
Document 1640
Till familj och vänner List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I. Eriksson, S.; Gutiérrez, O.A.; Bjerling, P.; Tomkinson, B. (2009) Development, evaluation and application of tripeptidyl-peptidase II sequence signatures. Archives of Biochemistry and Biophysics, 484(1):39-45 II. Lindås, A-C.; Eriksson, S.; Josza, E.; Tomkinson, B. (2008) Investigation of a role for Glu-331 and Glu-305 in substrate binding of tripeptidyl-peptidase II. Biochimica et Biophysica Acta, 1784(12):1899-1907 III. Eklund, S.; Lindås, A-C.; Hamnevik, E.; Widersten, M.; Tomkinson, B. Inter-species variation in the pH dependence of tripeptidylpeptidase II. Manuscript IV. Eklund, S.; Kalbacher, H.; Tomkinson, B. Characterization of the endopeptidase activity of tripeptidyl-peptidase II. Manuscript Paper I and II were published under maiden name (Eriksson). Reprints were made with permission from the respective publishers. Contents Introduction ..................................................................................................... 9 Enzymes ..................................................................................................... 9 Enzymes and pH dependence .............................................................. 11 Peptidases ................................................................................................. 12 Serine peptidases ................................................................................. 14 Intracellular protein degradation .............................................................. 15 The ubiquitin-proteasome system ........................................................ 15 Cytosolic peptide degradation ............................................................. 17 MHC class I antigen presentation ........................................................ 18 What is TPP II? ........................................................................................ 18 TPP II forms a gigantic complex ......................................................... 18 TPP II has a widespread distribution ................................................... 19 Why work with TPP II? ........................................................................... 20 TPP II is implicated in many cellular processes .................................. 20 TPP II has different substrate specificities .......................................... 21 TPP II is a potential drug target ........................................................... 22 Present investigation ..................................................................................... 23 What defines the primary structure of TPP II? (Paper I).......................... 23 Why does TPP II release tripeptides? (Paper II) ...................................... 25 Purification of TPP II (Paper II and III) ................................................... 27 How does the kinetic behaviour of TPP II vary with pH? (Paper III) ...... 29 What is the substrate specificity of the endopeptidase activity of TPP II? (Paper IV) ................................................................................................. 30 Conclusions ................................................................................................... 32 Future perspectives ....................................................................................... 33 Sammanfattning på svenska .......................................................................... 34 Acknowledgements ....................................................................................... 37 References ..................................................................................................... 39 Abbreviations 3T3-L1 Fibroblast cell line with capability to differentiate into adipocytes AAA-pNA Alanyl-alanyl-alanyl-paranitroanilide AAF-pNA Alanyl-alanyl-phenylalanyl-paranitroanilide BRCT Breast cancer type 1 susceptibility protein C-terminal CD8+ cytotoxic T-lymphocytes EL4 Mouse lymphoma cell line ER Endoplasmatic reticulum HEK293 Human embryonal kidney cells IMAC Immobilized metal ion affinity chromatography MHC Major histocompatibility complex Tpp2 the gene encoding tripeptidyl-peptidase II TPP II Tripeptidyl-peptidase II, species is indicated with lower-case letter, i.e. mTPP II, murine TPP II, hTPP II, human TPP II, dTPP II, TPP II from Drosophila melanogaster Introduction Proteins are some of the most diverse components of life. Consider the estimated number of species on earth, five million. In every genome, there are perhaps ten thousand genes encoding at least one protein. Splicing variants and posttranslational modifications add to the variety, leaving the total number of unique proteins somewhere in the vicinity of 1011. This might give the impression that we all consist of a chaotic soup of proteins. On the contrary, there is a high degree of organization in living organisms. The rate of production of each protein is fine-tuned from the gene level, where transcription is tightly controlled, to the likewise precise mRNA maturation and translation levels. Once formed the proteins are affected by a plethora of factors such as covalent modifications, cofactors and effectors, which can all determine the function of proteins and the activity of enzymes. The delicate balance between synthesis and degradation determines the concentration of each protein, in turn deciding the fate of the cell. Ultimately, proteins are degraded by a category of enzymes known as peptidases, an event that also can be highly selective. This thesis concerns one such peptidase, tripeptidylpeptidase II (TPP II), and how structure relates to function in this enzyme. Enzymes Enzymes are proteins that catalyse chemical reactions, i.e. increase the rates of transformation of one molecule into another, thereby directing the flow of metabolites in cells. Catalysis decreases the activation energy, Ea, of the reaction as depicted in Fig. 1. This can be achieved through stabilization of the transition state, as is apparent for triose phosphate isomerase (1), or by offering a different reaction route, e.g. as for pyridoxal phosphate dependent transamidases (2). 9 Ea Substrate Product Figure 1. Energy diagram for a simple reaction, the same reaction with lowered activation energy (Ea), and divided in two steps. For bimolecular or higher order reactions, i.e. reactions involving two or more reactants, one of the main contributions of the enzyme might be binding the reactants in a favourable manner. This increases the local concentration of reactants and is sometimes referred to as the proximity effect (3). The standard amino acid residues allow acid-base, covalent, nucleophilic and electrophilic catalysis to occur, although sometimes non-standard amino acids are needed, e.g. for glutathione peroxidase, where a selenocysteine plays an important role in catalysis (4). For catalysis of redox reactions, cofactors such as heme groups and NADH are often utilized (5, 6). k1 E+S k-1 ES k2 E+P Scheme 1 Enzymology, the study of enzyme catalysis, started in the late 19th century, when scientist began to analyse the rate of alcohol fermentation in yeast (7). It was established in the early 20th century that enzymes had hyperbolic substrate dependence, as illustrated in Fig. 2. For the simplest enzyme catalysed reaction, i.e. Scheme 1, where the enzyme (E) binds substrate (S) and releases the product (P), this behaviour can be described by the MichaelisMenten equation, developed by Leonor Michaelis and Maud Menten (8), [1]: , 10 [1] 0 5 10 v0 20 30 where KM, the Michaelis constant, is (k-1+k2)/k1. In a reaction where k2 is much smaller than k-1, KM represents the dissociation constant, KD, of the enzyme/substrate complex. For more complicated reactions, the Michaelis constant becomes correspondingly more complex, e.g. for epoxide hydrolase (9). Despite this, KM is usually taken to correspond to the affinity of the enzyme for the substrate, with a lower KM signifying a higher affinity. The rate at which an enzyme saturated with substrate can catalyse a reaction equals kcat, also named turnover number, which is the number of catalytic events (or turnovers) per unit time. In the example above, where k2 <<k-1, kcat is very close to k2 although, as for KM, increasing complexities in reactions pathways results in increased complexities in the expression for kcat. Since enzymes are seldom saturated with substrate in a cellular environment, kcat is rarely reached and might thus be viewed as a rather poor measure of the real efficacy of the enzyme. Instead, the catalytic efficiency kcat/KM is often used, as it describes how fast the rate of the catalysed reaction increases with increased substrate concentration at substrate concentrations below KM. 0 200 400 600 800 [Substrate] Figure 2. The dependence of the initial rate of enzyme catalyzed reactions on substrate concentration is usually hyperbolic. The figure is based on data from Paper III. Enzymes and pH dependence Physiological conditions are usually assumed to have a pH around 7.5, and it is hardly surprising to find that most cytosolic enzymes have their highest activity close to this pH. However, in certain situations it can be more favourable for the cell if enzymes have lower pH optima, e.g. the degradation enzymes of the acidic lysosome, which would wreak havoc in the cell if they were released into the cytosol. Many lysosomal enzymes have a pH opti11 mum around 5, which corresponds to the pH in the lysosome, but renders them far from efficient in the more neutral cytosol. Furthermore, pH dependency studies have been utilized in enzymology to characterize reactive groups. It has also been used to probe the microenvironment in the active site, investigating effects from distant residues (10). In this way, scientists have been able to probe the activities of enzymes with macroscopic detection techniques such as spectroscopy and fluorescence. In this work, pH dependency studies were conducted to investigate variations in substrate binding and rate of catalysis between enzymes from different species as well as between different substrates. Peptidases Peptidases are enzymes that catalyze the degradation of proteins by hydrolyzing peptide bonds. At neutral pH peptide bonds are kinetically very stable, with a t½ of at least 2 years (11). The reason for this stability is in part explained by the double bond nature of the peptide bond, where the electrons of the carbonyl bond are partially translocated to the amide bond by the electronegative nitrogen. This results in a sterical hindrance, as the planar amide bond is less accessible to water. The hydrolysis of peptide bonds proceeds through a tetrahedral intermediate in all peptidases studied (Fig. 3), but the nucleophile conducting the initial attack on the carbonyl carbon varies. Oxyanion hole + Tetrahedral intermediate Acyl enzyme Figure 3. Reaction scheme for hydrolysis of a peptide bond by a serine peptidase, showing the tetrahedral intermediate, the oxyanion hole and the formation and hydrolysis of the acyl enzyme. 12 The nomenclature of peptide binding and hydrolysis is illustrated in Fig. 4. The cleaved peptide bond is called the scissile bond, and the amino acid sidechains toward the N-terminus are called P1, P2, P3 etc., while the sidechains towards the C-terminus are named P1’, P2’, P3’ etc. The corresponding binding pockets in the enzyme are called S1, S2, S3 and S1’, S2’ and S3’, respectively (12). S3 S1 S 2’ P3 P1 P2’ + - P2 P1’ P3’ S 1‘ S 3’ S2 Figure 4. The nomenclature of substrate binding by peptidases. The scissile bond is indicated by an arrow, and the residues and binding pockets are marked according to (12). Peptidases are classified according to the nucleophile making the initial attack, or the residue or atom involved in activating water to act as a nucleophile, into four major groups. The most common type of peptidases are serine peptidases, which utilize a serine residue as the nucleophile, resulting in an acyl enzyme (Fig. 3). The serine residue is usually part of a catalytic triad, as discussed below (13). Serine peptidases have many important functions in the body, such as in the blood clotting cascade and in the digestive tract (14, 15). Like serine peptidases, cysteine peptidases also form a covalent bond with the substrate as part of the reaction mechanism, this time involving a cysteine residue. The active site of cysteine peptidases encompasses a cysteine and a histidine residue, which fill essentially the same role as the catalytic triad in serine peptidases (16). Caspases are examples of cysteine peptidases that are involved in regulation and execution of apoptosis (17) Metallopeptidases rely on a metal ion tightly bound in the active site, often zinc, which acts as a Lewis acid and activates a water molecule, and thereby can attack the scissile bond (18). Thus, no covalent intermediate with the enzyme is formed. In humans, a large family of metallopeptidases are responsible for degrading the extracellular matrix. The fourth group, aspartate peptidases, are active at acidic pH, due to the two aspartate residues in the active site (19, 20). Many aspartic peptidases are lysosomal, although some also occur in the stomach fluids (21). 13 Other variants of peptidases exist, although they are rare. One exception of note is the proteasome, which has an active site containing an N-terminal threonine residue. This peptidase will be further discussed below. In addition to this classification of peptidases by catalytic mechanism, they are also categorised by substrate cleavage mode. Exopeptidases cleave at a specific distance from either the free N- or C-terminus of the substrate, usually one, but sometimes as much as nine amino acid residues from the terminus (22). Aminopeptidases are exopeptidases dependent on a free Nterminus, while carboxypeptidases are dependent on a free C-terminus. Exopeptidases rely on an interaction with the terminus that contributes a large portion of the energy for substrate binding (Paper II). In contrast, endopeptidases do not have a specified distance to either terminus, but generally rely on binding energy from a longer stretch of substrate (12). It has been noted, however, that exopeptidases may have an endopeptidase activity, although with lower activity due to inefficient substrate binding (23-25, Paper IV). Serine peptidases The two main clans of serine peptidases are trypsin and subtilisin (26). These have a similar catalytic triad, composed of an aspartate, histidine and serine, positioned in the same orientation in the 3D structures (27, 28). However, the catalytic residues do not occur in the same order in the peptide chain, and there are few other similarities in the amino acid sequence or threedimensional structure. It has therefore been concluded that the two clans have evolved independently through convergent evolution (29). The function of the serine residue, as discussed above, is to perform the nucleophilic attack on the carbonyl carbon of the substrate scissile bond. The role of the histidine is to deprotonate the serine residue, and protonate the amine leaving group (Fig. 3). The role of the aspartate has been disputed; either it accepts a proton from the histidine via a low-barrier hydrogen bond (30), or simply positions the histidine for interaction with the catalytic serine and the tetrahedral intermediate (31, 32). However, even though the catalytic triad increases reaction rates six orders of magnitude, the total rate enhancement is 3-4 orders of magnitude greater (11, 33). The majority of this enhancement is thought to stem from transition state stabilization by the oxyanion hole (Fig. 3). Although there are other clans with a Ser-His-Asp-triad, such as the prolyl oligopeptidases (34), not all serine peptidases have this active site composition. For example, some viral proteases have a Ser-His-His triad (35), and the sedolisins, or carboxy serine peptidases, utilize a Ser-Glu-Asp triad (36). One example of the latter is tripeptidyl-peptidase I, a lysosomal peptidase with exo- and endopeptidase activity (25). Furthermore, a bacterial family of peptidases related to -lactamases have a Ser-Lys dyad, and some 14 serine peptidases even have the N-terminus positioned so that it becomes part of the active site (37). Subtilisin-like serine peptidases The clan of subtilisin-like serine peptidases, or subtilases, have been grouped into six families (38). The subtilisin, thermitase, proteinase K and lantibiotic peptidase families are only found in microorganisms, while the kexin family serve as proprotein convertases in eukaryotes. The pyrolysin family is more diverse, both in species occurrence and in sequence conservation. All members of the pyrolysin family have a C-terminal extension, but this extension does not always show convincing sequence conservation, which might question this grouping. The majority of subtilases are excreted endopeptidases, and the most thoroughly studied enzyme of this clan, subtilisin BPN’, has an extensive substrate binding region with eight binding pockets (39). The oxyanion hole is formed by a conserved asparagine residue and the peptide backbone of a serine residue (40). Intracellular protein degradation Protein synthesis is one of the most energy-consuming pathways in anabolism, corresponding to 25-30% of the cellular oxygen consumption in mammals (41). Uncontrolled degradation is likely to be detrimental to the organism. Even so, up to 30% of the ribosomal products are defective and need to be degraded (42). Cellular processes such as mitosis and apoptosis are tightly regulated by expression of many proteins, which need to be degraded at the correct instance. Hence, the degradation of the proteins inside a cell is a very selective process. There are two major pathways for protein degradation in the cell, the lysosome and the ubiquitin-proteasome system. Proteins are taken up by the lysosome either by macro- or microautophagy, where a part of the cytosol is completely surrounded by membrane and devoured (43). This self-eating process increases in starvation. Alternatively, proteins might be specifically targeted for degradation in the lysosome by chaperonemediated autophagy (44). The ubiquitin-proteasome system targets proteins for degradation in the cytosol or nucleus by polyubiquitylation , which results in peptides that are either processed to free amino acids, or presented by MHC class I (45). Degradation of proteins by the ubiquitin-proteasome system is much more selective, as described below. The ubiquitin-proteasome system Ubiquitin (Ub) is a small protein utilized in many processes in the cell, including signal transduction and endocytosis (46,47). The ubiquitylation is 15 initialized by the activation of Ub by E1, which forms a thioester bond with the C-terminus of Ub (48). The thioester bond is transferred to the active-site cysteine of an E2 protein, of which there are approximately forty in mammals. Selectivity is achieved when one of the thousand E3 proteins recognizes a misfolded, malfunctioning or superfluous protein and promotes the attachment of the Ub molecule on a -NH2-group of a lysine residue in the target (49). Elongation of the Ub-chain through E4 elongase leads to polyubiquitylation, usually via the Lys-48 of the Ub moieties, which targets the selected protein for degradation in the proteasome (50), (Fig. 5). The 26S proteasome is a 2.5 MDa complex consisting of the 20S core particle, which confers the proteolytic activity, and usually two 19S regulatory particles. Core particles consists of four rings with seven subunits each, 1-7 or 1-7 (51). The -subunits are positioned in the middle of the cylinder, whereas the -subunits form the top and bottom, blocking entry into the interior of the core particle prior to association with the regulatory particle (52). Regulatory particles consist of at least 19 subunits, 6 of which are ATPases, whose combined function is to recruit, deubiquitylate and unfold polyubiquitylated proteins in an ATP-dependent manner (53). Unfolded proteins are passed into the catalytic core particle, where degradation commences at the active sites in the 1, 2 and 5 subunits (54, 55). Specificity differs between the three active sites, conferring preference for aromatic, acidic or basic amino acids in the P1 position (56). Once the peptide products are small enough to exit, they diffuse out of the core particle, resulting in products 225 amino acids in length (57), (Fig. 5). 16 E2 E1 E3 E3 E2 Ub Target Ub E1 Target Ub Ub Target Ub Target 19S E4 20S Target Ub 19S Ub TOP LAP BH TPP II Ub Ub Ub Free amino acids Figure 5. Protein degradation by the ubiquitin-proteasome system and cytosolic peptidases. Ubiquitin is activated by E1 and transferred to an E2 protein. The protein to be degraded (target) is recruited by an E3 protein, and this in turn promotes the attachment of ubiquitin to the target protein. Following chain elongation by E4, the target protein is recognized by the proteasome, deubiquitylated and degraded into peptides. These are further degraded by peptidases such as thimet oligopeptidase (TOP), tripeptidyl-peptidase II (TPP II), leucine aminopeptidase (LAP) and bleomycin hydrolase (BH). Cytosolic peptide degradation Proteasomal degradation products are further degraded by a diverse array of peptidases in the cytosol (Fig. 5). Products of 9-17 amino acids can be degraded by endopeptidases such as the metallopeptidase thimet oligopeptidase (TOP) into peptides of 6-9 amino acids (58). The cysteine peptidase bleomycin hydrolase (BH) can process peptides of up to 42 amino acids endopeptidolytically as well as by amino- and carboxypeptidase activities (59, 60). TPP II, a cytosolic peptidase whose main activity is the removal of tripeptides from the N-terminus of peptides, has been shown to be required for degradation of some peptides that are more than 15 amino acids (61). Other aminopeptidases such as the metallopeptidases puromycin-sensitive aminopeptidase and leucine aminopeptidase (LAP) can conclude the process by removing single amino acids (62, 63). The end products of cytosolic peptide degradation are for the vast majority of peptides free amino acids that are 17 reused in protein synthesis, although some longer peptides are exported into the ER lumen for presentation by the MHC class I, see below. MHC class I antigen presentation One of the major immunological defence systems in the body is the surveillance of cells by CD8+ cytotoxic T-lymphocytes (CTL). The CTLs recognize antigens derived from intracellular proteins displayed on the cell surface by major histocompatibility complex I. Peptides of 8-11 amino acids are either imported directly from the cytosol, or generated in the ER by further trimming of products from the cytosolic protein degradation by the proteasome or subsequent peptidases. These peptides are assembled onto MHC Class I molecules by the multimeric peptide loading complex, and the MHC Class Iantigen complex is transported to the cell surface via the Golgi apparatus (64). By this mechanism, the immune system can recognize cells containing viral or microbial proteins, as well as cells displaying altered protein composition, such as in tumour malignancies (65). What is TPP II? When first reported, TPP II was found in rat liver, but has since been established as a more or less ubiquitously expressed cytosolic peptidase (66-68, Paper I). As the name indicate, the main activity results in the release of tripeptides from the free N-terminus of longer peptides, i.e. an aminopeptidase activity. In addition to this activity, an endopeptidase activity has also been reported (69). Although this activity is three orders of magnitude lower than the exopeptidase activity, for some substrates it is as efficient as the proteasome. The catalytic domain of TPP II is subtilisin-like, with an almost 200 amino acid residue insert between the catalytic aspartate and histidine (66). In addition, following the subtilase domain is an approximately 700residue extension, leaving the total peptide chain length at 1249 amino acids in mammals. TPP II forms a gigantic complex The TPP II monomer is difficult to study in isolation, as dimers are formed very rapidly (70). Dimers stack into long strings, which twist around each other in pairs to form a large complex with a total of 16-20 dimers (Fig. 6). This complex has a molecular mass of 4-6 MDa, depending on species, and is approximately 50 nm in length (70, 71). To put this into perspective, most proteins weigh less than 100 kDa and measure no more than a few nm. 18 Figure 6.The EM-structure of the dTPP II complex (EMD 1732, (72)). The complex consists of two strands of ten dimers each, joined together at the end dimers to form a 6 MDa particle. The crystal structure of the TPP II dimer from Drosophila melanogaster was recently solved (72). Unfortunately, the enzyme is crystallized in an inactive form, having a misplaced loop in the active site and a distorted catalytic triad resulting from a partial unwinding of the helix containing the serine residue. This might be the reason for the severely decreased activity, approximately one tenth, in dimers when dissociated compared to when in the complex (73). The reversible dissociation and association of the complex has been proposed to be a regulation mechanism, albeit a rather slow regulation (71, 73). Regardless if complex formation can be controlled by the cell, the low activity of the dimers probably serves to protect the cell from unhindered proteolysis, as the active site is rather well exposed in the dimer crystals. In the complex, however, the active site must be reached through a chamber system, which would effectively hinder any native proteins from reaching the active site (72). TPP II has a widespread distribution Since first discovered in rat liver, TPP II activity has been reported in various tissues in other mammals, chicken, fruit fly, plant and yeast (67, 74-76, Paper I). In fact, TPP II seems to have a ubiquitous distribution in animals and plants, which promotes the idea that TPP II has a housekeeping function 19 in the cell. Despite this, removal of the TPP II in Arabidopsis thaliana or Schizosaccharomyces pombe did not result in any physiological abnormalities (74, Paper I). In contrast, removal of the TPP2 gene in mice has a more drastic effect. One group reported a failure to generate TPP2-/- mice due to embryonic lethality in day 9.5 (77). Another group successfully generated TPP II deficient mice, although these mice had decreased life spans and several defects in the immune system (78). These findings are discussed in more detail below. Why work with TPP II? There are many reasons for studying the action of a specific enzyme. The knowledge might be used to explain the physiological role of the enzyme, or to elucidate the mechanism of binding and catalysis displayed by that enzyme. This knowledge is essential for drug discovery efforts, since mechanistic data is utilized to a large extent in modern drug design (79). For TPP II, all these apply; there are numerous reports on the involvement of TPP II in different physiological processes, but the exact role of TPP II is not known. Furthermore, the mechanism of substrate interaction is not fully understood, and TPP II could be a drug target for more than one disease. TPP II is implicated in many cellular processes The physiological function of TPP II has so far been proposed to be degradation of peptides released by the proteasome into tripeptides that can be efficiently degraded by aminopeptidases into free amino acids (66) (Fig. 5). However, this housekeeping function does not seem to be consistent with the involvement of TPP II in processes such as apoptosis and cancer, as described below. Since no specific substrate for TPP II has been identified, and the structure excludes the possibility of degradation of intact proteins, the exact role of TPP II in these processes remains to be determined. It is likely that a decrease in TPP II levels will result in an increased concentration of peptides in the cell, in conjunction with decreased rates of release of amino acids. This might result in secondary effects, such as decreased rates of protein production or disruption of protein-protein interactions (80, 81). TPP II in apoptosis The first indication that TPP II was involved in apoptosis control was a report that an activity inhibited by AAF-CMK promoted apoptosis induced by Shigella flexneri invasion, ATP and staurosporine (82). In contrast to this find, overexpression of TPP II in Burkitt’s lymphoma cells was found to impart protection from apoptosis (83). In combination with decreased proteasomal activity, increased TPP II activity developed during tumour growth 20 in lymphoma and melanoma cells, which was interpreted as an adaptation to avoid apoptosis (84). In addition to these findings, overexpression of TPP II in HEK293-cells has been found to increase growth rate and result in chromosomal aberrations and centrosome abnormalities (85). Knockdown of TPP II by siRNA led to decreased growth rates and polynucleated cells (85). Purportedly, TPP II allows cells to avoid apoptosis despite activated mitotic check points (86). It has been reported that TPP II translocates into the nucleus as part of the irradiation response (87, 88), although this is disputed (89). However, TPP II depletion in mice results in activation of apoptosis in T-cells, and premature senescence in fibroblasts, in conjunction with upregulation of p53 (78). Although the precise mechanism behind this is unclear, it is apparent that TPP II contributes to the survival of cells that would otherwise undergo apoptosis. TPP II in MHC class I antigen processing Proteasomal products are often trimmed before loading onto the MHC Class I complex. The role of TPP II in this process has been investigated in several studies. For example, the HIV Nef74-82 epitope has been reported to be generated by endopeptidolytic cleavage by TPP II (90). Other epitopes have also been reported to be dependent on TPP II (91, 92). For several epitopes, however, no involvement of TPP II could be observed (93-96), and sometimes a destructive role was noted (97). In general, TPP II seems to be important for processing longer proteasomal degradation products (14-17 amino acids and more), but not shorter ones (61, 98-100). TPP II has different substrate specificities It is not known which of the exo-and endopeptidase activities of TPP II that is important for the physiological role of TPP II in different situations. The exopeptidase activity is quite well-charactarized, whereas the endopeptidase activity has been less studied. In the exopeptidase activity, the N-terminal amino group is bound and a preference for aliphatic or aromatic amino acids in the P1 position has been noted (75, Paper II). The endopeptidase activity, although much slower, has been reported to favour basic amino acids in the P1 position (69). This apparent discrepancy raises questions about the substrate binding ability of TPP II, and whether the binding sites differ between the two activities. For example, the absence of a binding interaction with the N-terminal amino group of the substrate might open a possible binding interaction in a different portion of the active site. Alternatively, the active site for the endopeptidase activity might be located at a completely different part of the enzyme. However, no homology with any known peptidases has been found elsewhere, which renders this possibility unlikely. Nevertheless, studying the binding interactions for both the exo-and endopeptidase activi21 ties can give valuable information regarding the architecture of the active site, and thus be of aid in the development of specific inhibitors of TPP II. TPP II is a potential drug target It has been reported that a membrane-bound form of TPP II has the ability to degrade the neuropeptide cholecystokinin-8 (101). Since this peptide aids in transmission of satiety signals, it was hypothesised that inhibiting TPP II would decrease hunger or overeating. Indeed, injection of the specific TPP II inhibitor butabindide reduced food intake in mice (101). Since butabindide is unsuitable as a drug candidate for obesity because of its low stability in blood plasma, attempts have been made at producing more promising molecules, although none has reached clinical trials as of yet (102-104). There have been several attempts at producing cells and organisms with reduced concentrations of or completely depleted of TPP II (74, 77, 78, 98). One group has reported homozygos Tpp2-/- mice that were embryonic lethal (77). Heterozygotes, however, had a compromised adipogenesis, a phenomenon also observed in the worm Caernohabditis elegans (77). The decreased adipogenesis could not be linked to decreased food intake, and thus not to decreased degradation of cholecystokinin-8. Furthermore, in 3T3-L1 cells treated with RNAi against TPP II, decreased adipogenesis was also observed, and this could be reversed by expression of a part of the Cterminus, indicating that the function of TPP II in this context was independent of enzymatic activity (77). This curious report again links TPP II to obesity, although apparently by a completely different mechanism. Since TPP II has been shown to have an anti-apoptotic role in many different cancer cell types, it could be considered a drug target for cancer therapy. One group has reported that Z-GLA, a tripeptide with a blocked Nterminus, is a highly potent TPP II inhibitor (87). In combination with irradiation treatments, this molecule inhibited tumour growth. However, other groups have since failed to confirm the TPP II-inhibiting capabilities of ZGLA (78). This leaves the question whether TPP II would be a good drug target for cancer therapy open for further investigation. 22 Present investigation May you live in interesting times – T. Pratchett The overall aim of the present investigation was to gain insight into the function of TPP II, both on an enzymatic and a physiological level. With a vision to one day fully understand the cellular function of TPP II, and to provide knowledge for potential future drug development, these are the questions addressed in this thesis: What defines the primary structure of TPP II? Why does TPP II release tripeptides? How does the kinetic behaviour of TPP II vary with pH? What is the substrate specificity of the endopeptidase activity of TPP II? By answering these questions, I hope to make a contribution to the understanding of this intriguing enzyme. I have personally been fascinated by the dual exo/endopeptidase activity of TPP II, and how the substrate specificity seems to differ between these. It has been very interesting to first investigate one, then the other, trying to piece together how the active site might interact with the substrate in the different situations, and what importance this might have in the cellular environment. What defines the primary structure of TPP II? (Paper I) Tripeptidyl-peptidase II is a very large enzyme, not only with respect to the quaternary structure, but also the peptide chain itself is quite extensive, with approximately 1250 amino acid residues in mammals, and even longer in some species, such as insects (76). The catalytic subtilisin-like domain only constitutes 300-400 amino acids, and the rest of the sequence has little or no similarity to any known protein. It was previously known that the insert between the catalytic aspartate and histidine, henceforth referred to as the DHinsert, had an important role in complex formation (105). In addition, part of the C-terminal extension (amino acids 520 and onward) had been reported to affect such varying functions as fat metabolism and nuclear translocation 23 (77, 87). Hence, it appeared important to investigate sequence similarities, to see if there were any highly conserved regions that might have functional importance. Eleven regions of high evolutionary conservation were identified in a set of 16 sequences selected from 58 TPP II sequence homologues found during data harvesting. From these regions, signatures were created that were able to separate TPP II homologues from other subtilisin-like serine peptidases, such as pyrolysin (Paper I). Pyrolysin also contains a DH-insert and a Cterminal extension but does not have any of the functional traits that characterize TPP II (38). The signatures were very selective, and all TPP II homologues detected so far matched at least two signatures. Thus, these signatures can be used as a tool for identifying TPP II homologues, which might yield more accurate annotations in the future. Seven of the signatures covered regions of previously noted functional importance, such as the catalytic aspartate and histidine residues, as well as part of the BRCT domain reported to be important for nuclear translocation (87). Since the remaining four also contains amino acid residues that are conserved in TPP II homologues from all eukaryotic species, it was suspected that they also have a vital role in this enzyme (Paper I). Indeed, when the crystal structure of TPP II from Drosophila melanogaster was published (72), it could be noted that two of these conserved regions of unknown function were positioned in the interface between the catalytic domain and a C-terminal domain. Two highly conserved tryptophan residues from two different regions were in close proximity. Further investigations will be needed to elucidate whether these two residues, or other highly conserved residues, play an as yet unknown role in the structure or function of TPP II. TPP II homologues from human, mouse, rat, fruit fly and arabidopsis all have a very large molecular size and degrades peptides into tripeptides (71, 74, 76, 106). In addition, a peptidase sharing these characteristics was purified from the yeast Schizosacharomyces pombe, although this was not believed at the time to be a sequence homologue (107). However, we were able to prove that a genetically engineered S. pombe without the gene encoding a TPP II homologue did not contain the tripeptidyl-peptidase-like activity found in the unmodified yeast (Paper I). This demonstrated that there are functional TPP II sequence homologues in the three main kingdoms of eukaryotes, and thus we made the assumption that any sequence homologues found within this domain also shared a common function. During the search for TPP II sequence homologues, it was evident that all sequenced genomes from animals and plants contained at least one copy of a gene encoding TPP II. In fungi, only some genomes contained a homologue, while most protozoan did (Paper I). In our database search, we also came across an enzyme from the bacteria Blastopirellula marina annotated as a pyrolysin, which had very high sequence similarity to TPP II. Upon a phy24 B P de n dr ob in adi fe t B sta is vi m ns gn ar yi in C a“ K M r ex e py br A in in ro e ha Su vi tha (S Fu P ly r co li bt S yr d sin ce rin tii lli an re (H ilisi tro oly ” a s p s vi n in ic sia sa ( a B ( e) pie lic “T P fu ns P h ) en P I rio su ifo I” s) rm is) logenetic analysis, this enzyme was grouped with TPP II homologues from plants and so it was hypothesised that it was the result of a horizontal gene transfer (Fig. 7). Furthermore, another bacterial protein, from Salinospora tropica, annotated as a TPP II homologue was grouped together with pyrolysin. In both cases, the signatures developed in this work gave more accurate annotations. Further investigation revealed that the protein from B.marina did not appear to have any tripeptidyl-peptidase activity, which might be guessed from the lack of a conserved glutamate residue important for exopeptidase substrate binding (Paper II). It appears that since the TPP II-like activity did not confer any fitness advantage in these bacteria, it has slowly lost that function. H sa pi en s C sa ae ryz o R be om p S sa o b glo s i M s yd e ga n C el is ma U ns ecte N v is e nn itrip ster v N oga lan e iae Dm mb a g A Eukaryotes = Green Bacteria = Red Archea = Blue Figure 7. Neighbour joining was used to perform a phylogenetic analysis on TPP II homologues as well as other subtilases (cf. Paper I). Eukaryotic proteins are marked in green, bacterial in red and archeal in blue. For TPP II homologues, the name of the species in which the gene was found is given whereas for other subtilases, the name of the enzyme is given with the species in parenthesis. Why does TPP II release tripeptides? (Paper II) It was evident at an early stage that TPP II was dependent on a free Nterminus in the peptide substrate in order to bind and affect hydrolysis (68, 75). Thus, it was hypothesised that the positively charged N-terminal amino group interacted with one or more negatively charged amino acid residues, 25 forming a molecular ruler for “measuring” the substrate. It was found from sequence comparison that three acidic amino acid residues positioned in or around the potential S3 pocket were conserved throughout the known TPP II homologues at the time (Paper II). To see the possible spatial arrangement of these residues, a homology model was built based on the closest structure found at the time, subtilisin BPN’. Two of the residues, Glu-305 and Glu331, were found to be positioned favourable for interaction with substrate so as to provide a molecular ruler for the enzyme (Fig. 8). Glu-331 Glu-305 Ser-449 Asp-44 His-264 Figure 8. The active site of a homology model of mTPP II, with the catalytic triad and Glu-305 and Glu-331 shown as ball and stick, and the backbones of the S1 pocket-forming residues magenta. This model was constructed based on the crystal structure of dTPP II and subtilisin BPN’ as described in Paper III. To test if these glutamates indeed interacted with the N-terminus of substrates, mutated variants of murine TPP II (mTPP II) with the respective glutamate exchanged for glutamine or lysine were expressed in Pichia pastoris and purified by IMAC (Paper II). Only one mutant variant, E331Q, had sufficiently high enzymatic activity to allow kinetic parameters to be determined. It was found that KM was increased at least 100-fold in this mutant compared to the wild-type enzyme, suggesting a quite significant decrease in binding affinity for the substrate used. Furthermore, the inhibition of the E331Q mutant by an octapeptide inhibitor was essentially unaffected by the removal of the N-terminal amino group of this peptide. In contrast, for the wild-type mTPP II, this change caused a 1000-fold increase in IC50. The specific TPP II inhibitor butabindide, which was developed to bind the S3 pocket of TPP II, also had a drastically increased IC50. For the Glu-305 mutants, however, little or no activity could be detected, which might indicate a drastic change in the structure of the active site (Paper II). Indeed, at least for the E305Q mutant, a decrease in size was observed, which could reflect difficulties in complex formation. However, since the dissociated enzyme has 26 one tenth of the activity of the complex (73), this could not be the sole cause of the low activity in this mutant variant. When the crystal structure for dTPP II was solved, the glutamates corresponding to Glu-305 and Glu-331, i.e. Glu-312 and Glu-343, were found in a similar position (72). The glutamate corresponding to Glu-305 was found to form a hydrogen bond with a tyrosine residue in the C-terminal part of the enzyme, which might be the reason for the complete loss of activity when this residue was mutated in mTPP II. Mutant variants of dTPP II with glutamine instead of glutamate in these two positions could form the complex but were shown to have severely decreased activity, at least in part dependent on an increase in KM (72). This confirms our theory that a molecular ruler exists in TPP II, consisting of at least one glutamate. Other peptidases have also been reported to have a similar mechanism of substrate binding, such as prolyl tripeptidylpeptidase, dipeptidyl peptidase IV and ERAP1 (22, 23, 108). In conclusion, it is now evident that the substrate binding mechanism of mTPP II involves an interaction with Glu-331. This interaction positions the peptide substrate such that the third peptide bond is cleaved, and thus it constitutes a molecular ruler, ensuring that the product released is a tripeptide. Purification of TPP II (Paper II and III) In order to study the kinetics of the exo-and endopeptidase activities of TPP II, a new expression and purification system needed to be developed. The system used for the glutamate variants provided a source of TPP II where mutant variants could be expressed and purified without contaminations from endogenous, wild-type enzyme, which had been a problem when HEK293 cells were used (109). However, it did not produce enzyme of high enough purity to enable in-depth kinetic investigation of the exopeptidase activity, or any investigation at all into the endopeptidase activity. One of the obstacles was that the heterologously expressed, histidinetagged enzyme did not appear to bind to the IMAC column (Paper II). This resulted in low yield as well as low purification (Fig. 9). It was hypothesized that the N-terminally positioned histidine tag was embedded in the complex, thus unable to bind to the metal ions in the gel matrix. To improve binding, I therefore attempted to dissociate the complex with guanidine, urea and increased pH, respectively. While the first two treatments increased binding slightly, it was a very precarious balance not to denature the protein, since renaturation through dialysis was impossible. In addition, while an increase in pH to 8.5 did not destroy the enzyme, nor did it appear to increase binding to the IMAC column or even dissociate the complex. Following these results, IMAC was abandoned as a purification strategy. Since good results had been achieved using Escherichia coli as a host for dTPP II (70), expression of mTPP II was commenced in this organism. An 27 initial expression system was set up in BL21(DE3) cells using a pET vector. Subsequently, the first 200 base pairs were substituted for a version codon optimised for E. coli, and the bacterial strain was exchanged for Rosetta (DE3). However, the resulting expression was merely 5% of that achieved for dTPP II, and neither optimization of codons nor expression conditions increased expression yield more than marginally. It is possible that the mRNA structure of mammalian TPP II hinders rapid translation. Nevertheless, the E. coli system provided higher yield and easier handling conditions than the P. pastoris system previously used, and was thus utilized as the starting point for the new purification system. Unfortunately, the new expression system made binding to the anion exchange matrix difficult, since high nucleic acid content conferred high ionic strength as well as high density. To rectify this, a protocol based on polyethylene imide and ammonium sulfate precipitations were developed that allowed subsequent binding and purification. Two additional chromatography steps were added, hydrophobic interaction chromatography and gel filtration, resulting in 80% or higher purity (Paper III). In all, this system conferred a much higher yield and purity for wild-type mTPP II and mutants thereof than any previously published procedure (Fig. 9). Figure 9. Comparison between the two protocols used in this work, namely the Pichia pastoris expression system combined with IMAC (Paper II) and the E. coli expression system and three-step purification procedure described in Paper III. 28 How does the kinetic behaviour of TPP II vary with pH? (Paper III) To better understand the kinetic behaviour of TPP II, a pH dependence study was undertaken, comparing mTPP II, hTPP II and dTPP II. The results revealed a difference in the pH-optimum profiles of two substrates differing only in the P1 position, AAF-pNA or AAA-pNA. The suspotbstrate with alanyl in the P1 position had a much flatter pH profile without any clear optima. This effect was surmised to stem from differences in the kcatdependence on pH (Paper III). Throughout the investigation, it was obvious that the AAA-pNA substrate yielded both lower KM and kcat than AAF-pNA. This could be caused by non-productive binding, as the smaller substrate might have the possibility to bind in more than one conformation to the active site, although only one would be favourable for catalysis (Paper III). At a pH above 7.6 there was an increase in KM with pH for all enzyme/substrate combinations (Fig. 10). The increase in KM at higher pH could be due to deprotonation of the N-terminal amino group of the substrate. However, dTPP II had a minimum in KM for both substrates around pH 7-7.5. It was hypothesized that the S1 pocket in dTPP II was compressed by one or more protonated groups, as this would decrease the binding affinity at lower pH. 0.0 log KM 1.0 2.0 pH-dependence of KM 6.0 6.5 7.0 7.5 pH 8.0 8.5 Figure 10. The pH dependence of KM for mTPP II ( ) and dTPP II ( ) using AAFpNA as a substrate. Adapted from Paper III. 29 In a newly built homology model of mTPP II based on the crystal structure of dTPP II, it could be seen that the binding pocket for S1 was broad enough to allow a small residue like alanine to bind in a less defined manner than a bulkier one such as phenylalanine. It was hypothesised that His-267 (mTPP II numbering) in the vicinity of the catalytic triad could be responsible for the difference in pH-dependence between the two substrates. Furthermore, Asp-474 (dTPP II numbering), located behind the S1 pocket in dTPP II but not mTPP II or hTPP II was identified as a potential candidate responsible for the difference in pH-dependence in KM. In conclusion, the studies of pH-dependent kinetic parameters have revealed differences in catalysis and substrate binding both between species and between substrates. Finding the reasons behind these discrepancies would provide valuable information on the substrate binding region of TPP II. What is the substrate specificity of the endopeptidase activity of TPP II? (Paper IV) When the endopeptidase activity of TPP II was first discovered, it was proposed to favour basic residues in the P1 position of its substrates (69). That would separate it from the exopeptidase activity, which preferentially cleaves after aromatic or aliphatic amino acids (75). While it seemed plausible that the endopeptidase activity originated from the same active site as the exopeptidase, this did not explain the apparent discrepancy in substrate specificity. Since only seven cleavage sites in two different peptides had been found so far (69, 90), and little or no kinetic measurements had been carried out for the endopeptidase activity, we decided to make a more thorough examination of this activity. As a starting point, we used the peptide Nef69-87, which had been previously shown to be cleaved at two positions (90). Of these two positions, only one could be verified, and several previously unreported cleavage sites were found, both for the human and the murine enzymes (Paper IV). In addition to Nef69-87, the peptide LL37 was also used in the investigation, and was found to be cleaved at several positions, mostly after basic residues. Taken in total, the results suggest that the endopeptidase activity is less selective than previously reported (see Fig. 2 of Paper IV). The rate of hydrolysis of the endopeptidase substrates were, as previously reported, approximately four-five orders of magnitude lower than for the exopeptidase activity. This increase could be due to a much lower binding affinity, since the interaction with the N-terminal amino group reported in Paper II is most likely lost. Cleavage experiments using the E331Q mutant studied in Paper II could confirm this. 30 The specific TPP II inhibitor butabindide, which has a low-nanomolar KI when measuring the exopeptidase activity, was not nearly as efficient at inhibiting the endopeptidase activity (Paper IV). Together with the discrepancies in substrate specificity, this suggests a different binding mode of the substrate during endopeptidolytic cleavage. Butabindide was designed to bind to the same amino acid residues that would interact with the N-terminal amino group of the substrate during exopeptidolytic cleavage (101). This interaction was later confirmed by our group (Paper II). If, during endopeptidolytic cleavage, the substrate is bound in such a way as to not interact with these groups, it might evade this inhibition. In the homology model of mTPP II based both on the crystal structure of dTPP II and subtilisin, the substrate binding pocket is very broad, which explains the promiscuous substrate preferences, and possibly allows for an alternative substrate binding mode during the endopeptidase activity (Fig. 8). In conclusion, the endopeptidolytic substrate specificity does appear to be different from the exopeptidase specificity, possibly due to a different mode of binding. It also appears that the rate of endopeptidolytic hydrolysis is four-five orders of magnitude lower than for the exopeptidase, which questions the significance of this activity under normal physiological conditions. 31 Conclusions This work has provided several new insights into the structure and function of TPP II. While it was previously known that TPP II existed in a broad range of eukaryotic species, conserved sequence motifs have now been identified that can be used to find new homologues. The majority of these covered known regions of functional importance, and others have since been noted to participate in interactions between monomers. We hope that these signatures will facilitate more correct annotations of TPP II in the future. The substrate binding mechanism of the exopeptidase activity has been demonstrated to be dependent on Glu-331, a trait that is conserved between TPP II homologues. Thus, a molecular ruler exists in TPP II, a phenomenon that recurs in other peptidases cleaving peptides to a defined length. Differences in the pH dependence of catalysis and substrate binding have been revealed, both between species and between substrates. Further investigations into the reasons for these discrepancies will provide more detailed information regarding the active site configuration in TPP II. The substrate specificities seem to differ between the exo- and endopeptade activities, although the reason behind this could not be deduced in the current investigation. No specific physiological substrate for the endopeptidase activity has been proposed, and the catalytic rate with the peptides used in this investigation was very low. This argues against a role for the endopeptidase activity under normal physiological conditions. Variations in substrate binding dependent on pH, substrate, species or exo- or endopeptidase activity have been investigated in this work. The image created is one of a flexible binding cleft, which allows substrates to bind in modes incompatible with catalysis, or to ignore the molecular ruler mechanism described above. This type of investigations should provide valuable information for future drug development efforts, and facilitate the determination of the role of TPP II in physiological functions. 32 Future perspectives It is often the case that when a question is answered, several others appear as were they curious onlookers of a spectacle. The present work makes no exception, as a multitude of future research topics arose from the observations herein. For example, some of the evolutionarily conserved sequence motifs may provide excellent starting points for future investigations of the functions of TPP II. Mutagenesis studies of single amino acid residues, e.g. one or both of the conserved tryptophan residues would provide information regarding the importance of these residues on substrate binding, catalysis or complex formation. Since there have been reports of cellular functions being affected by non-catalytic parts of the enzyme, it should prove worthwhile to test the effects of these mutations in cell cultures a well. The substrate binding mechanism of the exopeptidase activity, although extensively studied, still holds some puzzles. Why is the binding of different substrates affected dissimilarly by pH in dTPP II, compared to mammalian TPP IIs? How can some substrates be bound in a non-productive manner? The study of mutant variants of TPP II should provide some answers to these questions, and thus provide further insights into the binding mechanism of TPP II. This, in turn, would provide valuable information for future drug discovery. The endopeptidase activity poses an even greater enigma: why is the substrate specificity different between this and the exopeptidase activity? To find an answer to this, the binding mode of endopeptidase substrates must be elucidated, perhaps through a combination of docking studies and assays with different peptide substrates. Furthermore, the kinetics of the endopeptidase activity is still virtually unexplored, and this would be a key element in the clarification of the physiological role of this activity, perhaps through endeavouring to create better FRET-substrates. 33 Sammanfattning på svenska Syftet med det här avhandlingsarbetet är att utforska struktur och funktion hos enzymet tripeptidylpeptidas II (TPP II), och hur samband mellan dessa ger TPP II dess speciella egenskaper. De flesta proteiner i cellen befinner sig i en jämvikt mellan nybildning och nedbrytning. Cytosolisk nedbrytning av proteiner är en specifik process, där proteiner märks med det lilla proteinet ubiquitin, vilket bildar långa kedjor som känns igen av 26S-proteasomen och bryts ned till peptider av olika längd (se Fig. 5). Peptiderna måste sedan brytas ned helt till sina beståndsdelar, aminosyror, så att de kan återanvändas i nya proteiner. För att åstadkomma detta finns en hel uppsjö av peptidaser, som antingen kan klyva peptiderna från ändan (exopeptidaser) eller mitt i (endopeptidaser). Exopeptidaser kan antingen klyva från N-terminalen av peptider (aminopeptidaser) eller från C-terminalen (karboxypeptidaser). TPP II är ett av de peptidaser som hjälper till att finfördela peptider, vilket den gör genom att klyva dem i tre aminosyror långa bitar. Dessa kan sedan snabbt sönderdelas till fria aminosyror av andra aminopeptidaser. TPP II är ett serinpeptidas, vilket betyder att den aktiva ytan har en katalytisk triad bestående av aspartat, histidin och serin. Uppbyggnaden av TPP II är väldigt speciell; peptidkedjan på 1249 aminosyror (hos däggdjur) veckas och bildar dimerer, som sedan bildar långa strängar. Två strängar vrids sedan om varandra och bildar ett komplex, vars totala molekylvikt uppgår till 4-6 MDa, se Fig. 6. Varför TPP II bildar ett så stort komplex är inte helt uppenbar, men det står klart att den aktiva ytan ligger undangömd i ett grottsystem, dit inga fullstora proteiner kan ta sig. Dimerer har en tiondel så hög aktivitet som det fullstora komplexet. Det är möjligt att denna komplexbindning har selekterats fram som ett sätt att skydda cellen från oreglerad proteinnedbrytning. TPP II förekommer hos alla djur och växter, och även i andra eukaryoter. Den uttrycks i många olika vävnader, varför man har antagit att den har en ”hushållningsfunktion” i att snabba på peptidnedbrytningen. Utöver denna funktion har TPP II även befunnits vara involverat i många andra av cellens funktioner, såsom apoptos, mitos och antigenprocessering, och fysiologiska processer som cancer, adipogenes och mättnadsrespons. Ännu vet man inte vilken mekanism som ger dessa effekter, och mer information kan behöva insamlas innan dess exakta funktion in vivo kan klarläggas. Eftersom TPP II visats vara involverat i dessa processer, anses enzymet vara ett potentiellt 34 mål för läkemedelsutveckling, något som också kräver detaljerade kunskaper om enzymets funktion och struktur. Det första delarbetet (Paper I) beskriver konstruktionen av sekvenssignaturer baserade på väl konserverade regioner i peptidsekvensen hos TPP IIhomologer från många olika arter. Dessa signaturer är tänkta som redskap för säkrare annotering av TPP II-homologer, men eftersom de är baserade på delar av proteinet som är i hög grad konserverade, är de också förmodligen regioner som har stor betydelse för struktur eller funktion. Av de elva signaturerna täckte sju redan kända funktionella eller strukturellt viktiga aminosyrarester, och av de övriga befinner sig två i gränsytan mellan aktiva domänen och C-terminalen, något som kan vara strukturellt viktigt. Genom försök i fissionsjäst, Schizosaccharomyces pombe, kunde vi visa att det enzym som tidigare visats vara en funktionell homolog till TPP II också är en sekvenshomolog, vilket innebär att funktionella sekvenshomologer nu dokumenterats i både djur, växter och svamp. Under sökandet efter TPP II-homologer hittade vi även ett bakteriellt enzym vars sekvens visade sig vara närbesläktat med TPP II från växter. Undersökningar visade att detta enzym troligen hade tappat den TPP II-lika aktiviteten. Exopeptidasaktiviteten hos TPP II klyver som sagt peptider i tre aminosyror långa bitar, från N-terminalen på substratpeptiden, som inte får vara blockerad. En hypotes som undersöktes i det andra delarbetet (Paper II) var att en eller flera negativt laddade grupper i TPP II interagerar med positivt laddade N-terminalen på peptiden. På så sätt kan ett avstånd på tre aminosyror bildas till den katalytiskt aktiva gruppen, en ”molekylär linjal”. Från sekvens- och strukturhomologi kunde två glutamatrester, Glu-305 och Glu331, utpekas som möjliga kandidater för den här interaktionen, och dessa muterades båda två till glutamin och lysin. Från aktivitetsmätningar kunde slutsatsen dras att bara en av de muterade varianterna, E331Q, hade tillräckligt hög aktivitet för att kinetiska parametrar skulle kunna bestämmas. Dessa avslöjade att KM ökat åtminstone hundrafalt vid utbytandet av Glu-331 mot en glutamatrest, vilket tyder på att denna är viktig för substratbindningen. Ytterligare försök, gjorda med en peptidhämmare med och utan N-terminal aminogrupp, visade på att E331Q band peptiden med och utan N-terminal ungefär lika bra, medan det för vildtypsenzymet var en skillnad i IC50 på 2-3 tiopotenser. Vidare undersökningar av exopeptidasaktiviteten har gjorts med avseende på pH-beroende hos TPP II från tre olika arter och med två olika substrat, AAF-pNA och AAA-pNA. Resultaten visade att ett av substraten, AAApNA, verkade kunna binda improduktivt till den aktiva ytan. Dessutom hade detta substrat en plattare pH-profil, utan ett distinkt maximum, för alla enzymer. Detta berodde på en lägre ökning i kcat med pH p.g.a. deprotonering av histidinet i den katalytiska triaden, vilket skulle kunna tyda på en negativ effekt från en aminosyra med samma pKa. Varför denna effekt skulle påverka det ena substratet mer än det andra är i nuläget svårt att svara på. Den 35 största skillnaden mellan enzymen ligger i pH-beroendet av KM, där dTPP II har ett tydligt minimum vid pH 7.6, medan de två mammala enzymen enbart ökar i KM över pH 7.6, inte under. En hypotes som formulerades var att detta berodde på en aspartatrest som bara återfinns i dTPP II. Endopeptidasaktiviteten hos TPP II är mycket mindre välkaraktäriserad än exopeptidasaktiviteten. I den sistnämnda finns en preferens för att klyva efter aromatiska och alifatiska aminosyrarester, medan det har rapporterats att endopeptidasaktiviteten klyver efter basiska aminosyrarester. Undersökningen som gjordes i delarbete fyra (Paper IV) kunde inte påvisa någon specifik preferens, och det verkar därför som om endopeptidasaktiviteten är mer promiskuös än man tidigare trott. Hastigheten för hydrolys med endopeptidasaktiviteten är mycket lägre, omkring 4-5 magnituder, än den för exopeptidasaktiviteten. Detta gör att den fysiologiska relevansen för denna aktivitet rimligen är låg, åtminstone i normalfallet. Det är dock möjligt att endopeptidasaktiviteten har betydligt högre affinitet för vissa naturliga substrat än för dem vi undesökt. Sammanfattningsvis har dessa undersökningar visat att det finns konserverade regioner i TPP II som är bevarade i alla eukaryoter, vilket förhoppningsvis ska göra TPP II-homologer lättare att hitta i framtiden. En av de bäst bevarade regionerna innefattar Glu-331, som är viktig för bindning av substrat, och utgör en ”molekylär linjal” i enzymet. För exopeptidasaktiviteten kunde även konstateras att vissa substrat kan binda på ett improduktivt sätt, vilket tyder på att substratet kan binda på mer än ett sätt till den aktiva ytan. Substratspecificiteten för endopeptidasaktiviteten kunde inte fastställas, vilket till stor del berodde på den låga aktiviteten och en promiskuös tendens hos enzymet. 36 Acknowledgements Jag skulle vilja utsträcka ett stort tack till…/I would like to thank… Birgitta, för att du alltid tagit dig tid, varit uppmuntrande och i största allmänhet varit en enastående handledare. Helena, för att du är en outsinlig kraft- och inspirationskälla. Mina medförfattare: Ann-Christin, för tiden tillsammans på labb Omar, keep on dancing like nobody’s watching. Pernilla, för att du delade med dig av din jäst-expertis. Hubert Kalbacher & crew, it was a pleasure working at your lab. Emil, för den korta men otroligt intensiva tiden! Micke, för att du delar med dig av din kunskap. Exjobbarna genom åren: Linnea, Vanda, Mahmudur och Sabina, för tappra insatser och goda skratt på labb. Doktorander (och doktorer) på biokem: Helena N (för sena nätter och tidiga morgnar i Grekland), Eric (för att du stod ut med min värmefixering), Diana (för att du alltid är så positiv!), Natalia (för att du lärde mig hur man lär ut), Johan (det är alltid roligt att jobba med dig), Angelica (för att du aldrig är rädd att fråga), Wei Zhang (I admire your energy), Göran, Cissi, Sophia och alla nya och gamla biokemister! Wei, for sharing office space and chitchat over the years! Korridorskamraterna på IMBIM: Sophia, Pia, Åsa, Erik, Per, Chi, Raza, Karin, Lisa, Andreas och alla andra som sett till att det varit liv och rörelse på labb. TA-personal på biokemi och IMBIM: Lillian, Inger, ”Nalle”, Barbro, Rehné, Marianne, Erika och alla andra som alltid hjälper till! Pappa, för att du alltid ställer upp och förstår allting… Mamma, som alltid är entusiastisk och försöker sätta dig in i vad jag håller på med. 37 Mina sedan-embryostadiet-vänner, Jossan och Ylva, för att ni finns där. Jossan speciellt för att du alltid är ärlig och rak och förväntar dig detsamma av mig. Ylva, för att du alltid obevekligen är dig själv. RotG-gruppen (Jonas, Magda, Fredrik och Toni, plus DD), för att ha gett mina tankar annat att kretsa kring än labbet. Everyone speaks Giant! Sist, men absolut inte minst, Lars, som försilvrar molnen på min himmel varje dag. Det här avhandlingsarbetet finansierades av Carl Tryggers Stiftelse, O.E. och Edla Johanssons Vetenskapliga stiftelse, Kemistsamfundet och Liljewalchs resestipendium 38 References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. Johnson, L. N., and Wolfenden, R. (1970) Changes in absorption spectrum and crystal structure of triose phosphate isomerase brought about by 2-phosphoglycollate, a potential transition state analogue, J. Mol. Biol 47, 93-100. Velick, S. F., and Vavra, J. (1962) A kinetic and equilibrium analysis of the glutamic oxaloacetate transaminase mechanism, J. Biol. Chem 237, 2109-2122. Knowles, J. R., and Parsons, C. A. (1969) Proximity effect in catalysed systems: a dramatic effect on ester hydrolysis, Nature 221, 53-54. Maiorino, M., Aumann, K. D., Brigelius-Flohé, R., Doria, D., van den Heuvel, J., McCarthy, J., Roveri, A., Ursini, F., and Flohé, L. (1995) Probing the presumed catalytic triad of selenium-containing peroxidases by mutational analysis of phospholipid hydroperoxide glutathione peroxidase (PHGPx), Biol. Chem. Hoppe-Seyler 376, 651-660. DeWitt, D. L., el-Harith, E. A., Kraemer, S. A., Andrews, M. J., Yao, E. F., Armstrong, R. L., and Smith, W. L. (1990) The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases, J. Biol. Chem 265, 5192-5198. Bellamacina, C. R. (1996) The nicotinamide dinucleotide binding motif: a comparison of nucleotide binding proteins, FASEB J 10, 1257-1269. Cornish-Bowden, A. (2004) Introduction to enzyme kinetics, in Fundamentals of Enzyme Kinetics Third., ss 23-70. Portland Press Ltd., London. Michaelis, L., and Menten, M. (1913) Kinetik der Invertinwirkung, Biochem. Zeits. 49, 333-369. Lindberg, D., Ahmad, S., and Widersten, M. (2010) Mutations in salt-bridging residues at the interface of the core and lid domains of epoxide hydrolase StEH1 affect regioselectivity, protein stability and hysteresis, Arch. Biochem. Biophys 495, 165-173. Russell, A. J., Thomas, P. G., and Fersht, A. R. (1987) Electrostatic effects on modification of charged groups in the active site cleft of subtilisin by protein engineering, J. Mol. Biol 193, 803-813. Carter, P., and Wells, J. A. (1988) Dissecting the catalytic triad of a serine protease, Nature 332, 564-568. Schechter, I., and Berger, A. (1967) On the size of the active site in proteases. I. Papain, Biochem. Biophys. Res. Commun 27, 157-162. Polgár, L. (2005) The catalytic triad of serine peptidases, Cell. Mol. Life Sci 62, 2161-2172. Beck, I. T. (1973) The role of pancreatic enzymes in digestion, Am. J. Clin. Nutr 26, 311-325. Luchtman-Jones, L., and Broze, G. J. (1995) The current status of coagulation, Ann. Med 27, 47-52. 39 16. Polgár, L. (1973) On the mode of activation of the catalytically essential sulfhydryl group of papain, Eur. J. Biochem 33, 104-109. 17. Alenzi, F. Q., Lotfy, M., and Wyse, R. (2010) Swords of cell death: caspase activation and regulation, Asian Pac. J. Cancer Prev 11, 271-280. 18. Vallee, B. L., and Auld, D. S. (1990) Active-site zinc ligands and activated H2O of zinc enzymes, Proc. Natl. Acad. Sci. U.S.A 87, 220-224. 19. Delpierre, G. R., and Fruton, J. S. (1965) Inactivation of pepsin by diphenyldiazomethane, Proc. Natl. Acad. Sci. U.S.A 54, 1161-1167. 20. Knowles, J. R. (1970) On the mechanism of action of pepsin, Philos. Trans. R. Soc. Lond., B, Biol. Sci 257, 135-146. 21. Dean, R. T. (1979) Lysosomes and protein degradation, Ciba Found. Symp 139-149. 22. Chang, S., Momburg, F., Bhutani, N., and Goldberg, A. L. (2005) The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a "molecular ruler" mechanism, Proc. Natl. Acad. Sci. U.S.A 102, 17107-17112. 23. Bermpohl, F., Löster, K., Reutter, W., and Baum, O. (1998) Rat dipeptidyl peptidase IV (DPP IV) exhibits endopeptidase activity with specificity for denatured fibrillar collagens, FEBS Lett 428, 152-156. 24. Polgár, L., and Csoma, C. (1987) Dissociation of ionizing groups in the binding cleft inversely controls the endo- and exopeptidase activities of cathepsin B, J. Biol. Chem 262, 14448-14453. 25. Ezaki, J., Takeda-Ezaki, M., Oda, K., and Kominami, E. (2000) Characterization of endopeptidase activity of tripeptidyl peptidase-I/CLN2 protein which is deficient in classical late infantile neuronal ceroid lipofuscinosis, Biochem. Biophys. Res. Commun 268, 904-908. 26. Rawlings, N. D., Morton, F. R., Kok, C. Y., Kong, J., and Barrett, A. J. (2008) MEROPS: the peptidase database, Nucleic Acids Res 36, D320-325. 27. Wright, C. S., Alden, R. A., and Kraut, J. (1969) Structure of subtilisin BPN' at 2.5 Ångström resolution, Nature 221, 235-242. 28. Kraut, J., Sieker, L. C., High, D. F., and Freer, S. T. (1962) Chymotrypsinogen: a three-dimensional fourier synthesis at 5 angstrom resolution, Proc. Natl. Acad. Sci. U.S.A 48, 1417-1424. 29. Alden, R. A., Wright, C. S., and Kraut, J. (1970) A hydrogen-bond network at the active site of subtilisin BPN', Philos. Trans. R. Soc. Lond., B, Biol. Sci 257, 119-124. 30. Frey, P. A., Whitt, S. A., and Tobin, J. B. (1994) A low-barrier hydrogen bond in the catalytic triad of serine proteases, Science 264, 1927-1930. 31. Bachovchin, W. W. (1985) Confirmation of the assignment of the low-field proton resonance of serine proteases by using specifically nitrogen-15 labeled enzyme, Proc. Natl. Acad. Sci. U.S.A 82, 7948-7951. 32. Ash, E. L., Sudmeier, J. L., De Fabo, E. C., and Bachovchin, W. W. (1997) A low-barrier hydrogen bond in the catalytic triad of serine proteases? Theory versus experiment, Science 278, 1128-1132. 33. Corey, D. R., and Craik, C. S. (1992) An investigation into the minimum requirements for peptide hydrolysis by mutation of the catalytic triad of trypsin, J. Am. Chem. Soc. 114, 1784-1790. 34. Rawlings, N. D., Polgar, L., and Barrett, A. J. (1991) A new family of serine-type peptidases related to prolyl oligopeptidase, Biochem. J 279 ( Pt 3), 907-908. 35. Chen, P., Tsuge, H., Almassy, R. J., Gribskov, C. L., Katoh, S., Vanderpool, D. L., Margosiak, S. A., Pinko, C., Matthews, D. A., and Kan, C. C. (1996) 40 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. Structure of the human cytomegalovirus protease catalytic domain reveals a novel serine protease fold and catalytic triad, Cell 86, 835-843. Wlodawer, A., Li, M., Dauter, Z., Gustchina, A., Uchida, K., Oyama, H., Dunn, B. M., and Oda, K. (2001) Carboxyl proteinase from Pseudomonas defines a novel family of subtilisin-like enzymes, Nat. Struct. Biol 8, 442-446. Brannigan, J. A., Dodson, G., Duggleby, H. J., Moody, P. C., Smith, J. L., Tomchick, D. R., and Murzin, A. G. (1995) A protein catalytic framework with an N-terminal nucleophile is capable of self-activation, Nature 378, 416-419. Siezen, R. J., and Leunissen, J. A. (1997) Subtilases: the superfamily of subtilisin-like serine proteases, Protein Sci 6, 501-523. Grøn, H., Meldal, M., and Breddam, K. (1992) Extensive comparison of the substrate preferences of two subtilisins as determined with peptide substrates which are based on the principle of intramolecular quenching, Biochemistry 31, 6011-6018. Robertus, J. D., Kraut, J., Alden, R. A., and Birktoft, J. J. (1972) Subtilisin; a stereandemical mechanism involving transition-state stabilization, Biochemistry 11, 4293-4303. Rolfe, D. F., and Brown, G. C. (1997) Cellular energy utilization and molecular origin of standard metabolic rate in mammals, Physiol. Rev 77, 731-758. Schubert, U., Antón, L. C., Gibbs, J., Norbury, C. C., Yewdell, J. W., and Bennink, J. R. (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes, Nature 404, 770-774. Abeliovich, H., Dunn, W. A., Kim, J., and Klionsky, D. J. (2000) Dissection of autophagosome biogenesis into distinct nucleation and expansion steps, J. Cell Biol 151, 1025-1034. Salvador, N., Aguado, C., Horst, M., and Knecht, E. (2000) Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state, J. Biol. Chem 275, 27447-27456. Salomons, F. A., Acs, K., and Dantuma, N. P. (2010) Illuminating the ubiquitin/proteasome system, Exp. Cell Res 316, 1289-1295. Schlesinger, D. H., Goldstein, G., and Niall, H. D. (1975) The complete amino acid sequence of ubiquitin, an adenylate cyclase stimulating polypeptide probably universal in living cells, Biochemistry 14, 2214-2218. Aguilar, R. C., and Wendland, B. (2003) Ubiquitin: not just for proteasomes anymore, Curr. Opin. Cell Biol 15, 184-190. Ciechanover, A., Heller, H., Katz-Etzion, R., and Hershko, A. (1981) Activation of the heat-stable polypeptide of the ATP-dependent proteolytic system, Proc. Natl. Acad. Sci. U.S.A 78, 761-765. Pickart, C. M., and Fushman, D. (2004) Polyubiquitin chains: polymeric protein signals, Curr Opin Chem Biol 8, 610-616. Pickart, C. M. (2000) Ubiquitin in chains, Trends Biochem. Sci 25, 544-548. Löwe, J., Stock, D., Jap, B., Zwickl, P., Baumeister, W., and Huber, R. (1995) Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution, Science 268, 533-539. Murata, S., Yashiroda, H., and Tanaka, K. (2009) Molecular mechanisms of proteasome assembly, Nat. Rev. Mol. Cell Biol 10, 104-115. Benaroudj, N., Tarcsa, E., Cascio, P., and Goldberg, A. L. (2001) The unfolding of substrates and ubiquitin-independent protein degradation by proteasomes, Biochimie 83, 311-318. 41 54. Köhler, A., Bajorek, M., Groll, M., Moroder, L., Rubin, D. M., Huber, R., Glickman, M. H., and Finley, D. (2001) The substrate translocation channel of the proteasome, Biochimie 83, 325-332. 55. Ruschak, A. M., Religa, T. L., Breuer, S., Witt, S., and Kay, L. E. (2010) The proteasome antechamber maintains substrates in an unfolded state, Nature 467, 868-871. 56. Bogyo, M., Shin, S., McMaster, J. S., and Ploegh, H. L. (1998) Substrate binding and sequence preference of the proteasome revealed by active-site-directed affinity probes, Chem. Biol 5, 307-320. 57. Kisselev, A. F., Akopian, T. N., Woo, K. M., and Goldberg, A. L. (1999) The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation, J. Biol. Chem 274, 3363-3371. 58. Saric, T., Graef, C. I., and Goldberg, A. L. (2004) Pathway for degradation of peptides generated by proteasomes: a key role for thimet oligopeptidase and other metallopeptidases, J. Biol. Chem 279, 46723-46732. 59. Kajiya, A., Kaji, H., Isobe, T., and Takeda, A. (2006) Processing of amyloid beta-peptides by neutral cysteine protease bleomycin hydrolase, Protein Pept. Lett 13, 119-123. 60. Zheng, W., Johnston, S. A., and Joshua-Tor, L. (1998) The unusual active site of Gal6/bleomycin hydrolase can act as a carboxypeptidase, aminopeptidase, and peptide ligase, Cell 93, 103-109. 61. Reits, E., Neijssen, J., Herberts, C., Benckhuijsen, W., Janssen, L., Drijfhout, J. W., and Neefjes, J. (2004) A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation, Immunity 20, 495-506. 62. Wagner, G. W., Tavianini, M. A., Herrmann, K. M., and Dixon, J. E. (1981) Purification and characterization of an enkephalin aminopeptidase from rat brain, Biochemistry 20, 3884-3890. 63. Cappiello, M., Alterio, V., Amodeo, P., Del Corso, A., Scaloni, A., Pedone, C., Moschini, R., De Donatis, G. M., De Simone, G., and Mura, U. (2006) Metal ion substitution in the catalytic site greatly affects the binding of sulfhydryl-containing compounds to leucyl aminopeptidase, Biochemistry 45, 3226-3234. 64. Yewdell, J. W., and Lev, A. (2007) Self-reporting peptides illuminate the MHC groove, Nat. Chem. Biol 3, 201-202. 65. Seliger, B. (2008) Molecular mechanisms of MHC class I abnormalities and APM components in human tumors, Cancer Immunol. Immunother 57, 1719-1726. 66. Tomkinson, B., and Lindås, A. (2005) Tripeptidyl-peptidase II: a multi-purpose peptidase, Int. J. Biochem. Cell Biol 37, 1933-1937. 67. Bålöw, R. M., and Eriksson, I. (1987) Tripeptidyl peptidase II in haemolysates and liver homogenates of various species, Biochem. J 241, 75-80. 68. Bålöw, R. M., Ragnarsson, U., and Zetterqvist, Ö. (1983) Tripeptidyl aminopeptidase in the extralysosomal fraction of rat liver, J. Biol. Chem 258, 11622-11628. 69. Geier, E., Pfeifer, G., Wilm, M., Lucchiari-Hartz, M., Baumeister, W., Eichmann, K., and Niedermann, G. (1999) A giant protease with potential to substitute for some functions of the proteasome, Science 283, 978-981. 70. Seyit, G., Rockel, B., Baumeister, W., and Peters, J. (2006) Size matters for the tripeptidylpeptidase II complex from Drosophila: The 6-MDa spindle form stabilizes the activated state, J. Biol. Chem 281, 25723-25733. 42 71. Macpherson, E., Tomkinson, B., Bålöw, R. M., Höglund, S., and Zetterqvist, Ö. (1987) Supramolecular structure of tripeptidyl peptidase II from human erythrocytes as studied by electron microscopy, and its correlation to enzyme activity, Biochem. J 248, 259-263. 72. Chuang, C. K., Rockel, B., Seyit, G., Walian, P. J., Schönegge, A., Peters, J., Zwart, P. H., Baumeister, W., and Jap, B. K. (2010) Hybrid molecular structure of the giant protease tripeptidyl peptidase II, Nat. Struct. Mol. Biol 17, 990-996. 73. Tomkinson, B. (2000) Association and dissociation of the tripeptidyl-peptidase II complex as a way of regulating the enzyme activity, Arch. Biochem. Biophys 376, 275-280. 74. Book, A. J., Yang, P., Scalf, M., Smith, L. M., and Vierstra, R. D. (2005) Tripeptidyl peptidase II. An oligomeric protease complex from Arabidopsis, Plant Physiol 138, 1046-1057. 75. Bålöw, R. M., Tomkinson, B., Ragnarsson, U., and Zetterqvist, Ö. (1986) Purification, substrate specificity, and classification of tripeptidyl peptidase II, J. Biol. Chem 261, 2409-2417. 76. Renn, S. C., Tomkinson, B., and Taghert, P. H. (1998) Characterization and cloning of tripeptidyl peptidase II from the fruit fly, Drosophila melanogaster, J. Biol. Chem 273, 19173-19182. 77. McKay, R. M., McKay, J. P., Suh, J. M., Avery, L., and Graff, J. M. (2007) Tripeptidyl peptidase II promotes fat formation in a conserved fashion, EMBO Rep 8, 1183-1189. 78. Huai, J., Firat, E., Nil, A., Million, D., Gaedicke, S., Kanzler, B., Freudenberg, M., van Endert, P., Kohler, G., Pahl, H. L., Aichele, P., Eichmann, K., and Niedermann, G. (2008) Activation of cellular death programs associated with immunosenescence-like phenotype in TPPII knockout mice, Proc. Natl. Acad. Sci. U.S.A 105, 5177-5182. 79. Nassar, N., Cancelas, J., Zheng, J., Williams, D. A., and Zheng, Y. (2006) Structure-function based design of small molecule inhibitors targeting Rho family GTPases, Curr Top Med Chem 6, 1109-1116. 80. Princiotta, M. F., Schubert, U., Chen, W., Bennink, J. R., Myung, J., Crews, C. M., and Yewdell, J. W. (2001) Cells adapted to the proteasome inhibitor 4-hydroxy- 5-iodo-3-nitrophenylacetyl-Leu-Leu-leucinal-vinyl sulfone require enzymatically active proteasomes for continued survival, Proc. Natl. Acad. Sci. U.S.A 98, 513-518. 81. Ferro, E. S., Hyslop, S., and Camargo, A. C. M. (2004) Intracellullar peptides as putative natural regulators of protein interactions, J. Neurochem 91, 769-777. 82. Hilbi, H., Puro, R. J., and Zychlinsky, A. (2000) Tripeptidyl peptidase II promotes maturation of caspase-1 in Shigella flexneri-induced macrophage apoptosis, Infect. Immun 68, 5502-5508. 83. Gavioli, R., Frisan, T., Vertuani, S., Bornkamm, G. W., and Masucci, M. G. (2001) c-myc overexpression activates alternative pathways for intracellular proteolysis in lymphoma cells, Nat. Cell Biol 3, 283-288. 84. Hong, X., Lei, L., and Glas, R. (2003) Tumors acquire inhibitor of apoptosis protein (IAP)-mediated apoptosis resistance through altered specificity of cytosolic proteolysis, J. Exp. Med 197, 1731-1743. 85. Stavropoulou, V., Xie, J., Henriksson, M., Tomkinson, B., Imreh, S., and Masucci, M. G. (2005) Mitotic infidelity and centrosome duplication errors in cells overexpressing tripeptidyl-peptidase II, Cancer Res 65, 1361-1368. 43 86. Stavropoulou, V., Vasquez, V., Cereser, B., Freda, E., and Masucci, M. G. (2006) TPPII promotes genetic instability by allowing the escape from apoptosis of cells with activated mitotic checkpoints, Biochem. Biophys. Res. Commun 346, 415-425. 87. Hong, X., Lei, L., Künert, B., Naredla, R., Applequist, S. E., Grandien, A., and Glas, R. (2007) Tripeptidyl-peptidase II controls DNA damage responses and in vivo gamma-irradiation resistance of tumors, Cancer Res 67, 7165-7174. 88. Preta, G., de Klark, R., and Glas, R. (2009) A role for nuclear translocation of tripeptidyl-peptidase II in reactive oxygen species-dependent DNA damage responses, Biochem. Biophys. Res. Commun 389, 575-579. 89. Tsurumi, C., Firat, E., Gaedicke, S., Huai, J., Mandal, P. K., and Niedermann, G. (2009) Viability and DNA damage responses of TPPII-deficient Myc- and Ras-transformed fibroblasts, Biochem. Biophys. Res. Commun 386, 563-568. 90. Seifert, U., Marañón, C., Shmueli, A., Desoutter, J., Wesoloski, L., Janek, K., Henklein, P., Diescher, S., Andrieu, M., de la Salle, H., Weinschenk, T., Schild, H., Laderach, D., Galy, A., Haas, G., Kloetzel, P., Reiss, Y., and Hosmalin, A. (2003) An essential role for tripeptidyl peptidase in the generation of an MHC class I epitope, Nat. Immunol 4, 375-379. 91. Diekmann, J., Adamopoulou, E., Beck, O., Rauser, G., Lurati, S., Tenzer, S., Einsele, H., Rammensee, H., Schild, H., and Topp, M. S. (2009) Processing of two latent membrane protein 1 MHC class I epitopes requires tripeptidyl peptidase II involvement, J. Immunol 183, 1587-1597. 92. Grauling-Halama, S., Bahr, U., Schenk, S., and Geginat, G. (2009) Role of tripeptidyl peptidase II in the processing of Listeria monocytogenes-derived MHC class I-presented antigenic peptides, Microbes Infect 11, 795-802. 93. Wherry, E. J., Golovina, T. N., Morrison, S. E., Sinnathamby, G., McElhaugh, M. J., Shockey, D. C., and Eisenlohr, L. C. (2006) Re-evaluating the generation of a "proteasome-independent" MHC class I-restricted CD8 T cell epitope, J. Immunol 176, 2249-2261. 94. Basler, M., and Groettrup, M. (2007) No essential role for tripeptidyl peptidase II for the processing of LCMV-derived T cell epitopes, Eur. J. Immunol 37, 896-904. 95. Marcilla, M., Villasevil, E. M., and de Castro, J. A. L. (2008) Tripeptidyl peptidase II is dispensable for the generation of both proteasome-dependent and proteasome-independent ligands of HLA-B27 and other class I molecules, Eur. J. Immunol 38, 631-639. 96. Bhutani, N., Venkatraman, P., and Goldberg, A. L. (2007) Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation, EMBO J 26, 1385-1396. 97. Preta, G., Marescotti, D., Fortini, C., Carcoforo, P., Castelli, C., Masucci, M., and Gavioli, R. (2008) Inhibition of serine-peptidase activity enhances the generation of a survivin-derived HLA-A2-presented CTL epitope in colon-carcinoma cells, Scand. J. Immunol 68, 579-588. 98. York, I. A., Bhutani, N., Zendzian, S., Goldberg, A. L., and Rock, K. L. (2006) Tripeptidyl peptidase II is the major peptidase needed to trim long antigenic precursors, but is not required for most MHC class I antigen presentation, J. Immunol 177, 1434-1443. 99. Lévy, F., Burri, L., Morel, S., Peitrequin, A., Lévy, N., Bachi, A., Hellman, U., Van den Eynde, B. J., and Servis, C. (2002) The final N-terminal trimming of a subaminoterminal proline-containing HLA class I-restricted antigenic peptide in the cytosol is mediated by two peptidases, J. Immunol 169, 4161-4171. 44 100. Kawahara, M., York, I. A., Hearn, A., Farfan, D., and Rock, K. L. (2009) Analysis of the role of tripeptidyl peptidase II in MHC class I antigen presentation in vivo, J. Immunol 183, 6069-6077. 101. Rose, C., Vargas, F., Facchinetti, P., Bourgeat, P., Bambal, R. B., Bishop, P. B., Chan, S. M., Moore, A. N., Ganellin, C. R., and Schwartz, J. C. (1996) Characterization and inhibition of a cholecystokinin-inactivating serine peptidase, Nature 380, 403-409. 102. Ganellin, C. R., Bishop, P. B., Bambal, R. B., Chan, S. M. T., Leblond, B., Moore, A. N. J., Zhao, L., Bourgeat, P., Rose, C., Vargas, F., and Schwartz, J. (2005) Inhibitors of tripeptidyl peptidase II. 3. Derivation of butabindide by successive structure optimizations leading to a potential general approach to designing exopeptidase inhibitors, J. Med. Chem 48, 7333-7342. 103. Ganellin, C. R., Bishop, P. B., Bambal, R. B., Chan, S. M., Law, J. K., Marabout, B., Luthra, P. M., Moore, A. N., Peschard, O., Bourgeat, P., Rose, C., Vargas, F., and Schwartz, J. C. (2000) Inhibitors of tripeptidyl peptidase II. 2. Generation of the first novel lead inhibitor of cholecystokinin-8-inactivating peptidase: a strategy for the design of peptidase inhibitors, J. Med. Chem 43, 664-674. 104. De Winter, H., Breslin, H., Miskowski, T., Kavash, R., and Somers, M. (2005) Inhibitor-based validation of a homology model of the active-site of tripeptidyl peptidase II, J. Mol. Graph. Model 23, 409-418. 105. Tomkinson, B., Ní Laoi, B., and Wellington, K. (2002) The insert within the catalytic domain of tripeptidyl-peptidase II is important for the formation of the active complex, Eur. J. Biochem 269, 1438-1443. 106. Tomkinson, B. (1994) Characterization of cDNA for murine tripeptidyl-peptidase II reveals alternative splicing, Biochem. J 304 ( Pt 2), 517-523. 107. Osmulski, P. A., and Gaczynska, M. (1998) A new large proteolytic complex distinct from the proteasome is present in the cytosol of fission yeast, Curr. Biol 8, 1023-1026. 108. Ito, K., Nakajima, Y., Xu, Y., Yamada, N., Onohara, Y., Ito, T., Matsubara, F., Kabashima, T., Nakayama, K., and Yoshimoto, T. (2006) Crystal structure and mechanism of tripeptidyl activity of prolyl tripeptidyl aminopeptidase from Porphyromonas gingivalis, J. Mol. Biol 362, 228-240. 109. Hilbi, H., Jozsa, E., and Tomkinson, B. (2002) Identification of the catalytic triad in tripeptidyl-peptidase II through site-directed mutagenesis, Biochim. Biophys. Acta 1601, 149-154. 45






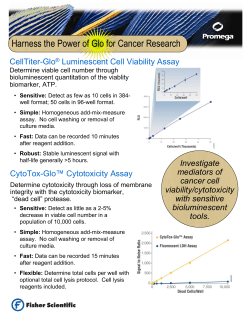


© Copyright 2026