., JR., SHORE, R.E., ANDERSSON, M., 2935 BUCHHOLZ, T.A., CAPANU, M., BERNSTEIN, L., LYNCH, C.F., MALONE, K.E., ANTON- 2936 CULVER, H., HAILE, R.W., ROSENSTEIN, B.S., REINER, A.S., THOMAS, D.C. and BERNSTEIN, 2937 J.L. (2008). “Dose to the contralateral breast from radiation therapy and risk of second primary breast 2938 cancer in the WECARE Study,” Int. J. Radiat. Oncol. Biol. Phys. 72(4), 1021–1030. 2939 TAO, Z., AKIBA, S., ZHA, Y., SUN, Q., ZOU, J., LI, J., LIU, Y., YUAN, Y., TOKONAMI, S., 2940 MORISHOMA, H., KOGA, T., NAKAMURA, S., SUGAHARA, T. and WEI, L. (2012). “Cancer and 2941 non-cancer mortality among inhabitants in the high background radiation area of Yangjiang, China 2942 (1979–1998),” Health Phys. 102(2), 173–181. 2943 2944 TAPIO, S. and JACOB, V. (2007). “Radioadaptive response revisited,” Radiat. Environ. Biophys. 46(1), 1– 12. 2945 TAWN, E.J., WHITEHOUSE, C.A. and MARTIN, F.A. (2000). “Sequential chromosome aberration analysis 2946 following radiotherapy - no evidence for enhanced genomic instability,” Mutat. Res. 465(1–2), 45–51. 2947 2948 2949 TAWN, E.J., WHITEHOUSE, C.A. and TARONE, R.E. (2004). “FISH chromosome aberration analysis on retired radiation workers from the Sellafield nuclear facility,” Radiat. Res. 162(3), 249–256. TAWN, E.J., WHITEHOUSE, C.A., WINTHER, J.F., CURWEN, G.B., REES, G.S., STOVALL, M., 2950 OLSEN, J.H., GULDBERG, P., RECHNITZER, C., SCHRODER, H. and BOICE, J.D., JR. (2005). 2951 “Chromosome analysis in childhood cancer survivors and their offspring—no evidence for radiation 2952 therapy-induced persistent genomic instability,” Mutat. Res. 583(2), 198–206. 98 NCRP SC 1-21 Draft of March 31, 2015 [MR] 2953 2954 NOT TO BE DISSEMINATED OR REFERENCED TAWN, E.J., WHITEHOUSE, C.A. and RIDDELL, A.E. (2006). “FISH chromosome analysis of plutonium workers from the Sellafield nuclear facility,” Radiat. Res. 165(5), 592–597. 2955 TAWN, E.J., REES, G.S., LEITH, C., WINTHER, J.F., CURWEN, G.B., STOVALL, M., OLSEN, J.H., 2956 RECHNITZER, C., SCHROEDER, H., GULDBERG, P. and BOICE, J.D., JR. (2011). “Germline 2957 minisatellite mutations in survivors of childhood and young adult cancer treated with radiation,” Int. J. 2958 Radiat. Biol. 87(3), 330–340. 2959 TAWN, E.J., CURWEN, G.B., REES, G.S. and JONAS, P. (2015). “Germline minisatellite mutations in 2960 workers occupationally exposed to radiation at the Sellafield nuclear facility,” J. Radiat. Protect. 35(1), 2961 21–36. 2962 TAYLOR, M., DEHAINAULT, C., DESJARDINS, L., DOZ, F., LEVY, C., SASTRE, X., COUTURIER, J., 2963 STOPPA-LYONNET, D., HOUDAYER, C. and GAUTHIER-VILLARS, M. (2007). “Genotype- 2964 phenotype correlations in hereditary familial retinoblastoma,” Hum. Mutat. 28(3), 284–293. 2965 TEDESCHI, B., CAPOROSSI, D.,VERNOLE, P., PADOVANI, L., APPOLLONI, M., ANZIDEI, P. and 2966 MAURO, F. (1995). “Do human lymphocytes exposed to the fallout of the Chernobyl accident exhibit an 2967 adaptive response? 2. Challenge with bleomycin,” Mutat. Res. 332(1–2), 39–44). 2968 THOMAS, R.S., CLEWELL, H.J., III, ALLEN, B.C., YANG, L., HEALY, E. and ANDERSEN, M.E. 2969 (2012). “Integrating pathway-based transcriptomic data into quantitative chemical risk assessment: A five 2970 chemical case study,” Mutat. Res. 746(2), 135–143. 2971 TOLLEFSEN, K.E., SCHOLZ, S., CRONIN, M.T., EDWARDS, S.W., DE KNECHT, J., CROFTON, K., 2972 GARCIA-REYERO, N., HARTUNG, T., WORTH, A. and PATLEWICZ, G.(2014). “Applying adverse 2973 outcome pathways (AOPs) to support integrated approaches to testing and assessment (IATA),” Regul. 2974 Toxicol. Pharmacol. 70(3), 629–640. 2975 TRAVIS, L.B., RABKIN, C.S., BROWN, L.M., ALLAN, J.M., ALTER, B.P., AMBROSONE, C.B., BEGG, 2976 C.B., CAPORASO, N., CHANOCK, S., DEMICHELE, A., FIGG, W.D., GOSPODAROWICZ, M.K., 2977 HALL, E.J., HISADA, M., INSKIP, P., KLEINERMAN, R., LITTLE, J.B., MALKIN, D., NG, A.K., 2978 OFFIT, K., PUI, C.H., ROBISON, L.L., ROTHMAN, N., SHIELDS, P.G., STRONG, L., TANIGUCHI, 2979 T., TUCKER, M.A. and GREENE, M.H. (2006). “Cancer survivorship— genetic susceptibility and 2980 second primary cancers: Research strategies and recommendations,” J. Natl. Cancer Inst. 98(1), 15–25. 2981 2982 2983 2984 TUCKER, J.D. (2008). “Low-dose ionizing radiation and chromosome translocations: A review of the major considerations for human biological dosimetry,” Mutat. Res. 659(3), 211–220. TUCKER, J.D. and LUCKINBILL, L.S. (2011). “Estimating the lowest detectable dose of ionizing radiation by FISH whole-chromosome painting,” Radiat. Res. 175(5), 631–637. 99 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 2985 TUCKER, J.D., TAWN, E.J., HOLDSWORTH, D., MORRIS, S., LANGLOIS, R., RAMSEY, M.J., KATO, 2986 P., BOICE, J.D., JR., TARONE, R.E. and JENSEN, R.H. (1997a). “Biological dosimetry of radiation 2987 workers at the Sellafield nuclear facility,” Radiat. Res. 148(3), 216–226. 2988 2989 TUCKER, J.D., EASTMOND, D.A. and LITTLEFIELD, L.G. (1997b). “Cytogenetic end-points as biological dosimeters and predictors of risk in epidemiological studies,” IARC Sci. Publ. (142), 185–200. 2990 TURKER, M.S. CONNOLLY, L, DAN, C., LASAREV, M., GAUNY, S., KWOH, E. and KRONENBERG, 2991 A. (2009). “Comparison of autosomal mutations in mouse kidney epithelial cells exposed to iron ions in 2992 situ or in culture,” Radiat. Res. 172(5), 558–566. 2993 TURKER, M.S. GRYGORYEV, D., DAN, C., ECKELMANN, B., LASAREV, M., GAUNY, S., KWOH, E. 2994 KRONENBERG, A. (2013). “Autosomal mutations in mouse kidney epithelial cells exposed to high- 2995 energy protons in vivo or in culture,” Radiat. Res. 179(5), 521–529. 2996 TYBURSKI, J.B., PATTERSON, A.D., KRAUSZ, K.W., SLAVIK, J., FORNACE, A.J., JR., GONZALEZ, 2997 F.J. and IDLE, J.R. (2008). “Radiation metabolomics. 1. Identification of minimally invasive urine 2998 biomarkers for gamma-radiation exposure in mice,” Radiat. Res. 170(1), 1–14. 2999 TYBURSKI, J.B., PATTERSON, A.D., KRAUSZ, K.W., SLAVIK, J., FORNACE, A.J., JR., GONZALEZ, 3000 F.J. and IDLE, J.R. (2009). “Radiation metabolomics. 2. Dose- and time-dependent urinary excretion of 3001 deaminated purines and pyrimidines after sublethal gamma-radiation exposure in mice,” Radiat. Res. 3002 172(1), 42–57. 3003 3004 3005 ULLRICH, R.L. (1984). “Tumor induction in BALB/c mice after fractionated or protracted exposures to fission-spectrum neutrons,” Radiat. Res. 97(3), 587–597. UNSCEAR (1993). United Nations Scientific Committee on the Effects of Atomic Radiation. Sources and 3006 Effects of Ionizing Radiation. UNSCEAR 1993 Report to the General Assembly, with Scientific 3007 Annexes, E.94.IX.2 (United Nations Publications, New York). 3008 UNSCEAR (1994). United Nations Scientific Committee on the Effects of Atomic Radiation. Sources and 3009 Effects of Ionizing Radiation. UNSCEAR 1994 Report to the General Assembly, with Scientific 3010 Annexes, E.94.IX.11 (United Nations Publications, New York). 3011 UNSCEAR (2000). United Nations Scientific Committee on the Effects of Atomic Radiation. Sources and 3012 Effects of Ionizing Radiation. UNSCEAR 2000 Report to the General Assembly, with Scientific 3013 Annexes. Volume II: Effects (United Nations Publications, New York). 3014 UNSCEAR (2001). United Nations Scientific Committee on the Effects of Atomic Radiation. Hereditary 3015 Effects of Radiation. UNSCEAR 2001 Report to the General Assembly, with Scientific Annex, 3016 http://www.unscear.org/unscear/en/publications/2001.html (accessed 2014), E.01.IX.2 (United Nations 3017 Publications, New York). 100 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 3018 UNSCEAR (2008a). United Nations Scientific Committee on the Effects of Atomic Radiation. “Annex A. 3019 Epidemiological studies of radiation and cancer,” pages 13 to 322 in Effects of Ionizing Radiation, 3020 UNSCEAR 2006 Report to the General Assembly, with Scientific Annexes. Volume I: Report to the 3021 General Assembly, Scientific Annexes A and B (United Nations Publications, New York). 3022 UNSCEAR (2008b). United Nations Scientific Committee on the Effects of Atomic Radiation. “Annex B. 3023 Epidemiological evaluation of cardiovascular disease and other non-cancer diseases following radiation 3024 exposure,” pages 325 to 383 in Effects of Ionizing Radiation, UNSCEAR 2006 Report to the General 3025 Assembly, with Scientific Annexes. Volume I: Report to the General Assembly, Scientific Annexes A 3026 and B (United Nations Publications, New York). 3027 UNSCEAR (2011). United Nations Scientific Committee on the Effects of Atomic Radiation. “Health effects 3028 due to radiation from the Chernobyl accident,”Annex D in Sources and Effects of Ionizing Radiation. 3029 UNSCEAR 2008 Report to the General Assemblywith Scientific Annexes, Volume II, 3030 http://www.unscear.org/docs/reports/2008/11-80076_Report_2008_Annex_D.pdf. (accessed 2014), 3031 E.11.IX.3 (United Nations Publications, New York). 3032 UNSCEAR (2012). United Nations Scientific Committee on the Effects of Atomic Radiation Biological 3033 Mechanisms of Radiation Actions at Low Doses. A White Paper to Guide the Scientific Committee's 3034 Future Programme of Work, Report V.12-57831 (United Nations Publications, New York). 3035 3036 3037 3038 3039 UPTON, A.C. (1990). “Carcinogenic effects of low-level ionizing radiation,” J. Natl. Cancer Inst. 82(6), 448– 449. VAKIFAHMETOGLU, H., OLSSON, M. and ZHIVOTOVSKY, B. (2008). “Death through a tragedy: Mitotic catastrophe,” Cell Death Differ. 15(7), 1153–1162. VANDENABEELE, P., GALLUZZI, L., VANDEN BERGHE, T. and KROEMER, G. (2010). “Molecular 3040 mechanisms of necroptosis: An ordered cellular explosion,” Nat. Rev. Mol. Cell Biol. 11(10), 700–714. 3041 VINKEN, M. (2013). “The adverse outcome pathway concept: A pragmatic tool in toxicology,” Toxicology 3042 3043 3044 3045 312, 158–165. VOGELSTEIN, B., PAPADOPOULOS, N., VELCULESCU, V.E., ZHOU, S., DIAZ, L.A., JR. and KINZLER, K.W. (2013). “Cancer genome landscapes,” Science 339(6127), 1546–1558. VOZILOVA, A.V., SHAGINA, N.B., DEGTEVA, M.O., EDWARDS, A.A., AINSBURY, E.A., MOQUET, 3046 J.E., HONE, P., LLOYD, D.C., FOMINA, J.N. and DARROUDI, F. (2012). “Preliminary FISH-based 3047 assessment of external dose for residents exposed on the Techa River,” Radiat. Res. 177(1), 84–91. 3048 3049 3050 3051 VRIJHEID, M. (2014). “The exposome: A new paradigm to study the impact of environment on health,” Thorax 69(9), 876–878. WAKEFORD, R. (2012). “Radiation effects: Modulating factors and risk assessment—an overview,” Ann. ICRP 41(3–4), 98-107. 101 NCRP SC 1-21 Draft of March 31, 2015 [MR] 3052 3053 3054 3055 NOT TO BE DISSEMINATED OR REFERENCED WAKEFORD, R. and LITTLE, M.P. (2003). “Risk coefficients for childhood cancer after intrauterine irradiation: A review,” Int. J. Radiat. Biol. 79(5), 293–309. WAKEFORD, R. and TAWN, E.J. (2010). “Editorial: The meaning of low dose and low dose-rate,” J. Radiol. Protect. 30(1), 1–3. 3056 WANG, Z.Y., BOICE, J.D., JR., WEI, L.X., BEEBE, G.W., ZHA, Y.R., KAPLAN, M.M., TAO, Z.F., 3057 MAXON, H.R., III, ZHANG, S.Z., SCHNEIDER, A.B., TAN, B., WESSELER, T.A., CHEN, D., 3058 ERSHOW, A.G., KLEINERMAN, R.A., LITTLEFIELD, L.G. and PRESTON, D. (1990). “Thyroid 3059 nodularity and chromosome aberrations among women in areas of high background radiation in China,” 3060 J. Natl. Cancer Inst. 82(6), 478–485. 3061 WARD, J.F. (1998). “Nature of lesions formed by ionizing radiation,” pages 65 to 84 in DNA Damage and 3062 Repair: Volume II: DNA Repair in Higher Eukaryotes. Nickoloff, J.A. and Hoekstra, M.F., Eds. (Humana 3063 Press, Totowa, New Jersey). 3064 WATSON, G.E., LORIMORE, S.A. and WRIGHT, E.G. (1996). “Long-term in vivo transmission of alpha- 3065 particle-induced chromosomal instability in murine haemopoietic cells,” Int. J. Radiat. Biol. 69(2), 175– 3066 182. 3067 WATSON, G.E., POCOCK, D.A., PAPWORTH, D., LORIMORE, S.A. and WRIGHT, E.G. (2001). “In vivo 3068 chromosomal instability and transmissible aberrations in the progeny of haemopoietic stem cells induced 3069 by high- and low-LET radiations,” Int. J. Radiat. Biol. 77(4), 409–417. 3070 WEINBERG, R.A. (2013). The Biology of Cancer, 2nd ed. (Garland Science, New York). 3071 WESTCOTT, P.M., HALLIWILL, K.D., TO, M.D., RASHID, M., RUST, A.G., KEANE, T.M., 3072 DELROSARIO, R., JEN, K.Y., GURLEY, K.E., KEMP, C.J., FREDLUND, E., QUIGLEY, D.A., 3073 ADAMS, D.J. and BALMAIN, A. (2014). “The mutational landscapes of genetic and chemical models of 3074 Kras-driven lung cancer,” Nature, doi: 10.1038/nature13898 3075 3076 WHITEHOUSE, C.A. and TAWN, E.J. (2001). “No evidence for chromosomal instability in radiation workers with in vivo exposure to plutonium,” Radiat. Res. 156(5), 467–475. 3077 WIESE, C., GAUNY, S.S., LIU, W.C., CHERBONNEL-LASSERRE, C.L. and KRONENBERG, A. (2001). 3078 “Different mechanisms of radiation-induced loss of heterozygosity in two human lymphoid cell lines 3079 from a single donor,” Cancer Res. 61(3), 1129–1137. 3080 3081 WILD, C.P., SCALBERT, A. and HERCEQ, Z. (2013). “Measuring the exposome: A powerful basis for evaluating environmental exposures and cancer risk,” Environ. Mol. Mutagen 54(7), 480–499. 3082 WILDING, C.S., CURWEN, G.B., TAWN, E.J., SHENG, X., WINTHER, J.F., CHAKRABORTY, R. and 3083 BOICE, J.D., JR. (2007). “Influence of polymorphisms at loci encoding DNA repair proteins on G2 3084 chromosomal radiosensitivity,” Environ. Mol. Mutagen 48(1), 48–57. 102 NCRP SC 1-21 Draft of March 31, 2015 [MR] 3085 NOT TO BE DISSEMINATED OR REFERENCED WILSON, P.F., NAGASAWA, H., FITZEK, M.M., LITTLE, J.B. and BEDFORD, J.S. (2010). “G2-phase 3086 chromosomal radiosensitivity of primary fibroblasts from hereditary retinoblastoma family members and 3087 some apparently normal controls,” Radiat. Res. 173(1), 62–70. 3088 3089 3090 WINTHER, J.F. and OLSEN, J.H. (2012). “Does cancer treatment in childhood induce transgenerational genetic damage?,” J. Clin. Oncol. 30(3), 225–226. WINTHER, J.F., OLSEN, J.H., WU, H., SHYR, Y., MULVIHILL, J.J., STOVALL, M., NIELSE, A., 3091 SCHMIEGELOW, M. and BOICE, J.D., JR. (2012). “Genetic disease in the children of Danish survivors 3092 of childhood and adolescent cancer,” J. Clin. Oncol. 30(1), 27–33. 3093 3094 WINTON, D.J., PEACOCK, J.H. and PONDER, B.A. (1989). “Effect of gamma radiation at high- and lowdose rate on a novel in vivo mutation assay in mouse intestine,” Mutagenesis 4(5), 404–406. 3095 WONG, F.L., BOICE, J.D., JR., ABRAMSON, D.H., TARONE, R.E., KLEINERMAN, R.A., STOVALL, 3096 M., GOLDMAN, M.B., SEDDON, J.M., TARBELL, N., FRAUMENI, J.F., JR. and LI, F.P. (1997). 3097 “Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk,” JAMA 278(15), 1262–1267. 3098 3099 3100 YAMADA, M., WONG, F.L., FUJIWARA, S., AKAHOSHI, M. and SUZUKI, G. (2004). “Noncancer disease incidence in atomic bomb survivors, 1958–1998,” Radiat. Res. 161(6), 622–632. YU, Y., OKAYASU, R., WEIL, M.M., SILVER, A., MCCARTHY, M., ZABRISKIE, R., LONG, S., COX, 3101 R. and ULLRICH, R.L. (2001). “Elevated breast cancer risk in irradiated BALB/c mice associates with 3102 unique functional polymorphism of the Prkdc (DNA-dependent protein kinase catalytic subunit) gene,” 3103 Cancer Res. 61(5), 1820–1824. 3104 YU, C.L., TUCKER, M.A., ABRAMSON, D.H., FURUKAWA, K., SEDDON, J.M., STOVALL, M., 3105 FRAUMENI, J.F., JR. and KLEINERMAN, R.A. (2009). “Cause-specific mortality in long-term 3106 survivors of retinoblastoma,” J. Natl. Cancer Inst. 101(8), 581–591. 3107 ZABLOTSKA, L.B., BAZYKA, D., LUBIN, J.H., GUDZENKO, N., LITTLE, M.P., HATCH, M., FINCH, 3108 S., DYAGIL, I., REISS, R.F., CHUMAK, V.V., BOUVILLE, A., DROZDOVITCH, V., KRYUCHKOV, 3109 V.P., GOLOVANOV, I., BAKHANOVA, E., BABKINA, N., LUBARETS, T., BEBESHKO, V., 3110 ROMANENKO, A. and MABUCHI, K. (2013). “Radiation and the risk of chronic lymphocytic and other 3111 leukemias among Chornobyl cleanup workers,” Environ. Health Perspect. 121(1), 59–65. 3112 3113 3114 ZEIDAN, Y.H. and HANNUN, Y.A. (2010). “The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation,” Curr. Mol. Med. 10(5), 454–466. ZHANG, Y., ZHOU, J., BALDWIN, J., HELD, K.D., PRISE, K.M., REDMOND, R.W. and LIBER, H.L. 3115 (2009). “Ionizing radiation-induced bystander mutagenesis and adaptation: quantitative and temporal 3116 aspects,” Mutat. Res. 671(1–2), 20–25. 103 NCRP SC 1-21 Draft of March 31, 2015 [MR] NOT TO BE DISSEMINATED OR REFERENCED 3117 ZHANG, Q., MATZKE, M., SCHEPMOES, A.A., MOORE, R.J., WEBB-ROBERTSON, B.J., HU, Z., 3118 MONROE, M.E., QIAN, W.J., SMITH, R.D. and MORGAN, W.F. (2014). “High and low doses of 3119 ionizing radiation induce different secretome profiles in a human skin model,” PLoS One 9(3), 92332. 3120 ZHOU, H., RANDERS-PEHRSON, G., WALDREN, C.A., VANNAIS, D., HALL, E.J. and HEI, T.K. 3121 (2000). “Induction of a bystander mutagenic effect of alpha particles in mammalian cells,” Proc. Natl. 3122 Acad. Sci USA 97(5), 2099–2104. 104


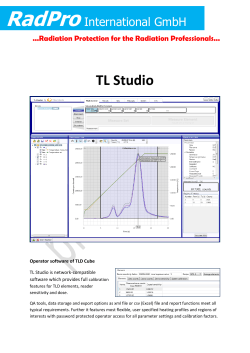







© Copyright 2026