
The Metabolism of Androstenone and Other Steroid
The Metabolism of Androstenone and Other Steroid Hormone Conjugates in Relation to Boar Taint by Heidi M. Laderoute A Thesis presented to The University of Guelph In partial fulfillment of requirements for the degree of Master of Science in Animal and Poultry Science with Toxicology Guelph, Ontario, Canada © Heidi M. Laderoute, April, 2015 ABSTRACT THE METABOLISM OF ANDROSTENONE AND OTHER STEROID HORMONE CONJUGATES IN RELATION TO BOAR TAINT Heidi M. Laderoute University of Guelph, 2015 Advisor: Dr. E.J. Squires Increased public interest in the welfare of pigs reared for pork production has led to an increased effort in finding new approaches for controlling the unpleasant odour and flavour from heated pork products known as boar taint. Therefore, this study investigated the metabolism of androstenone and the enzymes involved in its sulfoconjugation in order to further understand the pathways and genes involved in the development of this meat quality defect. Leydig cells that were incubated with androstenone produced 3-ketosulfoxy-androstenone, providing direct evidence, for the first time, that sulfoconjugation of this steroid does occur in the boar. In addition, human embryonic kidney cells that were overexpressed with porcine sulfotransferase (SULT) enzymes showed that SULT2A1, but not SULT2B1, was responsible for sulfoconjugating androstenone. These findings emphasize the importance of conjugation in steroid metabolism and its relevance to boar taint is discussed. ACKNOWLEDGEMENTS I would like to gratefully and sincerely thank my advisor, Dr. E. James Squires, for providing me with the opportunity to be a graduate student and for introducing me to the world of boar taint. This project would not have been possible without your guidance, encouragement, and patience over the last few years. I would also like to thank Dr. James Raeside and Dr. John Cant for serving on my committee and taking the time to provide me with valuable advice, information, and support on numerous aspects of my project. I am very grateful to Yanping Lou for her technical expertise and friendship. I wouldn’t have finished my studies without her ability to troubleshoot and keep the HPLC ‘Frankenstein monster’ alive. I am also grateful to Heather Christie for the time she spent assisting with early morning Leydig cell isolations and for her advice on different experimental approaches. I would also like to thank Dr. Dyanne Brewer from the Mass Spectrometry Facility at the University of Guelph for her help with sample analysis and interpretation. My thanks and appreciation also goes to my friends and colleagues in the Department of Animal and Poultry Science. To Dr. Matthew Gray, thank you for your patience in answering my many questions, for helping to isolate hepatocytes, and for teaching me how to run a Western blot. To my lab mates, both past and present, thank you for the well-needed distractions and for reminding me that it was okay to take breaks. Finally, thank you to my family and my amazing husband Chris. Your love and support were vital in keeping me sane throughout the writing process. iii TABLE OF CONTENTS ACKNOWLEDGEMENTS ........................................................................................... III TABLE OF CONTENTS ............................................................................................... IV LIST OF FIGURES ...................................................................................................... VII LIST OF TABLES .......................................................................................................... IX LIST OF ABBREVIATIONS ......................................................................................... X CHAPTER 1: LITERATURE REVIEW ....................................................................... 1 1.1 INTRODUCTION ........................................................................................................... 1 1.2 ANDROSTENONE BIOSYNTHESIS ................................................................................. 4 1.2.1 Steroidogenesis ................................................................................................... 4 1.2.2 Biological Function ............................................................................................ 8 1.2.3 Accumulation in Adipose Tissue ....................................................................... 10 1.3 ANDROSTENONE METABOLISM ................................................................................ 11 1.3.1 Phase I Metabolism .......................................................................................... 11 1.3.2 Phase II Metabolism ......................................................................................... 13 1.3.2.1 Sulfoconjugation ........................................................................................ 13 1.3.2.2 Glucuronidation ......................................................................................... 20 1.3.3 Enterohepatic Circulation ................................................................................ 24 1.4 CONTROLLING BOAR TAINT ..................................................................................... 25 1.4.1 Early Slaughter ................................................................................................. 25 1.4.2 Sperm Sorting ................................................................................................... 26 1.4.3 Immunocastration ............................................................................................. 27 1.4.4 Genetic Selection .............................................................................................. 28 iv CHAPTER 2: HYPOTHESIS AND RESEARCH OBJECTIVES ............................ 30 2.1 HYPOTHESIS ............................................................................................................. 30 2.2 RESEARCH OBJECTIVES ............................................................................................ 30 CHAPTER 3: METABOLISM OF ANDROSTENONE IN PRIMARY CULTURED PORCINE LEYDIG CELLS AND HEPATOCYTES............................................. 32 3.1 ABSTRACT ................................................................................................................ 32 3.2 INTRODUCTION ......................................................................................................... 33 3.3 MATERIALS AND METHODS ...................................................................................... 35 3.3.1 Reagents ........................................................................................................... 35 3.3.2 Research Animals ............................................................................................. 35 3.3.3 Leydig cell isolation ......................................................................................... 35 3.3.4 Hepatocyte isolation ......................................................................................... 36 3.3.5 Biosynthesis studies .......................................................................................... 37 3.3.6 High-Performance Liquid Chromatography (HPLC) ...................................... 37 3.3.7 Steroid Conjugate Analyses .............................................................................. 38 3.3.8 Mass Spectrometry ........................................................................................... 39 3.3.9 Data Analysis.................................................................................................... 40 3.4 RESULTS ................................................................................................................... 40 3.5 DISCUSSION .............................................................................................................. 49 CHAPTER 4: THE SULFOCONJUGATION OF ANDROSTENONE AND DEHYDROEPIANDROSTERONE BY HUMAN AND PORCINE SULT2A1 AND SULT2B1 ENZYMES ....................................................................................... 56 4.1 ABSTRACT ................................................................................................................ 56 v 4.2 INTRODUCTION ......................................................................................................... 57 4.3 MATERIALS AND METHODS ...................................................................................... 59 4.3.1 Materials........................................................................................................... 59 4.3.2 Plasmid Constructs ........................................................................................... 60 4.3.3 Cell Culture Transfection and Enzyme Expression .......................................... 61 4.3.4 HPLC ................................................................................................................ 62 4.3.5 Steroid Conjugate Analyses .............................................................................. 63 4.3.6 Western Blot Analysis ....................................................................................... 63 4.3.7 Data Analysis.................................................................................................... 64 4.4 RESULTS ................................................................................................................... 65 4.5 DISCUSSION .............................................................................................................. 77 CHAPTER 5: GENERAL DISCUSSION AND CONCLUSIONS ............................ 83 5.1 DISCUSSION AND FUTURE CONSIDERATIONS ............................................................ 83 5.2 CONCLUSIONS .......................................................................................................... 89 LITERATURE CITED .................................................................................................. 90 vi LIST OF FIGURES Figure 1.1 Steroid hormone biosynthesis pathway. ............................................................ 6 Figure 1.2 Steroid hormone sulfoconjugation .................................................................. 14 Figure 1.3 Potential sulfoconjugation pathways of androstenone. ................................... 16 Figure 1.4 Steroid sulfonate synthesis pathway ................................................................ 19 Figure 1.5 Steroid hormone glucuronidation .................................................................... 21 Figure 3.1 Characteristic HPLC profiles of conjugated and unconjugated steroids from 8 hour incubations of radiolabelled [3H]-androstenone with cells from the testis and liver ....................................................................................................................... 41 Figure 3.2 Mass spectra of conjugated metabolites of androstenone produced by the testes and liver ................................................................................................................ 44 Figure 3.3 Relative peak areas, as determined by HPLC analysis, of metabolites produced from incubations of radiolabelled [3H]-androstenone with Leydig cells over time. ............................................................................................................................... 45 Figure 3.4 Relative peak areas, as determined by HPLC analysis, of metabolites produced from incubations of radiolabelled [3H]-androstenone with hepatocytes over time. ............................................................................................................................... 46 Figure 3.5 Relative peak areas, as determined by HPLC analysis, of metabolites produced from incubations of radiolabelled [3H]-androstenone with Leydig cells over time when considering only sulfoconjugated and unconjugated steroids. .................... 47 Figure 3.6 The production of 3-keto-sulfoxy androstenone and 3-enol sulfonate from androstenone ......................................................................................................... 52 vii Figure 4.1 Amino acid sequence alignment of SULT2A1 enzymes from human and pig species ................................................................................................................... 66 Figure 4.2 Amino acid sequence alignment of SULT2B1 enzymes from human and pig species ................................................................................................................... 67 Figure 4.3 Typical Western blot of human and porcine SULT proteins. ......................... 70 Figure 4.4 Characteristic HPLC profiles of conjugated and unconjugated steroids from HEK cells expressing SULT enzymes and incubated with radiolabelled [3H ]androstenone (A) and [1,2,6,7-3H(N)]-DHEA (B). .............................................. 71 Figure 4.5 Relative peak areas, as determined by HPLC analysis, of androstenone sulfonate (A) and DHEAS (B) production over time for human and porcine SULTs. .................................................................................................................. 72 Figure 4.6 The production of androstenone sulfonate by porcine SULT2A1 (A) and human SULT2A1 (B) with increasing androstenone concentrations. .................. 73 Figure 4.7 The production of androstenone sulfonate by human SULT2B1a (A) and human SULT2B1b (B) with increasing androstenone concentrations. ................ 74 Figure 4.8 The production of DHEAS by porcine SULT2A1 (A) and human SULT2A1 (B) with increasing androstenone concentrations. ................................................ 75 Figure 4.9 The production of DHEAS by human SULT2B1a (A) and human SULT2B1b (B) with increasing androstenone concentrations. ................................................ 76 Figure 4.10 Kinetic model for partial substrate inhibition by DHEA............................... 81 viii LIST OF TABLES Table 3.1 Absolute mean peak areas of metabolites produced by Leydig cells and hepatocytes ............................................................................................................ 48 Table 4.1 Gene-specific primers used for PCR of human and porcine SULTs ................ 60 Table 4.2 Percent identities of human and porcine SULT amino acid sequences ............ 68 Table 4.3 Kinetic constants for porcine and human SULTs ............................................. 77 ix LIST OF ABBREVIATIONS AI artificial insemination AKR aldo-keto reductase APS adenosine 5’-phosphosulfate ATP adenosine triphosphate Aβ andien-β-synthetase BSA bovine serum albumin CAR constitutive androstane receptor CYP cytochrome P450 CYParo cytochrome P450 aromatase CYPscc cytochrome P450 side chain cleavage DHEA dehydroepiandrosterone DHEAS dehydroepiandrosterone sulfonate DHT dihydrotestosterone DMSO dimethyl sulfoxide EGTA ethylene glycol tetraacetic acid ESI electrospray ionization FBS fetal bovine serum FXR farnesoid X receptor G1P glucose-1-phosphate GABAA γ-aminobutyric acid type A GnRH gonadotropin releasing hormone HEK human embryonic kidney HEPES hydroxy ethyl piperazine ethane sulfonic acid HPLC high-performance liquid chromatography HSD hydroxysteroid dehydrogenase x LC-MS liquid chromatography-mass spectrometry LH luteinizing hormone NMDA N-methyl-D-aspartate NMR nuclear magnetic resonance OATP organic anion transporting polypeptide PAGE polyacrylamide gel electrophoresis PAPS 3’-phosphoadenosine 5’-phosphosulfate PCR polymerase chain reaction PSE pale, soft, exudative PVDF polyvinylidene fluoride PXR pregnane X receptor QTL quantitative trait loci Q-TOF quadrupole time-of-flight RIA radioimmunoassay RIPA radio-immunoprecipitation assay SDR short-chain dehydrogenase/reductase SDS sodium dodecyl sulfate SLC solute carrier SNP single nucleotide polymorphism SOAT sodium-dependent organic anion transporter SULT sulfotransferase UDP uridine diphosphate UDPG uridine diphosphate glucose UDPGA uridine diphosphate glucuronic acid UGT uridine diphosphate glucuronosyltransferase UTP uridine triphosphate xi CHAPTER 1: LITERATURE REVIEW 1.1 Introduction In the swine industry, male piglets are surgically castrated within the first week of life in order to reduce aggression and eliminate the unpleasant odour and flavour from heated pork products known as boar taint. 5α-androst-16-en-3-one (androstenone) and 3methylindole (skatole) are the two main compounds responsible for this meat quality defect which occurs in approximately 5 to 15% of uncastrated (intact) male pigs (Patterson, 1968; Lundström et al., 1988; Babol and Squires, 1999; Diaz et al., 1999). Although castration reduces the levels of these compounds, this method is less than ideal for both the producer and consumer. From an economic standpoint, uncastrated boars are more profitable because they have better feed conversion efficiency, faster growth, and greater lean yields than castrates (Gower, 1971; Babol and Squires, 1995; Andersson et al., 1997). In addition, intact boars excrete less nitrogen and have a higher level of unsaturated fatty acids, which directly benefits the environment and the health of consumers, respectively (Wood et al., 1986; Barton-Gade, 1987; Huber et al., 2013). From an ethical standpoint, there is a growing concern for animal welfare since the vast majority of male pigs are castrated without the use of anaesthesia and analgesia. Consequently, this has been shown to induce physiological and behavioural reactions indicative of pain (Giersing et al., 2006; Albrecht, 2013). During castration, pain-related call types can be identified and it has been shown that piglets castrated without the use of anaesthesia produce almost twice as many ‘screams’ as piglets castrated with anesthesia 1 (Marx et al., 2003). In addition, castrated piglets are less active while awake and show more pain related behaviours such as trembling and tail wagging when compared to sham-castrated piglets (Hay et al., 2003). Some of these behaviours last for at least four days following castration. As a result of these welfare concerns, the European Union has prohibited castration without anaesthesia since 2012, and intends to ban surgical castration altogether by 2018 (EC Declaration, 2010). Therefore, there is increased pressure placed on the pig industry worldwide to find alternative methods for reducing boar taint without using castration. Before new methods can be developed, however, factors affecting the synthesis and metabolism of skatole and androstenone have to be well understood. Androstenone is a 16-androstene steroid that is produced in the Leydig cells of the testes together with androgens, estrogens, and other 16-androstene steroids (Gower, 1972). Its production is regulated by the hypothalamic-pituitary axis where luteinizing hormone (LH) is released from the pituitary in response to gonadotropin releasing hormone (GnRH) secreted from the hypothalamus (Zamaratskaia and Squires, 2009). Since androstenone is produced in association with testicular steroids that stimulate growth and fertility, the challenge is to reduce the level of androstenone without affecting the other testicular steroids (Moe et al., 2007b). Due to its lipophilic properties, androstenone readily accumulates in adipose tissue and the cut off levels used for sensory perception of androstenone range between 0.5 μg/g and 1.0 μg/g in fat (Desmoulin et al., 1982; Claus et al., 1994). However, consumer acceptability is highly variable and depends on the intensity of boar taint in raw material, the manufacturing process, the temperature at which eaten, and the consumers’ sensitivity (Desmoulin et al., 1982; 2 Annor-Frempong et al., 1997). While some consumers can detect androstenone at very low concentrations, others are anosmic to it (Wysocki and Beauchamp, 1984; Keller et al., 2007). Bremner et al. (2003) reviewed several studies on androstenone sensitivity and found that between 7.6 and 75% of consumers are anosmic, while Weiler et al. (2000) reported values between 48 and 70% as being anosmic, depending on geographic location and gender. The ability to detect androstenone is genetically determined and is related to the OR7D4 human odour receptor; however women are generally more sensitive (Wysocki and Beauchamp, 1984; Weiler et al., 2000; Keller et al., 2007). While the majority of sensitive consumers describe the odour of androstenone as urine/perspirationlike, it has been reported that up to 8% of highly sensitive consumers liked the smell of androstenone, describing it as sweet, fruity, or perfume-like (Griffiths and Patterson, 1970; Font i Furnols et al., 2003; Andresen, 2006; Robic et al., 2008). Unlike androstenone, most people are able to detect skatole and describe it as having a fecal-like or naphthalene odour and bitter taste (Weiler et al., 1997, 2000; Andresen, 2006). Its threshold value for consumer acceptance is between 0.20 μg/g and 0.25 μg/g in fat (Andresen, 2006). Skatole is produced when bacteria metabolize tryptophan in the large intestine (Jensen et al., 1995). Some skatole is excreted with the feces while the remaining amount is absorbed through the gut wall and is either metabolized in the liver or absorbed into adipose tissue (Lundström and Zamaratskaia, 2006; Tajet et al., 2006; Zamaratskaia and Squires, 2009). Because the amount of skatole produced is primarily dependent on the availability of tryptophan and the composition of intestinal bacteria, it can be controlled through dietary means (Giersing et al., 2006; Lundström and Zamaratskaia, 2006; Zamaratskaia and Squires, 2009). Including certain 3 types of fibers, such as raw potato starch or sugar beet pulp in the diet, for example, has been shown to reduce skatole levels in the fat and plasma of boars (Van Oeckel et al., 1998; Whittington et al., 2004; Zamaratskaia et al., 2005; Chen et al., 2007b). In addition, undigested carbohydrates have been shown to increase fecal wet and dry weight and decrease intestinal transit time, which reduces the amount of skatole absorbed through the intestinal wall (Drochner, 1993; Wang et al., 2004; Zamaratskaia and Squires, 2009). In addition to diet, fat skatole levels can be reduced by rearing pigs on slatted floors rather than concrete, by using wet feeding instead of dry, by keeping pigs clean, and by providing unlimited access to drinking water (as reviewed by Bonneau, 1998). Because skatole can largely be controlled through diet and other environmental factors, and because androstenone is strongly associated with testicular steroids and the occurrence of boar taint, the remainder of this review will focus on biochemical and genetic factors involved in the regulation of androstenone in boars. 1.2 Androstenone Biosynthesis 1.2.1 Steroidogenesis The production of androstenone and other testicular steroids is controlled by the neuroendocrine system. LH is secreted by the pituitary gland in response to GnRH and binds to receptors on the Leydig cells to increase steroidogenesis (Claus et al., 1994; Zamaratskaia et al., 2007). Consequently, androstenone, androgens, and estrogens are secreted following a closely parallel pattern (Andresen, 2006). Although a temporary activation of the hypothalamic-pituitary-gonadal axis occurs at approximately two to four weeks of age, biosynthesis of androstenone and other testicular steroids is low in young pigs (Gower, 1972; Bonneau, 1982; Kwan et al., 1985; Sinclair et al., 2001). As boars 4 reach sexual maturity, at approximately 14 to 15 weeks of age, there is a surge of testosterone and androstenone production, which is quickly followed by an increase in the concentration of androstenone in fat (Claus et al., 1994; Gunn et al., 2004; Zamaratskaia et al., 2008a,b). These steroids continue to gradually increase and reach their highest levels at approximately two years of age (Booth, 1975). Therefore, steroidogenesis depends to a large extent on sexual maturity and consequently on the age and weight of the animal (Bonneau, 1982). All steroid hormones are synthesized from cholesterol and thus have very similar structures (Figure 1.1). In addition, steroid synthesis is primarily carried out by cytochrome P450 (CYP) enzymes, a family of heme-containing mono-oxygenases located on intracellular membranes (Rose et al., 1997). The first step in steroidogenesis involves the side chain cleavage of cholesterol to produce pregnenolone by the enzyme cytochrome P450 side chain cleavage (CYPscc), also known as CYP11A1 (Payne and Youngblood, 1995; Robic et al. 2008, 2014). This occurs in the inner mitochondrial membrane (Hébert and Cooke, 1992; Payne and Youngblood, 1995). Pregnenolone is then transported to the smooth endoplasmic reticulum where it can be converted into progesterone, 17α-hydroxypregnenolone, or 5,16-androstadien-3β-ol (androstadienol) (Robic et al., 2014). Both pregnenolone and progesterone are precursors for the three major groups of steroid hormones, corticoids, androgens, and estrogens, as well as the 16-androstene steroids (Katkov and Gower, 1970; Saat et al., 1972; Brooks and Pearson, 1986; Robic et al., 2014). Androgens and estrogens are necessary for spermatogenesis and sexual behaviour (Gunn et al., 2004). In addition, they favour the development of lean tissue and influence 5 3α-androstenol 3α-HSD 5α-R 3β-HSD Aβ Aldosterone 3β-androstenol CYP11 3β-HSD Androstenone Androstadienone Androstadienol Aβ 11β-HSD CYP11 CYP21 3β-HSD CYPSCC 11-dehydro corticosterone Corticosterone 11-DOC Progesterone CYP17 CYP17 Pregnenolone Cholesterol 11β-HSD CYP11 CYP21 3β-HSD Cortisone Cortisol 11-deoxycortisol 17α-OHprogesterone CYP17 17α-OHpregnenolone CYP17 CYParo CYParo 3β-HSD 17β-HSD Estrone 17β-HSD 19-OH-∆4-AD Androstenedione DHEA 17β-HSD CYParo CYParo 3β-HSD Estradiol 19-OH-Testo Testosterone Androstenediol Figure 1.1 Steroid hormone biosynthesis pathway. CYPscc, cytochrome P40 side chain cleavage; Aβ, andien β-synthetase; 3βAdapted Brooks and Pearson, 1986 and Robic et5α-R, al., 2014 HSD,from 5α-reductase; 3α-HSD, 3α-hydroxysteroid dehydrogenase; CYP21, cytochrome P450 3β-hydroxysteroid dehydrogenase; 21; 11-DOC, 11-deoxycorticosterone; CYP11, cytochrome P450 11; 11β-HSD, 11β-hydroxysteroid dehydrogenase; CYP17, cytochrome P450 17; CYParo, cytochrome P450 aromatase; 19-OH-∆4-AD, 19-hydroxy androstenedione; 17β-HSD, 17βhydroxysteroid dehydrogenase; 19-OH-Testo, 19-hydroxy testosterone (adapted from Brooks and Pearson, 1986; Robic et al., 2014). 6 metabolism, which helps to explain the improved carcass qualities seen in boars compared to castrates (Babol et al., 1998; Gunn et al., 2004). Testosterone, the main androgen in males, can be synthesized through two metabolic pathways. In the first pathway, 17α-hydroxyprogesterone is converted to androstenedione by CYP17, which is then transformed into testosterone via the reduction of the C-17 keto group to a hydroxyl group (Cooke and Gower, 1977; Ruokonen, 1978; Shimizu, 1979; Brooks and Pearson, 1986; Robic et al., 2014). The alternate pathway involves the oxidation of 17αhydroxypregnenolone to dehydroepiandrosterone (DHEA), also by CYP17, which then undergoes oxidation and isomerisation to yield androstenedione or androstenediol (Brooks and Pearson, 1986). Cytochrome P450 aromatase (CYParo), also known as CYP19A, expressed in Leydig cells, is responsible for converting testosterone to estradiol (Payne and Youngblood, 1995). The conversion of pregnenolone into the 16-androstene steroids can also occur through two pathways; however, unlike androgen and estrogen synthesis, neither pathway requires prior C-17 hydroxylation of pregnenolone or progesterone (Brophy and Gower, 1972; Gower, 1972; Shimizu, 1979). In the first pathway, pregnenolone is converted into progesterone, which is then converted into androsta-4,16-dien-3-one (androstadienone). Androstadienone is then metabolized to androstenone via 5α-reductase, which is further converted into either 5α-androst-16-en-3α-ol (3α-androstenol) or 5α-androst-16-en-3β-ol (3β-androstenol) (Brophy and Gower, 1972; Bonneau, 1982). In the second and more predominant pathway, pregnenolone is irreversibly converted to androsta-5,16-dien-3β-ol (androstadienol) by the andien-β-synthetase (Aβ) enzyme complex. Androstadienol is further metabolized to androstadienone by 3β-hydroxysteroid dehydrogenation and 7 isomerization of the double bond from the B ring to the A ring (Hébert and Cooke, 1992; Sinclair, 2004). The Aβ enzyme complex responsible for 16-androstene production includes CYP17 and cytochrome b5 (CYB5) (Ahmad and Gower, 1968; Meadus et al., 1993; Moe et al., 2007a; Robic et al., 2008, 2014). CYB5, a membrane bound protein, stimulates the activity of CYP17 by acting as an allosteric modulator to promote the association of CYP17 with its electron donor (Zamaratskaia et al., 2007). CYB5 is also particularly important in the synthesis of androstenone, as pigs with high CYB5 activity have high levels of androstenone in fat (Davis and Squires, 1999; Leung et al., 2010). In addition, a single nucleotide polymorphism (SNP) has been found in the untranslated region of the CYB5 gene which is associated with a lower fat androstenone level (Lin et al., 2005). 1.2.2 Biological Function Androstenone synthesized in the testis is released into the spermatic vein and then passes into the systemic circulation where the majority is taken up by the submaxillary glands and adipose tissue, and the remaining portion is metabolized (Katkov et al., 1972; Bonneau, 1982; Lundström and Zamaratskaia, 2006; Zamaratskaia and Squires, 2009). The androstenone taken up by the submaxillary glands becomes concentrated by binding to a specific low molecular mass protein called pheromaxein (Booth, 1987; Austin et al., 2004; Andresen, 2006). Once bound, androstenone is reduced to physiologically active 3α-androstenol and, to a lesser extent, 3β-androstenol (Katkov et al., 1972; Booth, 1987). The function of pheromaxein is considered essential since the 16-androstenes are highly lipophilic and occur in very high concentrations in submaxillary glands and saliva 8 (Booth, 1980a,b, 1987; Booth and von Glos, 1991; Austin et al., 2004; Andresen, 2006). When a boar comes in contact with a female, he starts to salivate profusely and thus excretes a large amount of androstenone and 3α-androstenol into the environment (Bonneau, 1982; Booth, 1987; Sinclair, 2004). These androstenes consequently stimulate the typical mating stance seen in sows that are in estrous, allowing the boar to mount and copulate (Bonneau, 1982; Brooks and Pearson, 1986; Lundström and Zamaratskaia, 2006). Therefore, although the 16-androstenes do not possess any androgenic activity, androstenone and 3α-androstenol do act as pheromones to stimulate reproductive functions in the female pig (Andresen, 1976, 2006). In addition to being present in the submaxillary glands, androstenone and 3α-androstenol have also been found in boar sweat glands which are assumed to also play a role in boar courtship behaviour (Stinson and Patterson, 1972; Perry et al., 1980; Bonneau, 1982). Several other possible pheromonal actions of the 16-androstene steroids have been investigated. For example, androstenone has been shown to suppress aggressive behaviour in young pigs and it is thought that the level of androstenes in saliva may be involved in establishing hierarchy (Andresen, 1976; McGlone et al., 1987). In addition, it has been shown that androstenone induces the release of oxytocin in estrous pigs, which may affect the fertility of the female by influencing the sperm transport rate (Mattioli et al., 1986). There is also evidence suggesting that 3α-androstenol is involved as a priming pheromone which acts to accelerate puberty in gilts (Hughes, 1982; Kirkwood et al., 1983; Pearce et al., 1988). 9 1.2.3 Accumulation in Adipose Tissue Androstenone is highly lipophilic and accumulates in adipose tissue in much higher concentrations than the other testicular hormones (Brooks and Pearson, 1986; Claus et al., 1994; Robic et al., 2008). However, storage in adipose tissue is reversible, since fat androstenone levels decrease following castration (Bonneau, 1982; Brooks and Pearson, 1986; Claus et al., 1994). The concentration of androstenone in adipose tissue is directly related to the concentration in blood, which is consequently determined by a boar’s genetic capacity to both synthesize and metabolize 16-androstenes (Zamaratskaia et al., 2008a). In plasma, androstenone concentration is variable, ranging from a few nanograms to at least 60 ng/ml, and levels greater than 15 ng/ml have been associated with high androstenone concentrations in adipose tissue (Andresen, 1976; Sinclair et al., 2001; Andresen, 2006). Therefore, increased production, decreased metabolism, or both will lead to a higher amount of androstenone in the blood, which will consequently lead to an increased concentration in adipose tissue (Sinclair et al., 2001). In many species, steroids are transported to target tissues partly bound to specific proteins such as sex hormone binding globulin (SHBG) (Andresen, 2006; Zamaratskaia et al., 2008a). The binding of androstenone to plasma proteins would increase its polarity and reduce the transfer of the steroid into adipose tissue (Andresen, 2006; Zamaratskaia et al., 2008a). However, SHBGs have not been detected in porcine plasma, and it is possible that androstenone only binds non-specifically to serum components such as αglobulins and albumin (Cook et al., 1977; Zamaratskaia et al., 2008a). Since androstenone binding capacity does not vary greatly between animals, this non-specific binding is unlikely to affect androstenone accumulation in adipose tissue (Zamaratskaia et al., 2008a). 10 1.3 Androstenone Metabolism The metabolism of androstenone and other steroids occurs primarily in the kidney, liver, and testes through both phase I and phase II reactions (Hobkirk, 1985; Sinclair, 2004). Phase I metabolism includes oxidation, reduction, and hydrolysis reactions while phase II metabolism involves the conjugation of a hydrophilic group, such as sulfonate or glucuronic acid, to hydroxyl groups that already exist on the steroid or are added on during phase I metabolism (Sinclair, 2004; Rasmussen et al., 2012a,b). Consequently, the biological properties of steroids and other compounds undergoing these metabolic biotransformations are dramatically affected. Typically, phase I and phase II reactions have been thought of as deactivation or detoxification mechanisms; however the biological significance of these reactions is more complex than this (Sinclair, 2004; Rasmussen et al., 2012b). 1.3.1 Phase I Metabolism The key enzymes involved in the phase I metabolism of androstenone are hydroxysteroid dehydrogenases (HSDs) (Doran et al., 2004). HSDs are oxidoreductases that belong to two distinct protein superfamilies. The short-chain dehydrogenase/reductase (SDR) family includes 3β-HSD, 11β-HSD, and 17β-HSD, while the aldo-keto reductase (AKR) family includes 3α-HSD and 20α-HSD (Pawlowski et al., 1991; Krozowski, 1994; Miura et al., 1994; Jörnvall et al., 1995; Penning, 2003). The reduction and oxidation activities of HSDs allow isoforms to function as molecular switches that will either inactivate a hormone or produce an active ligand, thus regulating the amount of hormone that binds to a steroid hormone receptor (Penning, 1997, 2003). 11 In the case of androstenone, 3α- and 3β- HSD reduce the 3-keto group to a 3α- or 3βhydroxyl group to produce either 3α-androstenol or 3β-androstenol, respectively (Figure 1.1) (Doran et al., 2004; Sinclair et al., 2005a,b). Androstenone was also shown to be transformed into 6-hydroxy androstenone in axillary microflora; however the enzyme responsible for this is unknown (Austin and Ellis, 2003). Consequently, these compounds are less likely to accumulate in adipose tissue due to the hydrophilic nature of the hydroxyl groups (García-Regueiro and Diaz, 1989). Although hepatic and testicular phase I metabolism differ in terms of the percentage of produced metabolites, the rate of 3β-androstenol formation, and therefore 3β-HSD activity, is three to six times higher than that of 3α-androstenol, regardless of metabolic site (Brophy and Gower, 1972; Doran et al., 2004, 2014; Sinclair et al., 2005a; Zamaratskaia and Squires, 2009). 3β-HSD is a particularly important enzyme as it is involved in the biosynthesis of all steroid hormones and controls critical steroid hormone related actions in the adrenal cortex, gonads, liver, fat, and other peripheral target tissues (Mason et al., 1998; Kershaw and Flier, 2004; Rasmussen et al., 2013). Numerous 3β-HSD isoforms have been identified in several species including three in humans, four in rats, and six in mice; however only one isoform has been found in pigs (Simard et al., 1993; Abbaszade et al., 1997; McBride et al., 1999; Von Teichman et al., 2001; Rasmussen et al., 2013). Several investigations have shown that low expression and activity of 3β-HSD in liver and testis is associated with reduced clearance and increased accumulation of androstenone in adipose tissue (Doran et al., 2004; Nicolau-Solano et al., 2006; Chen et al., 2007a). Conversely, high 3β-HSD expression and activity enhance androstenone metabolism and thus decrease androstenone accumulation in adipose tissue (Chen et al., 2007a; 12 Rasmussen et al., 2012b). Nicolau-Solano and Doran (2008) established that androstenone, as well as testosterone and estrone sulfate, induce expression of hepatic 3βHSD; however, this was dependent on substrate concentration and the age and weight of the boar. Conversely, Chen et al. (2007a) suggested that the induction of 3β-HSD expression may be a result of the absence of steroids following castration. External factors, such as diet and environmental toxins, have also been shown to affect the activity of 3β-HSD (Fink-Gremmels and Malekinejad, 2007; Hu et al., 2010; Rasmussen et al., 2012b, 2013). The exact mechanisms regulating this enzyme in porcine liver and testis, however, are still unclear. 1.3.2 Phase II Metabolism Phase II conjugation reactions play an important role in the biotransformation of hormones, neurotransmitters, and xenobiotic compounds (Falany, 1997). These reactions generate metabolites that are more water-soluble and thus more readily excreted in the urine and bile (Falany, 1997). Sulfoconjugation and glucuronidation are the primary phase II reactions responsible for the metabolism of androstenes, androgens, and estrogens. 1.3.2.1 Sulfoconjugation Sulfoconjugation, or sulfonation, is a major phase II biotransformation reaction that is important in the synthesis, transport, and metabolism of steroids (Falany, 1997; Weinshilboum et al., 1997). Sulfonation of steroids involves the transfer of a sulfo group (SO3-) from a donor molecule to the hydroxyl group of an acceptor molecule to form a 13 sulfonate conjugate (Figure 1.2) (Falany et al., 1995; Strott, 2002). Due to phase I metabolism, hydroxyl groups may be present at multiple positions on the steroid nucleus; however hydroxyl groups at positions 3, 17, and 21 are the most common sites for sulfonation to occur (Adams and McDonald, 1981; Strott, 1996). The donor molecule required for sulfonation is 3’-phosphoadenosine 5’-phosphosulfate (PAPS), and all mammalian tissues are able to carry out its synthesis (Robbins et al., 1956; Falany et al., 1995). PAPS is synthesized in the cytosol from inorganic sulfate and adenosine triphosphate (ATP) via a two-step process involving the enzymes ATP sulfurylase and adenosine 5’-phosphosulfate (APS) kinase (Falany, 1997; Strott, 2002). Sources of inorganic sulfate required for PAPS synthesis are acquired through the diet or from the catabolism of sulfur-containing amino acids (Falany, 1997). Sulfonation reactions are mediated by a family of enzymes termed sulfotransferases (SULTs) which are found in many tissues including liver, kidney, testis, ovary, intestinal tract, lung, and brain (Roberts and Lieberman, 1970; Gasparini et al., 1976; Hobkirk, 1985; Falany et al., 1995). Two broad classes of SULTs have been identified in human and animal tissues: (1) membrane-bound SULTs that are localized in the Golgi apparatus of most cells and are responsible for the sulfonation of + Steroid + SULT PAPS Steroid Sulfonate Figure 1.2 Steroid hormone sulfoconjugation. PAPS, 3’-phosphoadenosine 5’phosphosulfate; SULT, sulfotransferase; PAP, 3’- phosphoadenosine 5’-phosphate. 14 PAP macromolecular endogenous structures such as glycosaminoglycans, glycoproteins, and tyrosines in proteins and peptides; and (2) cytosolic SULTs which metabolize relatively small molecules including steroid hormones, xenobiotics, and neurotransmitters (Falany et al., 1995; Falany, 1997; Glatt and Meinl, 2004). To date, six distinct cytosolic SULT gene families have been identified in mammals; however only the SULT1 and SULT2 families are capable of sulfonating steroids (Strott, 2002; Blanchard et al., 2004; Neunzig et al., 2014). The SULT1 family primarily conjugates phenolic molecules including estrogens, thyroid hormones, catecholamines, and xenobiotics (Falany et al., 1989; Strott, 2002). The SULT2 family is primarily involved with the conjugation of hydroxyl groups of steroids with saturated A rings, including 3α-androstenol and 3β-androstenol (Falany et al., 1994; Falany, 1997; Falany and Rohn-Glowacki, 2013). Previously, it was thought that since androstenone did not contain any hydroxyl groups, it could not undergo sulfonation; however hydroxylated forms of androstenone have been produced by axillary microflora and porcine hepatocytes (Austin and Ellis, 2003; Sinclair et al., 2005b; Chen et al., 2007b). In addition, a large proportion of androstenone sulfonate has been found in the plasma of boars (Sinclair and Squires, 2005). It has also been suggested that androstenone may undergo enolisation of the 3-keto group prior to sulfoconjugation (Sinclair et al., 2005a,b, 2006). Since steroid enols are relatively unstable, sulfoconjugation may serve to stabilize this form of steroid (Sinclair et al., 2005a,b). The potential pathways of androstenone sulfoconjugation are shown in Figure 1.3. The SULT2 family consists of SULT2A1 and SULT2B1 subfamilies. In pigs, SULT2A1 is thought to be the key enzyme in the hepatic and testicular phase II metabolism of androstenone (Sinclair 2004, Sinclair et al., 2005b, 2006). This enzyme is 15 Androstenone ENO Hydroxy-androstenone 3-enol SULT SULT SULT 3-enol sulfonate Hydroxy-androstenone sulfonate 3-keto-4-sulfate Figure 1.3 Potential sulfoconjugation pathways of androstenone. ENO, enolase; SULT, sulfotransferase (adapted from Sinclair2011 et al., 2005b; Desnoyer, 2011). Adapted from Sinclair et al., 2005b and Desnoyer, capable of sulfonating both 3α- and 3β-androstenol and its expression and activity have been negatively correlated with both fat and plasma androstenone concentrations in boars (Falany et al., 1989, 1994; Sinclair and Squires, 2005; Sinclair et al., 2006). SULT2A1 activity may be affected by genetic polymorphisms, as several SNPs in humans have resulted in decreased levels of enzyme activity and protein (Igaz et al., 2002; Thomae et al., 2002). In addition, porcine SULT2A1 has been shown to be down-regulated by various nuclear receptors including the constitutive androstane receptor (CAR), the pregnane X receptor (PXR), and the farnesoid X receptor (FXR) (Gray and Squires, 2013). This has the potential to decrease steroid sulfoconjugation and increase androstenone accumulation in adipose tissue. SULT2B1 has also been negatively 16 correlated to fat androstenone concentration and has shown selectivity for 3βhydroxysteroids (Moe et al., 2007a,b; Meloche and Falany, 2001). Therefore, SULT2B1 may also play an important role in the phase II metabolism of androstenone, although direct evidence has yet to be generated. In pigs, only one SULT2B1 enzyme exists; however in humans this gene encodes two isoforms, SULT2B1a and SULT2B1b, which are generated by alternate splicing of the first exon (Her et al., 1998; Meloche and Falany, 2001; Kohjitani et al., 2006). In the blood, the concentration of sulfoconjugated steroids is several times higher than the unconjugated or free form (Saat et al., 1972; Gasparini et al., 1976; Neunzig et al., 2014). In fact, Sinclair and Squires (2005) showed that up to 69-72% of androstenone, 3α-androstenol, and 3β-androstenol are present in their sulfoconjugated forms in the peripheral and testicular vein plasma of mature boars. This may be attributable to an increase in serum protein binding, which would dramatically increase the half-life and decrease metabolic clearance rates of steroid sulfonates (Ruder et al., 1972; Glatt and Meinl, 2004). Protein binding also provides a soluble inactive transport mechanism to target tissues where the unconjugated steroid could be regenerated via hydrolysis of the sulfonate group by the action of membrane bound sulfatase enzymes (Falany, 1997; Glatt, 2000; Sinclair et al., 2005a). Therefore, high levels of sulfoconjugated steroids in the blood could act as a reservoir that serves to regulate the availability of active steroid hormones at both systemic and local levels (Hobkirk, 1993; Glatt and Meinl, 2004). In this way, the level of free androstenone that accumulates in adipose tissue can be controlled. These steroid sulfonates are also capable of acting as substrates for steroidogenic enzymes in the steroid-sulfonate synthesis pathway (Figure 17 1.4) (Falany et al., 1995; Sinclair, 2004). This pathway allows steroid sulfonates to be metabolized in the same way as unconjugated steroids, with compounds retaining their sulfonate groups throughout the entire series of biotransformations, originating from cholesterol sulfonate (Roberts et al., 1964; Gasparini et al., 1976). Cholesterol can be sulfoconjugated at its 3β-hydroxyl group and subsequently converted into pregnenolone sulfonate by side-chain cleavage via CYPscc (Hochberg et al., 1974; Tuckey, 1990). Pregnenolone sulfonate can then be metabolized to 17-hydroxypregnenolone sulfonate via CYP17 and to androstadienol sulfonate (Ruokonen, 1978; Gasparini et al., 1976; Neunzig et al., 2014). Further, 17-hydroxypregnenolone sulfonate can be converted to dehydroepiandrosterone sulfonate (DHEAS) (Lebeau et al., 1964). Since any of these steroid sulfonates can be hydrolyzed to their unconjugated forms by sulfatase enzymes, the steroid-sulfonate synthesis pathway may serve as an alternate route for the formation of free, active steroid hormones. Sulfonation typically increases water solubility and prevents steroid sulfates from binding to and activating steroid receptors (Falany, 1997; Strott, 2002). In addition, steroid sulfonates are unable to passively diffuse through cell membranes, since these compounds remain ionized at physiological pH values (Falany, 1997; Glatt and Meinl, 2004). However, cell uptake of steroid sulfonates can occur via the membrane bound solute carrier (SLC) group of transporter proteins found in a number of tissues including kidney, liver, lung, and testis (Hagenbuch and Meier, 2003; Harteneck, 2013). For example, the organic anion transporting polypeptide (OATP) family has been shown to have broad and overlapping substrate specificities for steroid conjugates as well as bile 18 Sulfonate Pathway Free Steroid Pathway SULT Cholesterol sulfonate CYPscc Cholesterol CYPscc Aβ SULT Androstadienol sulfonate Pregnenolone sulfonate Pregnenolone CYP17 CYP17 SULT 17α-OH-pregnenolone sulfonate CYP17 17α-OH-pregnenolone CYP17 SULT DHEAS DHEA Figure Steroid sulfonate synthesis pathway. SULT, sulfotransferase; CYPscc, Figure 1.41.4 Steroid sulfonate synthesis pathway. SULT, sulfotransferase; CYPscc , cytochrome P450 chain cleavage; andien-β-synthetase; CYP17, cytochrome cytochrome P450 sideside chain cleavage; Aβ,Aβ, andien-β-synthetase; CYP17, cytochrome P450 17;17; DHEAS, dehydroepiandrosterone sulfate; DHEA, dehydroepiandrosterone P450 DHEAS, dehydroepiandrosterone sulfonate; DHEA, dehydroepiandrosterone (adapted from Hobkirk, 1985; Sinclair, 2004). (adapted from Hobkirk, 1985; Sinclair, 2004). 19 salts, thyroid hormones, and xenobiotics (Meier et al., 1997; Kullak-Ublick et al., 2001; Hagenbuch and Meier, 2003; Petzinger and Geyer, 2006). Further, one member of the sodium-dependent organic anion transporter (SOAT) family appears to be specific for the transport of a number of steroid sulfonates including pregnenolone sulfonate and DHEAS (Geyer et al., 2006, 2007; Fietz et al., 2013). Although knowledge of the physiological role of steroid sulfonates is limited, recent studies have shown that they are involved in many different processes that do not require regeneration of the unconjugated steroid (Sinclair, 2004; Neunzig et al., 2014). Cholesterol sulfonate has a stabilizing role in cell membranes, supports platelet adhesion, and can regulate the activity of a variety of functional proteins (as reviewed by Strott and Higashi, 2003); pregnenolone sulfonate has been shown to regulate the γ-aminobutyric acid type A (GABAA) and the N-methyl-Daspartate (NMDA) receptors in the brain (Bowlby, 1993; Park-Chung et al., 1999); and DHEAS induces a non-classical signalling pathway in spermatogenic cells (Shihan et al., 2013). The physiological role of androstenone sulfonate has not been investigated. 1.3.2.2 Glucuronidation Glucuronidation is another major phase II biotransformation process involved in steroid metabolism. Steroid glucuronidation involves the transfer of glucuronic acid from uridine diphosphate (UDP) glucuronic acid (UDPGA) to a hydrophobic steroid molecule that has one or more electrophilic groups (Figure 1.5) (Bélanger et al., 1998; Shipkova and Wieland, 2005). UDPGA is synthesized via a two-step process. First, uridine diphosphate glucose (UDPG) is produced from glucose-1-phosphate (G1P) and uridine triphosphate (UTP) by the action of UDPG pyrophosphorylase. Second, UDPG is oxidized by UDPG dehydrogenase to produce UDPGA. Since glycogen is the ultimate 20 precursor of UDPGA, glucuronidation capacity is dependent on the levels of this compound (Miettinen and Leskinen, 1970). Like sulfonation, glucuronidation can occur at multiple sites on the steroid molecule due to functional groups added during phase I metabolism. However, the most common sites for glucuronidation to occur are at the 3αand 21-hydroxyl groups of C21 steroids and the 3-(C18) or 3α-(C19) and 17β-hydroxyl groups of C18 and C19 steroids (Makin and Trafford, 1972). Steroids with 3β-hydroxyl groups such as DHEA are generally excreted as sulfonates (Mackenzie et al., 1992). Glucuronidation reactions are catalyzed by uridine diphosphate glucuronosyltransferase (UGT) enzymes found in the endoplasmic reticulum of a number of tissues including liver, kidney, intestine, skin, and testes (Miettinen and Leskinen, 1970; Dutton, 1978; Burchell and Coughtrie, 1989). Mammalian UGTs have been classified into four major families based on protein sequence similarity: UGT1, UGT2, UGT3, and UGT8 (Mackenzie et al., 1997, 2005). The UGT1 family primarily conjugates bilirubin, estrogens, bile acids and numerous other drugs and xenobiotics (Ebner and Burchell, 1993; Senafi et al., 1994; Hum et al., 1999). The UGT2 family is divided into three subfamilies: UGT2A, whose members are specific to the olfactory UGT + Steroid + UDPGA UDP Steroid Glucuronide Figure 1.5 Steroid hormone glucuronidation. UDPGA, uridine diphosphate glucuronic acid; UGT, uridine diphosphate glucuronosyltransferase; UDP, uridine diphosphate. 21 epithelium and have glucuronidation activity towards several odorants (Lazard et al., 1991; Jedlitschky et al., 1999); UGT2B, whose members conjugate steroids, bile acids, fatty acids, carboxylic acids, phenols and carcinogens (Mackenzie et al., 1997; Turgeon et al., 2001); and UGT2C, which has only been identified in rabbits (Parkinson, 2001). The UGT3 family uses UDP-N-acetylglucosamine, UDP-glucose, or UDP-xylose instead of UDPGA as a glycosyl source and conjugates steroid hormones, bile acids, and xenobiotics (Mackenzie et al., 2008, 2011; Meech and Mackenzie, 2010). The UGT8 family contains only one member, UGT8A1, which uses UDP-galactose instead of UDPGA as a glycosyl source to conjugate ceramides and bile acids (Sprong et al., 1998; Meech et al., 2015). The UGT enzymes have wide and overlapping substrate specificities; however enzymes belonging to the UGT2B family are mainly responsible for the glucuronidation of steroids (Mackenzie et al., 1992; Turgeon et al., 2001). In humans, UGT2B7, UGT2B15, and UGT2B17 have shown a high capacity to conjugate androgens (Bélanger et al., 2003). UGT2B7 conjugates 5α-reduced metabolites of mineralocorticoids, glucocorticoids, progestins, and androgens, as well as 5β-reduced C19 and C21 steroids (Bélanger et al., 2003; Girard et al., 2003). UGT2B15 specifically conjugates the 17hydroxy position of 5α-reduced androgens, while UGT2B17 conjugates both the 3- and 17-hydroxy positions (Beaulieu et al., 1996; Bélanger et al., 2003). Although glucuronidation of androstenone, 3α-androstenol, and 3β-androstenol is possible, specific UGT enzymes responsible for these conjugations have not been identified. As with sulfoconjugation, glucuronidation results in an increase in polarity and facilitates excretion via the bile or urine (Mackenzie et al., 1992). In addition, steroid 22 glucuronidation may regulate the availability of active steroid hormones by serving as a storage pool from which the free steroid can be regenerated via glucuronidase enzymes (Bradlow, 1970; Hobkirk, 1985; Guillemette et al., 1996). However, high levels of certain steroid glucuronides have been associated with hepatotoxicity. 17β-estradiol glucuronide and testosterone glucuronide, for example, have been shown to inhibit bile flow, thus inducing cholestasis (Gosland et al., 1993; Clarke and Burchell, 1994). In contrast to sulfonation, a glucuronide synthesis pathway does not seem to exist as steroid glucuronides generally do not act as substrates for the enzymes involved in steroidogenesis (Roy, 1970; Sinclair, 2004). However, Rittmaster (1993) has suggested that the conversion of dihydrotestosterone (DHT) glucuronide to 3α-androstanediol 17glucuronide, and of 3α-androstanediol 3-glucuronide to androsterone glucuronide is possible. Although UGT enzymes are found in multiple extrahepatic tissues, the 16androstenes have not been detected as glucuronide conjugates in either the peripheral or testicular vein plasma of boars (Sinclair and Squires, 2005; Sinclair et al., 2005a). This led to the suggestion that the glucuronidation of 16-androstene steroids serves primarily as a mechanism to facilitate metabolic clearance (Sinclair et al., 2005b). This is supported by the high concentrations of androgen, estrogen, and androstene glucuronides found in the urine and bile, as well as the high levels of UGT enzymes found in the liver and kidney (Gower and Patterson, 1970; Sinclair et al., 2005b). In terms of biological roles in relation to boar taint, steroid glucuronidation has received very little attention, and much more research is needed. 23 1.3.3 Enterohepatic Circulation Most steroids and other compounds conjugated in the liver are excreted in the bile (Winter and Bokkenheuser, 1987). When these compounds reach the duodenum, they are exposed to the action of deconjugating enzymes secreted by the intestinal wall as well as by the intestinal microflora. The deconjugated compounds are then either reabsorbed in the ileum and caecum or eliminated in the feces (Eriksson, 1971; Groh et al., 1993). The intestinal wall only produces glucuronidase enzymes while intestinal bacteria, primarily Bacteroides, Escherichia coli, and Clostridiae, synthesize both glucuronidases and sulfatases (Adlercreutz and Martin, 1980; Macdonald et al., 1983). Gastrointestinal disorders, antibiotic treatment, and diet can all influence the composition of the intestinal microflora and thus impact the quantity of steroids that are reabsorbed (Van Eldere et al., 1990; Groh et al., 1993). Consequently, there is a large interest in finding ways to reduce androstenone concentrations through dietary additives that bind androstenone in the intestine (for review see Jen, 2009). Increasing the conjugation of steroids by enhancing the activation of SULTs and UGTs in the intestine, or decreasing the deconjugation activity of the intestine and bacteria would potentially increase the amount of steroids excreted in feces and decrease the amount of androstenone available to accumulate in adipose tissue. Following reabsorption, steroids are reconjugated by SULTs and UGTs present in the intestinal wall and liver (Falany, 1997). From here, the reconjugated steroids may once again undergo enterohepatic circulation or be sent to the kidney and excreted in the urine (Groh et al., 1993). In addition, these steroids may also re-enter the systemic circulation and become available to tissues that are capable of removing the conjugate groups. Although deconjugation is considered essential for efficient reabsorption, 24 evidence has suggested that some conjugated steroids such as estrone sulfonate, estrone glucuronide, pregnenolone sulfonate, and deoxycorticosterone sulfonate are capable of being reabsorbed without cleavage of the conjugate group (Eriksson, 1971; Sim et al., 1983; Sim and Back, 1985). However, much more research is needed to fully understand and confirm these endocrinological processes. 1.4 Controlling Boar Taint Surgical castration has been the traditional method used to control boar taint; however economical and ethical drawbacks have prompted researchers to find feasible, low-cost alternatives to castration that do not negatively affect other carcass characteristics. Some of these alternatives include early slaughter, sperm sorting, immunocastration, and genetic selection. 1.4.1 Early Slaughter Early slaughter involves slaughtering boars at a younger age and at a lighter weight before they have reached sexual maturity. Since androstenone is generally not produced and accumulated in adipose tissue until puberty, early slaughter may reduce the frequency of boar taint (Gunn et al., 2004). However, the correlation of fat androstenone concentration and carcass weight is low and most studies have shown that slaughtering at a lower weight does not entirely eliminate boar taint (Gunn et al., 2004; Zamaratskaia and Squires, 2009). In addition, the carcasses of young pigs often do not have enough meat to return a profit for producers. There are also welfare issues associated with the early slaughter of boars. Entire males are more aggressive than castrates, particularly when 25 mixed with unfamiliar pigs on farms, during transport, and during pre-slaughter lairage, which increases the risk for skin damage and leg injuries (Gunn et al., 2004; Giersing et al., 2006). Further, the physical exertion associated with fighting may mobilise muscle glycogen stores and lead to a higher frequency of pale, soft, exudative (PSE) meat which has a lower economic value (Warriss and Brown, 1985; Gunn et al., 2004). 1.4.2 Sperm Sorting Sperm sorting involves separating the X- and Y-chromosome bearing spermatozoa using flow cytometry (Johnson et al., 2005). The X-chromosome bearing spermatozoa may then be used in artificial insemination (AI) to produce predominately female pigs for pork production. Since boar taint is not a problem in sows, sperm sorting effectively reduces this issue (Gunn et al., 2004). In addition, the need for castration is greatly reduced and animal welfare is potentially increased. However, current sorting methods are not efficient enough to produce the high number of sperm needed for AI as conventional techniques require approximately 3 billion sperm per dose of semen, and the flow cytometry technique currently allows the sorting of only 10 to 15 million sperm per hour (Johnson, 1991, 2000; Gunn et al., 2004). One procedure has been developed that deposits spermatozoa deep into the uterine horn and reduces the number of sperm required per dose (Martinez et al., 2001, 2002). However, the catheter used in this technique may cause damage to the cervix and uterine mucosa, potentially compromising subsequent fertility and reducing the welfare of the sows (Giersing et al., 2006; Vazquez et al., 2008). In addition to welfare issues, the raising of only females for pork production may not be economically advantageous to producers as female pigs have poorer feed efficiency and produce less lean meat than intact boars (Thun et al., 2006). 26 1.4.3 Immunocastration Immunocastration involves vaccination with Improvac against GnRH which effectively castrates boars near the end of the grower-finisher phase of production (Dunshea et al., 2001). Consequently, this vaccination inhibits the production of LH and FSH from the pituitary, prevents testicular development, reduces the production of testicular steroid hormones and accumulation of skatole, and decreases aggressive behaviour (Dunshea et al., 2001; Metz et al., 2002; Cronin et al., 2003; Giersing et al., 2006). Pigs are vaccinated twice, at least four weeks apart, with the second booster injection given four to six weeks before slaughter. Prior to the second injection, pigs retain the production benefits of entire males (Dunshea et al., 2001; Cronin et al., 2003; Jaros et al., 2005). In addition, immunocastrates have been shown to grow faster to slaughter weight and have higher feed conversion efficiency than surgical castrates (Dunshea et al., 2001; Cronin et al., 2003; Zamaratskaia et al., 2008b). One US economic model estimated that these benefits may result in an additional net income of $5.32 per pig, after the cost and implementation of the vaccine (Buhr et al., 2013). Although immunocastration can be used to reduce androstenone concentrations in fat and decrease aggressive behaviour without negatively affecting performance, several disadvantages exist. For example, handling of heavy pigs that need to be captured, restrained, and injected twice is not practical for some pig producers (Einarsson, 2006; Giersing et al., 2006). Assuming that no pigs are missed, some boars will still have fat androstenone concentrations that are too high due to individual variation in immunological response to the vaccine (Einarsson, 2006). Another disadvantage involves the safety of humans. Since the Improvac vaccine is not species-specific, workers who 27 are accidentally self-injected may develop antibodies against GnRH and consequently become sterile (Prunier et al., 2006; Squires, 2010); however, two injections are needed. In addition, consumer acceptability of products from immunocastrated pigs may not be favourable and needs to be further investigated (Lundström and Zamaratskaia, 2006; Squires, 2010). Despite these drawbacks, Improvac has been approved for use in over 60 countries (Buhr et al., 2013). 1.4.4 Genetic Selection Genetic selection involves identifying genetic factors that control boar taint and implementing them in breeding programs to select pigs with low levels of androstenone (Moe et al., 2009). Since there are large differences in androstenone concentrations between breeds, with more Meishan, Duroc, and Piétrain boars showing higher androstenone levels than Landrace, Large White, Hampshire, and Yorkshire boars, it was suggested that there may be genetic differences in the tissue levels of androstenone (Xue et al., 1996; Tajet et al., 2006; Zamaratskaia and Squires, 2009). In addition, the heritability of fat androstenone is relatively high, ranging from 0.25 to 0.88, suggesting that genetic selection for low boar taint is possible (Jonsson and Andresen, 1979; Willeke et al., 1980; Sellier and Bonneau, 1988; Sellier et al., 2000). Previous attempts at selection against high fat androstenone were successful, however they were also associated with decreased growing performance in boars and delayed puberty in gilts due to a lower production of androgens and estrogens (Bonneau, 1982, 1998; Willeke et al., 1987; Sellier and Bonneau, 1988; Squires, 2006). Therefore, before starting selection, animals that have a decreased genetic capacity to accumulate androstenone in fat while maintaining normal levels of testicular steroids need to be identified using genetic 28 markers (Squires, 2006; Zamaratskaia and Squires, 2009). This would make it possible to select intact boars with little or no boar taint that otherwise grow as normal boars. Identification of quantitative trait loci (QLT) and investigation of SNPs in candidate genes are two common approaches used for developing genetic markers (Squires, 2006; Zamaratskaia and Squires, 2009). Research in this area has identified multiple SNPs in the CYP members CYP2E1, CYP21, CYP2D6 and CYP2C49 that were found to be significantly associated with fat androstenone but not testosterone or estrogens (Moe et al., 2009). In addition, a SNP in CYB5 was found to be significantly associated with lower fat androstenone concentrations (Lin et al., 2005). Further, Squires et al. (2014) showed that numerous SNPs in Duroc, Landrace, and Yorkshire breeds were significantly associated with androstenone and skatole levels. Therefore, these SNPs may be used to reduce androstenone without negatively affecting phenotypes related to growth and reproduction, however much more research is needed (Moe et al., 2009). 29 CHAPTER 2: HYPOTHESIS AND RESEARCH OBJECTIVES The traditional method of surgical castration used to control boar taint has come under scrutiny and economical, ethical, and legal issues are pushing for reliable alternatives. In order to develop new and effective boar taint control methods, a complete understanding and identification of the physiological mechanisms regulating fat androstenone accumulation and the genes controlling these processes are needed. Since androstenone accumulation is influenced by phase II metabolism, the focus of this research will be on the biosynthesis of androstenone and other steroid hormones in the liver and testis, with particular emphasis on sulfonate and glucuronide conjugates. 2.1 Hypothesis It is generally thought that androstenone does not undergo sulfoconjugation since it does not possess any hydroxyl groups; however, several researchers have provided evidence suggesting otherwise. In addition, both porcine SULT2A1 and SULT2B1 have been identified as important enzymes involved in steroid hormone sulfoconjugation. Therefore, it is expected that both porcine Leydig cells and hepatocytes will sulfoconjugate androstenone through the action of SULT2A1 and SULT2B1 enzymes. 2.2 Research Objectives Objective 1: To investigate the phase I and phase II metabolism of androstenone in both Leydig cells and hepatocytes. Previous research has suggested that androstenone, 3α-androstenol, and 3βandrostenol undergo sulfoconjugation, but not glucuronidation, in porcine Leydig cells; 30 however these results were achieved indirectly (Sinclair et al., 2005a; Desnoyer, 2011). In addition, 3β-androstenol was shown to be glucuronidated by porcine hepatocytes while studies investigating the sulfoconjugation capabilities of these cells have been conflicting (Sinclair et al., 2005b; Chen et al., 2015). Therefore, the conjugation abilities of Leydig cells and hepatocytes towards androstenone and its metabolites were investigated. Objective 2: To further characterize the roles of porcine SULT2A1 and SULT2B1 in androstenone sulfoconjugation. SULT2A1 has been identified as an important enzyme involved in the sulfoconjugation of 16-androstene steroids and its activity has been negatively correlated with adipose androstenone concentration (Sinclair et al., 2006). Similarly, decreased expression of SULT2B1 in porcine liver and testes has been associated with high androstenone concentrations (Moe et al., 2007a). Therefore, the sulfoconjugation abilities of porcine SULT2A1 and SULT2B1 towards androstenone were further investigated. Objective 3: To compare the sulfonation activity of human and porcine sulfotransferases toward androstenone and other steroid hormones. Pigs share many similar anatomical and physiological characteristics with humans making them a valuable experimental model for multiple research applications (Patterson et al., 2008; Swindle et al., 2012). Since many sulfotransferases are known to conjugate drugs and xenobiotics, similarities between human and porcine enzymes may further justify the increased use of pigs in research, particularly in preclinical toxicological testing of pharmaceuticals (Swindle et al., 2012). 31 CHAPTER 3: METABOLISM OF ANDROSTENONE IN PRIMARY CULTURED PORCINE LEYDIG CELLS AND HEPATOCYTES 3.1 Abstract Increased public interest in the welfare of pigs reared for pork production has led to an increased effort in finding alternatives to castration for controlling the unpleasant odour and flavour from heated pork products known as boar taint. The purpose of this study was to investigate the testicular and hepatic metabolism of androstenone in the boar. Leydig cells and hepatocytes were isolated from five mature Yorkshire boars and incubated with radiolabelled androstenone for 10 min, 1h, 4h, 8h, and 12h. Entire steroid profiles were analyzed by high-performance liquid chromatography (HPLC) and liquid chromatography-mass spectrometry (LC-MS). Sulfoconjugated steroids were produced by the Leydig cells but not hepatocytes, while glucuronides were produced by hepatocytes but not Leydig cells. Approximately 85% of androstenone was converted into sulfoconjugate metabolites in Leydig cell incubations after 8 hours. This sulfoconjugate fraction included androstenol sulfonate and 3-keto-sulfoxy androstenone. These findings provided direct evidence of the testicular production of a sulfoconjugated form of androstenone in the boar. In addition, the high proportion of sulfoconjugates produced by the Leydig cells emphasizes the importance of steroid conjugation and supports the theory that these steroids could act as a reservoir which serves to regulate the amount of unconjugated steroid hormones available for accumulation in adipose tissue. Keywords: androstenone, boar taint, glucuronidation, sulfoconjugation 32 3.2 Introduction In the swine industry, male piglets are surgically castrated within the first week of life in order to reduce aggression and eliminate the unpleasant odour and flavour from heated pork products known as boar taint. However, increasing welfare concerns are putting pressure on pork producers to find alternatives to castration. Before new methods can be developed, factors controlling and contributing to this meat quality defect have to be well understood. 5α-androst-16-en-3-one (androstenone), the primary compound responsible for boar taint (Patterson, 1968), is a 16-androstene steroid that is produced in the Leydig cells of the testes and readily accumulates in adipose tissue (Desmoulin et al., 1982; Claus et al., 1994). Since androstenone is produced together with testicular steroids that stimulate growth and fertility, the challenge is to reduce the level of androstenone without affecting the other testicular steroids (Gower, 1972; Moe et al., 2007b). This may be accomplished by either selectively reducing the rate of androstenone biosynthesis or increasing the rate of its metabolism (Doran et al., 2004). Androstenone metabolism occurs primarily in the kidney, liver, and testes through phase I and phase II reactions (Hobkirk, 1985; Sinclair, 2004). The phase I enzymes, 3α- and 3β-hydroxysteroid dehydrogenase (HSD), reduce androstenone to 3α-androstenol or 3β-androstenol, respectively (Doran et al., 2004; Sinclair et al., 2005a,b). These compounds may then be conjugated by the action of phase II enzymes, such as sulfotransferases (SULTs) or uridine diphosphate glucuronosyltransferases (UGTs), which result in the generation of metabolites that are more water-soluble and thus more likely to be excreted via the urine and bile (Falany, 1997). 33 Previous research has indirectly shown that porcine Leydig cells produce a relatively large amount of steroid sulfoconjugates, including androstenone sulfonate, but do not produce steroid glucuronides (Sinclair and Squires, 2005; Sinclair et al., 2005a; Desnoyer, 2011). The sulfoconjugation of androstenone likely occurs through prior enolisation of the 3-keto group (Sinclair et al., 2005a,b, 2006). Previous studies have also shown that porcine hepatocytes produce steroid glucuronides; however there is conflicting data regarding the ability of these cells to produce sulfoconjugated steroids (Sinclair et al., 2005b; Chen et al., 2015). One of the problems with steroid metabolism studies is a lack of appropriate analytical methods (Chen et al., 2015). Radioimmunoassays (RIAs), for example, can give erroneous results if there is antibody cross-reactivity and antibodies may not exist for all steroid metabolites of interest (Soldin and Soldin, 2009; Stanczyk and Clarke, 2010). In addition, gas chromatography-mass spectrometry (GC-MS) requires tedious extraction and derivatization procedures for each sample and thus steroid conjugates cannot be analyzed directly (Penning et al., 2010). The use of mass spectrometry with electrospray ionization (ESI) can now avoid many of these problems (Penning et al., 2010; Gouveia et al., 2013). In addition, mass spectrometry with a quadrupole time-offlight (Q-TOF) mass analyzer provides a relatively high sensitivity and accuracy for mass detection and has a virtually unlimited mass range (Gouveia et al., 2013). The purpose of this study was to investigate the metabolism of androstenone in primary cultured porcine Leydig cells and hepatocytes using appropriate analytical techniques that minimized sample processing. 34 3.3 Materials and methods 3.3.1 Reagents Radiolabelled [3H]-androstenone (10 Ci/mmol) was obtained from Moravek Biochemicals (Brea, CA, USA). Nonradioactive steroids were purchased from Steraloids Inc. (Newport, RI, USA). Organic solvents of analytical grade were obtained from Fisher Scientific (Toronto, ON, Canada). All other chemicals used for the isolation and incubation of Leydig cells and hepatocytes were of analytical grade and were purchased from Sigma–Aldrich Canada, Ltd. (Oakville, ON, Canada). 3.3.2 Research Animals A total of five mature intact Yorkshire boars weighing between 120 and 190kg were obtained from the Arkell Swine Research Facility at the University of Guelph and were used in accordance with the guidelines of the Canadian Council of Animal Care and the University of Guelph Animal Care Policy. Boars were electrically stunned and exsanguinated, followed by immediate surgical excision of the testes and liver for further processing. 3.3.3 Leydig cell isolation Primary Leydig cells were isolated as previously described (Sinclair et al., 2005a), with modifications. Briefly, testes were rinsed with cold water, decapsulated, and sliced into pieces approximately 1 cm2 × 1 mm thick. Tissue (120 – 150 g) was digested at 37°C for 45 – 60 minutes by constant stirring in 500 ml of culture medium (TC 199 without phenol red) containing 0.1% L-glutamine, 2.2% sodium bicarbonate, 1% bovine serum 35 albumin (BSA), 1% glucose, 1% collagenase, 0.05% DNase, and 0.05% trypsin inhibitor. Following digestion, tissue homogenate was passed through nylon mesh (155 µm and 80 µm) and the filtrate was centrifuged at 400 × g for 10 minutes at 4°C. The cell pellet was then resuspended in approximately 80 ml of media and layered onto discontinuous Percoll gradients consisting of 21, 26, 34, 40, and 60% interfaces, as previously described (Sinclair et al., 2005a). Cells were counted using a hemocytometer and cell viability was determined with a trypan blue exclusion test. Typical Leydig cell viability was 90%. 3.3.4 Hepatocyte isolation Primary hepatocytes were isolated as previously described (Sinclair et al., 2005b) with modifications. Briefly, the largest vein in one liver lobe was catheterized and perfused with Hanks’ balanced salt solution, pH 7.4, (without Ca2+, Mg2+, HCO3--, and phenol red) containing 1 mM ethylene glycol tetraacetic acid (EGTA) and 10mM hydroxy ethyl piperazine ethane sulfonic acid (HEPES) for approximately 10 minutes and then with Hanks’ balanced salt solution, pH 7.4, containing 10 mM HEPES for approximately 5 minutes at 37°C with a flow rate of 25 ml/minute. The blanched lobe was then digested with Williams’ Medium E, pH 7.4, containing 10 mM HEPES and 0.5 mg/ml Type I collagenase for approximately 30 minutes. After digestion, the hepatic lobe was dissected under sterile conditions to liberate the hepatocytes from the collagen matrix. Dissociated hepatocytes were collected in Williams’ Medium E, pH 7.4, supplemented with 10 mM HEPES, 0.02 units/ml insulin, 1% penicillin/streptomycin, and 10% fetal bovine serum (FBS). The cells were then filtered through sterile gauze and centrifuged at 15 × g for three minutes at room 36 temperature. Cells were rinsed by resuspension and centrifugation in collection medium and the cell pellet was resuspended in collection medium. Cells were counted using a hemocytometer and cell viability was determined with a trypan blue exclusion test. Typical hepatocyte viability was 85%. 3.3.5 Biosynthesis studies Resuspended Leydig cells and hepatocytes were inoculated at a concentration of approximately 2.0 × 106 cells/ml in Erlenmeyer flasks containing 6 – 12 ml of the culture mediums described above. Cells were then incubated with radiolabelled [3H]androstenone (20 µM, 17.9 µCi/µmol) dissolved in ethanol for 12 hours at 37ºC under 5% CO2 and 95% air in a shaking water bath with 100 oscillations/minute. Aliquots of 2 – 4 ml were removed in triplicate at 10 min, 1h, 4h, 8h, and 12h, transferred to pre-cooled test tubes, and centrifuged at 400 × g for five minutes at 4ºC. Supernatants were frozen at -20º until further use. Medium containing [3H]-androstenone that had not been inoculated with cells was used as a control. 3.3.6 High-Performance Liquid Chromatography (HPLC) Thawed supernatants from cell incubations were diluted 1:1 (v/v) with acetonitrile and centrifuged for 10 minutes at room temperature to precipitate protein. The supernatant was analyzed for steroids using reversed phase HPLC by injecting a 100µl aliquot onto a Luna 5µ C18(2) HPLC column (250 × 4.60 mm) (Phenomenex, Torrance, CA, USA). Elution of radiolabelled steroids was monitored by a β-RAM model 2 isotope detector (IN/US Systems, Tampa, FL, USA). The HPLC mobile phase, with a flow rate 37 of 1.0 ml/minute, consisted of an isocratic flow with acetonitrile/water (33:67, v/v) for 8 minutes, a linear gradient from 33 – 60% acetonitrile for the next 17 minutes, an isocratic flow with 100% acetonitrile for the next 5 minutes, and an isocratic flow with acetonitrile/water (33:67, v/v) for the remaining 10 minutes. In this system, free steroids eluted between 36 and 38 minutes and conjugated steroids eluted starting at approximately 20 minutes. The identities of free and conjugated steroids were further characterized by manually collecting entire peak fractions from the HPLC followed by further processing as outlined below. 3.3.7 Steroid Conjugate Analyses Collected fractions were dried using a Savant Speed Vac centrifugal vacuum concentrator and conjugated steroids present in the fraction were deconjugated as previously described (Raeside et al., 1999), with modifications. Briefly, dried fractions were incubated overnight with 5 ml of trifluoroacetic acid/ethyl acetate (1/100, v/v) at 45ºC. This process chemically removes sulfonate groups from steroids, if present. To remove glucuronide groups, separate fractions were incubated overnight with 1.0 ml of 0.5 M sodium acetate buffer, pH 5.0, containing 2500 units/ml of β-glucuronidase (type B-1, from bovine liver) at 37ºC. Steroids treated with β-glucuronidase were extracted twice with 5 ml of diethyl ether and supernatants were pooled. Samples from both treatments were dried under nitrogen at 45ºC, reconstituted with 1.0 ml of 85% acetonitrile, filtered with GHP Acrodisc syringe filters (0.2 μm) (Pall Corp., Ville St. Laurent, Quebec, Canada), and analyzed with HPLC using the same gradient system as above. 38 3.3.8 Mass Spectrometry Liquid chromatography-mass spectrometry was performed on entire supernatant samples from 8 hour cell incubations that contained only unlabelled androstenone and which were diluted 1:1 (v/v) with acetonitrile, as above. Steroids were analyzed on an Agilent 1200 HPLC liquid chromatograph interfaced with an Agilent UHD 6530 Q-Tof mass spectrometer at the Mass Spectrometry Facility of the Advanced Analysis Center at the University of Guelph. A 50 µl sample volume was injected onto a Poroshell 120 2.7µ C18 column (150 × 4.6 mm) (Agilent Technologies, Mississauga, ON, Canada). The HPLC mobile phase system consisted of an isocratic flow of 0.1% formic acid/acetonitrile with 0.1% formic acid (98:2, v/v) for 1 minute, a linear gradient from 2 – 100% acetonitrile with 0.1% formic acid for the next 19 minutes, an isocratic flow with 100% acetonitrile with 0.1% formic acid for 3.5 minutes, followed by re-equilibration for the final 10 minutes. The first 2 and last 5 minutes of gradient were sent to waste and not the spectrometer. The flow rate was maintained at 0.4 ml/minute. The mass spectrometer electrospray capillary voltage was maintained at 4.0 kV and the drying gas temperature was 250ºC with a flow rate of 8 L/minute. Nebulizer pressure was 30 psi and the fragmentor was set to 160. Nitrogen was used as a nebulizing, drying, and collisioninduced gas. The mass-to-charge ratio was scanned across the range of 50 – 1400 m/z in 4GHz positive or negative-ion auto MS/MS mode. Two precursor ions per cycle were selected for fragmentation. The instrument was externally calibrated with ESI TuneMix (Agilent Technologies). Chromatograms were analyzed with Agilent Qualitative Analysis B.06.00 software defining compounds by the Molecular Feature algorithm and generating possible compound formulas including elements C, H, O, N, S, and Cl. 39 3.3.9 Data Analysis Statistical analysis was carried out using a Student’s t-test using Microsoft Excel (2013). The statistical significance level was defined as P < 0.05. All data were expressed as means ± standard error. 3.4 Results Characteristic HPLC chromatographs of conjugated and unconjugated peaks in media from Leydig cells and hepatocytes incubated with [3H]-androstenone are shown in Figure 3.1. Three major peaks from Leydig cell incubations and two major peaks from hepatocytes were observed. The peak with a retention time (RT) of approximately 36 minutes, found in incubations from both cell types, was identified as unconjugated androstenone based on a comparison with authentic steroid standard and confirmed via LC-MS with an m/z- of 271.2 (data not shown). Authentic steroid standards of 3αandrostenol and 3β-androstenol eluted at approximately the same time as androstenone when analyzed by HPLC, suggesting that the peak observed at 36 minutes contained a mixture of unconjugated steroids. The presence of these steroids produced by both Leydig cells and hepatocytes was also confirmed by LC-MS; however, since these steroids have an identical mass (m/z- = 273.2), α- and β- linkages were not differentiated. The steroid peak with an RT of 20.25 minutes found in incubations with Leydig cells, but not hepatocytes (Figure 3.1A,C), corresponded to the RT of 5α-androstenone following fraction collection and solvolysis treatment (Figure 3.1B). No change in retention time was observed with this fraction following treatment with β-glucuronidase. Therefore, this peak was thought to represent at least one sulfoconjugated steroid. The third major peak observed (RT 2.7 minutes) was present in Leydig cell and hepatocyte incubations. 40 Figure 3.1 Characteristic HPLC profiles of conjugated and unconjugated steroids from 8 hour incubations of radiolabelled [3H]-androstenone with cells from the testis and liver. RT for a reference standard was approximately 36-37 minutes. (A) Leydig cells; (B) Leydig cells following fraction collection and solvolysis of the peak with an RT of 20.25 minutes; (C) hepatocytes. 41 However, there was no change in RT following either solvolysis or β-glucuronidase treatment. LC-MS analysis of this peak showed a number of polymer-related compounds. None of these compounds had mass-to-charge ratios that corresponded to those expected for androstenone metabolites. In addition, since this peak was also seen in control samples, it was thought to be an artifact rather than a product of androstenone metabolism, although its production over time by Leydig cells was significantly higher (p < 0.05) when compared to control samples (Figure 3.3A). Further analysis of conjugated androstenone metabolites from Leydig cells via LC-MS carried out in negative ionization mode identified an ion at an RT of 16.09 minutes with a mass-to-charge ratio (m/z) of 353.18 (Figure 3.2A). This is identical to the predicted m/z of androstenol sulfate. The ionized sulfate group can also be seen with m/z96.96. A second ion with an RT of 18.09 minutes and m/z- 367.16 was also identified, which has an identical m/z to a sulfonated form of androstenone previously reported as 3keto-4-sulfoxy-androstenone (Desnoyer, 2011) (Figure 3.2B). Therefore, it was concluded that the HPLC peak identified at RT 20.25 minutes contained both androstenol sulfate and a sulfonated form of androstenone. Steroid glucuronides were not found in incubations with Leydig cells. LC-MS analysis of hepatocyte incubations was carried out in positive ionization mode and an ion at RT 12.78 minutes with m/z+ 450.17 was identified (Figure 3.2C). This m/z corresponds to that of androstenol glucuronide, although this peak was not observed during HPLC analysis. Steroid sulfonates were not found in hepatocyte incubations. Time course data for the production of metabolites by Leydig cell and hepatocyte incubations with radiolabelled androstenone are shown in Figure 3.3 and Figure 3.4, 42 respectively. Relative peak area, representing relative radioactivity and steroid content, was significantly different (p < 0.05) in Leydig cell incubations when compared with control samples for all three major HPLC peaks. The peak representing the artifact increased from 52.1% ± 3.6 after 10 minutes in Leydig cell incubations to a maximum of 83.5% ± 2.0 after 4 hours. This was similar to data from hepatocyte incubations, which showed a relative peak area of 85.9% ± 3.5 after four hours; however this was not significantly different (p > 0.05) when compared to the control sample. When considering only the sulfoconjugated and unconjugated fractions produced from Leydig cell incubations, peak area was greatest for unconjugated steroids (98.9% ± 0.8), with a small amount present in the conjugated form (1.1% ± 0.8) after 10 minutes (Figure 3.5). The peak area percentage of unconjugated steroids decreased to 79.5% ± 6.9 after 1 hour, followed by a decrease to 28.9% ± 12.9 after 4 hours, and to 14.3% ± 8.1 after 8 hours. At 12 hours, the unconjugated fraction increased to 23.7% ± 13.2, however this was not statistically significant (p > 0.05). Corresponding absolute peak areas, representing absolute radioactivity and steroid content, for Leydig cell and hepatocyte incubations are shown in Table 3.1. Peak area was not significantly different (p > 0.05) for peak 1 (RT 2.7 min), identified as an artifact, between Leydig cell and hepatocyte incubations at any incubation times, with the exception of the 12 hour time point. For peak 3 (RT 36.9), significantly higher (p < 0.05) peak areas were observed with hepatocyte incubations when compared to Leydig cells for 0.17, 1, 4, and 8 hour incubation periods. This difference was not due to the production of sulfoconjugated metabolites by Leydig cells as the peak areas from hepatocyte incubations were still significantly higher when absolute areas of peak 2 and 3 were combined for Leydig cells (data not shown). 43 Figure 3.2 Mass spectra of conjugated metabolites of androstenone produced by the testes and liver. Samples from Leydig cells were analyzed in negative ionization mode and those from hepatocytes were analyzed in positive ionization mode. (A) androstenol sulfonate (m/z- = 353.1785) with ionized sulfate group (m/z- = 96.9598) produced by Leydig cells; (B) 3-keto-sulfoxy androstenone (m/z- = 367.1600) produced by Leydig cells; (C) androstenol glucuronide (m/z+ = 450.1687) produced by hepatocytes. 44 A Relative Peak Area (%) B C Time (hours) Figure 3.3 Relative peak areas, as determined by HPLC analysis, of metabolites produced from incubations of radiolabelled [3H]-androstenone with Leydig cells over time. (A) identified artifact (RT 2.7 min); (B) sulfoconjugated fraction (RT 20.2 min); (C) unconjugated androstenone and androstenols (RT 36.9 min); (●) Leydig cell sample; (■) control sample. Values are plotted as mean ± standard error for five independent experiments performed in triplicate (n = 5). Significant differences compared to the control (p < 0.05) were evaluated using a Student’s t-test and are denoted by (*). 45 Relative Peak Area (%) A B Time (hours) Figure 3.4 Relative peak areas, as determined by HPLC analysis, of metabolites produced from incubations of radiolabelled [3H]-androstenone with hepatocytes over time. (A) identified artifact (RT 2.47 min); (B) unconjugated androstenone and androstenols (RT 37.05 min); (●) hepatocyte sample; (■) control sample. Values are plotted as mean ± standard error for five independent experiments performed in triplicate (n = 5). Significant differences compared to the control (p < 0.05) were evaluated using a Student’s t-test and are denoted by (*). 46 Relative Peak Area (%) A B Time (hours) Figure 3.5 Relative peak areas, as determined by HPLC analysis, of metabolites produced from incubations of radiolabelled [3H]-androstenone with Leydig cells over time when considering only sulfoconjugated and unconjugated steroids. (A) identified sulfoconjugated fraction (RT 20.2 min); (B) unconjugated androstenone and androstenols (RT 36.9 min); (●) Leydig cell sample; (■) control sample. Values are plotted as mean ± standard error for five independent experiments performed in triplicate (n = 5). Significant differences compared to the control (p < 0.05) were evaluated using a Student’s t-test and are denoted by (*). 47 48 Leydig Control 76.04 ± 4.032 86.19 ± 5.061 102.1 ± 3.085 110.3 ± 7.892 141.5 ± 7.031 0.2382 ± 0.09832 0.2495 ± 0.1046 0.3734 ± 0.1574 0.6177 ± 0.1954 0.4008 ± 0.1758 391.9 ± 44.68 357.6 ± 30.30 287.4 ± 54.17 226.4 ± 35.43 284.3 ± 50.78 Leydig cells 76.13 ± 7.107 128.7 ± 16.18 229.1 ± 20.44 246.5 ± 7.929 236.0 ± 16.41 * 0.4543 ± 0.1583 11.08 ± 2.662 39.48 ± 6.076 78.64 ± 13.76 51.10 ± 16.44 75.30 ± 4.027 * 51.12 ± 7.909 * 13.12 ± 6.061 * 5.889 ± 2.350 * 13.36 ± 6.553 110.1 ± 6.883 * 78.58 ± 7.643 * 34.46 ± 8.484 * 25.33 ± 5.727 * 11.14 ± 3.920 ND ND ND ND ND 77.14 ± 4.361 139.9 ± 19.36 228.5 ± 30.95 273.6 ± 22.82 297.8 ± 28.30 * Hepatocytes 300.7 ± 9.749 247.3 ± 37.40 134.5 ± 53.33 39.47 ± 20.16 21.68 ± 8.503 ND ND ND ND ND 63.78 ± 0.9153 59.27 ± 4.515 65.95 ± 2.728 76.28 ± 11.20 127.0 ± 26.20 Hepatocyte Control Note. The data presented in this table represent means ± standard error for five independent experiments performed in triplicate (n = 5). ND, no detectable metabolite. Significant differences between Leydig cells and hepatocytes (p < 0.05) were evaluated using a Student’s t-test and are denoted by (*). Time (hours) Peak 1 (RT 2.7 min) 0.17 1 4 8 12 Peak 2 (RT 20.2 min) 0.17 1 4 8 12 Peak 3 (RT 36.9 min) 0.17 1 4 8 12 Mean Peak Area (arbitrary units × 104) Table 3.1 Absolute mean peak areas of metabolites produced by Leydig cells and hepatocytes 3.5 Discussion In the present study, HPLC and LC-MS were used to investigate the metabolism of androstenone in porcine Leydig cells and hepatocytes as in vitro models. The results showed that the Leydig cells produced steroid sulfonates, but not glucuronides, whereas the hepatocytes produced androstenol glucuronide, but no sulfonates. The production of 16-androstene glucuronides by hepatocytes, but not Leydig cells, has been shown in previous studies (Sinclair et al., 2005a; Chen et al., 2015) and corresponds to the theory that glucuronidation serves primarily as a mechanism to facilitate metabolic clearance (Sinclair et al., 2005b). This is further supported by the high concentrations of androgen, estrogen, and androstene glucuronides found in the urine and bile, as well as the high levels of UGT enzymes found in the liver and kidney (Gower and Patterson, 1970; Sinclair et al., 2005b). The observed androstenol glucuronide in the present study was only detected by LC-MS analysis and not by HPLC. This is presumably due to differences in levels of detection between these two techniques and suggests that glucuronidation activity was relatively low in hepatocytes compared to sulfoconjugation activity in Leydig cells. This is further supported by the differences observed between Leydig cells and hepatocytes when comparing absolute peak areas. The combined total peak area, and thus radioactivity, for metabolites produced from hepatocyte incubations was significantly higher compared to that of Leydig cells after 0.17, 1, 4, and 8 hour incubation periods. One explanation is that the Leydig cells may have taken up and retained more of the radiolabelled substrate compared to hepatocytes which may reflect the activity/viability of these cells. However, radioactivity was not measured in the cell pellets. 49 Previous studies have also shown the production of steroid sulfonates by Leydig cells (Saat et al., 1972, 1974; Ruokonen and Vihko, 1974a,b; Booth, 1975; Sinclair et al., 2005a); however results from porcine hepatocytes have been conflicting. Sinclair et al. (2005b) reported that approximately 25% of androstenone was converted into androstenone, 3α-androstenol, and 3β-androstenol sulfonates in hepatocyte incubations following deconjugation, derivatization, and GC-MS analysis of steroid metabolites. In contrast, Chen et al. (2015) did not detect any steroid sulfonates in porcine hepatocytes incubated with androstenone, which agrees with results from the present study. These authors suggested that sulfoconjugation may not have occurred because in vitro cell culture systems have lower sulfate concentrations when compared with in vivo systems. Alternatively, any number of problems could have occurred during hepatocyte isolation which may have led to cell contamination or cell lysis. This may also further explain why androstenol glucuronide was found by LC-MS but not HPLC analysis. Steroid sulfonates produced by Leydig cells included androstenol sulfonate and a sulfonated form of androstenone. Time course data for these sulfonates reached a maximum production level after 8 hours of incubation with approximately 85.7% of androstenone converted to sulfonated steroid metabolites. This high proportion of sulfoconjugated steroids is similar to that reported in previous studies using porcine Leydig cells incubated with androstenone (Sinclair et al., 2005a), as well as 16androstene, androgen, and estrogen steroid sulfonate levels reported in porcine plasma (Saat et al., 1974; Raeside and Renaud, 1983; Sinclair and Squires, 2005). In the present study, α- and β- linkages were not differentiated for the androstenol sulfonate identified, however, previous studies have consistently shown β-androstenol sulfonate as the 50 dominant metabolite in porcine Leydig cells, hepatocytes, and plasma (Saat et al., 1974; Sinclair et al., 2005a,b; Sinclair and Squires, 2005). Therefore, the metabolite identified as androstenol sulfonate most likely consists of a relatively high ratio of β-androstenol sulfonate to α-androstenol sulfonate. Previously, it was thought that since androstenone did not contain any hydroxyl groups, it could not undergo sulfonation; however hydroxylated forms of androstenone have been produced by axillary microflora and porcine hepatocytes (Austin and Ellis, 2003; Sinclair et al., 2005b; Chen et al., 2007b). In addition, a large proportion of androstenone sulfonate has been found in the plasma of boars, although these results were obtained indirectly (Sinclair and Squires, 2005). Androstenone is thought to be sulfoconjugated by prior enolisation of the 3-keto group (Sinclair et al., 2005a,b, 2006) which involves the removal of a hydrogen atom from a carbon that is replaced on an oxygen atom to form a hydroxyl group (Toullec, 1982). The exchange of a proton can occur between either positions 2 and 3 or positions 3 and 4; however, it has generally been thought that the direction of enolisation of 3-keto-5α-steroids is towards position 2 (Berkoz et al., 1962; Gardi et al., 1962). This newly formed hydroxyl group can then be acted upon by sulfotransferase enzymes to produce 3-enol androstenone sulfonate (Figure 3.6). However, this metabolite, with an expected mass of 352.5, was not observed in the present study. Instead, a metabolite with m/z- 367.2 was found which corresponds to a compound identified as 3-keto-4-sulfoxy androstenone in a similar study (Desnoyer, 2011). In addition, the present study showed that this steroid conjugate possessed a sulfate group with m/z- 96.96. The m/z of a sulfate ion can vary depending on where it was attached on the parent compound and this feature has previously been used to 51 Androstenone Formula: C19H28O Mass: 272.214 ENO 3-enol Formula: C19H28O Mass: 272.214 Hydroxy-androstenone Formula: C19H28O2 Mass: 288.209 SULT SULT 3-enol sulfonate Formula: C19H28O4S Mass: 352.171 3-keto-sulfoxy androstenone Formula: C19H28O5S Mass: 368.166 Figure 3.6 The production of 3-keto-sulfoxy androstenone and 3-enol sulfonate from androstenone. ENO, enolase; SULT, sulfotransferase (adapted from Sinclair et al., 2005b; Desnoyer, 2011). 52 identify the structures of sulfated metabolites (Yi et al., 2006). For example, an ion with m/z [M-H-80]- represents a neutral loss of SO3 and indicates that the metabolite was produced by the sulfoconjugation of a phenol or an enol with the sulfate attached to an sp2 carbon. This characteristic ion would have been expected if 3-enol androstenone sulfonate was observed in the present study. An ion with m/z 97, which was observed in the current study, represents the formation of a bisulfate anion (HSO4−) and indicates that the sulfate group was attached to an sp3 carbon and the sulfated product produced by the sulfoconjugation of an aliphatic alcohol (Yi et al. 2006). Therefore, the sulfated steroid observed in the current study with m/z- 367.2 would have been the result of the addition of a hydroxyl group onto androstenone at a position other than C3 followed by sulfoconjugation of this group to produce 3-keto-sulfoxy androstenone (Figure 3.6). Hydroxylated forms of androstenone have been shown to be produced by porcine hepatocytes (Sinclair et al., 2005b). In addition, Austin and Ellis (2003) identified 6hydroxy androstenone as a metabolite produced by axillary microflora. Hydroxylation of other steroids has also been shown to occur at the C6 position (Zimniak et al., 1991; Miyata et al., 1994); however, there are multiple sites where this reaction can occur due to the various enzymes that may be involved (Waxman, 1988; Martucci and Fishman, 1993; Gilep et al., 2011; Robic et al., 2014). Therefore, the position of the added hydroxyl group, and consequently the sulfate group, remains unknown. Unfortunately, many steroid conjugates are not yet commercially available as pure standards for chromatography, making identification of compounds from steroid metabolism difficult; however, the use of other analytical techniques such as nuclear magnetic resonance (NMR) spectroscopy has been previously used to determine the position of 53 sulfoconjugation in other compounds (Schaber et al., 2001; Ibrahim et al., 2003; Lerch et al., 2003) and may be a useful technique for future studies. Like glucuronidation, sulfoconjugation has typically been regarded as a mechanism to facilitate metabolic clearance (Sinclair, 2004; Rasmussen et al., 2012b). However, the presence of SULTs in the testis and other extrahepatic tissues (Roberts and Lieberman, 1970; Gasparini et al., 1976; Hobkirk, 1985; Falany et al., 1995), as well as the relatively high proportion of 3-keto-sulfoxy-androstenone produced by the Leydig cells in the current study, indicates otherwise. Since sulfoconjugated steroids can be deconjugated via the action of membrane bound sulfatase enzymes (Falany, 1997; Glatt, 2000), sulfoconjugation may regulate the availability of active steroid hormones by creating a reservoir from which unconjugated steroids could be regenerated by target tissues (Hobkirk, 1993; Glatt and Meinl, 2004). In addition, sulfoconjugation reactions generate metabolites that are more water-soluble and thus less likely to accumulate in adipose tissue (Falany, 1997). Therefore, the testicular production of sulfoconjugated androstenone has been suggested to impact the level of unconjugated androstenone that is available to accumulate in fat, which ultimately plays a role in determining whether or not a boar will develop boar taint (Sinclair et al., 2005a). It is important to note the techniques used for steroid conjugate analysis in the present study. Steroids are typically isolated using liquid/solid, liquid/liquid, or column extraction procedures prior to HPLC and MS analysis (Corpéchot et al., 1981; Guazzo et al., 1996; Rizea Savu et al., 1996; Bean and Henion, 1997; Chatman et al., 1999; Raeside et al., 1999). In addition, steroids may be derivatized in order to increase the sensitivity of detection (Higashi and Shimada, 2004; Penning et al., 2010). These methods are time- 54 consuming and result in indirect and potentially inaccurate data. In the present study, media samples from Leydig cell and hepatocyte incubations were precipitated with acetonitrile and entire samples were successfully analyzed using both HPLC and LC-MS. When steroids were analyzed by LC-MS, sulfoconjugated steroids tended to ionize better in negative mode, while glucuronidated steroids ionized better in positive mode. Therefore, a novel method for the analysis of sulfoconjugated and glucuronidated steroids is proposed in the current study. The ability to analyze an entire steroid profile from one sample is particularly relevant for studies that have a limited amount of sample, as well as those that require a rapid turnaround time (Soldin and Soldin, 2009). In summary, this study used effective HPLC and LC-MS techniques to investigate the metabolism of androstenone in isolated porcine Leydig cells and hepatocytes. This study provided new data on pig Leydig cell steroid metabolism. In particular, direct evidence was provided, for the first time, for the production of a sulfoconjugated form of androstenone by porcine Leydig cells. This metabolite was suggested to be 3-ketosulfoxy-androstenone, which may be formed via prior hydroxylation of androstenone. The study also investigated androstenone metabolite production over time and determined an optimal incubation time of 8 hours for the production of sulfoconjugated steroids. Future studies should focus on investigating the mechanism responsible for androstenone sulfoconjugation as well as confirming the structural form of androstenone sulfonate. 55 CHAPTER 4: THE SULFOCONJUGATION OF ANDROSTENONE AND DEHYDROEPIANDROSTERONE BY HUMAN AND PORCINE SULT2A1 AND SULT2B1 ENZYMES 4.1 Abstract Porcine sulfotransferase 2A1 (pSULT2A1) has previously been identified as a key enzyme involved in the testicular and hepatic metabolism of androstenone. pSULT2B1 may also be important, although no direct evidence exists for its involvement. The purpose of this study was to investigate the sulfoconjugation activity of pSULT2B1 and to further characterize and compare human and porcine sulfotransferase enzymes. pcDNA 3.1 vectors expressing pSULT2A1, pSULT2B1, human (h) SULT2A1, hSULT2B1a, and hSULT2B1b enzymes were transfected into human embryonic kidney (HEK) cells. Transfected cells were then incubated with either androstenone or dehydroepiandrosterone (DHEA) in both time-course and enzyme kinetics studies. The production of androstenone sulfonate and DHEA sulfonate increased over time for all enzymes with the exception of pSULT2B1. Enzyme kinetics analysis showed that androstenone and DHEA were poor substrates for the human orthologs, hSULT2B1a and hSULT2B1b. Comparisons between human and porcine SULT2A1 showed substantially different substrate affinities for androstenone (Km 5.8 ± 0.6 µM and 74.1 ± 15.9 µM, respectively) and DHEA (Km 9.4 ± 2.5 µM and 3.3 ± 1.9 µM, respectively). However, these enzymes did show relatively similar sulfonation efficiencies for DHEA (Vmax/Km 50.5 and 72.9 for hSULT2A1 and pSULT2A1, respectively). The findings in this study provide direct evidence, for the first time, suggesting that pSULT2B1 is not involved with androstenone metabolism and does not sulfonate DHEA. In addition, these results 56 suggest that data obtained from studies utilizing the pig as a human model be interpreted with caution, particularly in preclinical toxicological testing of pharmaceuticals where many sulfotransferase enzymes are required to activate drugs and where species differences may lead to significantly different responses in certain experiments. Keywords: boar taint, enzyme kinetics, human, pig, sulfotransferase 4.2 Introduction Sulfoconjugation, or sulfonation, plays an important role in the biotransformation of hormones, neurotransmitters, and xenobiotic compounds (Falany, 1997; Weinshilboum et al., 1997). These reactions generate metabolites that are more watersoluble and thus more easily excreted in the urine and bile (Falany, 1997). In the boar, this is a particularly relevant metabolic process, as the sulfoconjugation of 16-androstene steroids will influence the occurrence of boar taint, an unpleasant odour and flavour from heated pork products. Androstenone, the primary compound responsible for this meat quality defect, as well its metabolites 3α-androstenol and 3β-androstenol, have been shown to occur largely in their sulfoconjugated form, reaching levels of up to 72% relative to their unconjugated form in the peripheral and testicular vein plasma of mature boars (Sinclair and Squires, 2005). Sulfonation of steroids involves the transfer of a sulfo group (SO3-) from a donor molecule to the hydroxyl group of an acceptor molecule to form a sulfonate conjugate (Falany et al., 1995; Strott, 2002). The donor molecule required for sulfonation is 3’phosphoadenosine 5’-phosphosulfate (PAPS), and all mammalian tissues are able to carry out its synthesis (Robbins et al., 1956; Falany et al., 1995). Sulfonation reactions are 57 mediated by a family of enzymes termed sulfotransferases (SULTs) which are found in many tissues including liver, kidney, testis, ovary, intestinal tract, lung, and brain (Roberts and Lieberman, 1970; Gasparini et al., 1976; Hobkirk, 1985; Falany et al., 1995). To date, six distinct cytosolic SULT gene families have been identified in mammals; however only the SULT1 and SULT2 families are capable of sulfonating steroids (Strott, 2002; Blanchard et al., 2004; Neunzig et al., 2014). Porcine (p) SULT2A1, in particular, has been identified as a key enzyme involved in the testicular and hepatic metabolism of androstenone (Sinclair et al., 2005b, 2006). This enzyme is capable of conjugating a number of different steroids although it is most reactive towards dehydroepiandrosterone (DHEA), an important precursor steroid in androgen and estrogen synthesis (Falany et al., 1989, 2006). pSULT2B1 has also been suggested to play a role in androstenone metabolism as it has been negatively correlated to fat androstenone concentration and has shown selectivity for 3β-hydroxysteroids (Moe et al., 2007a,b; Meloche and Falany, 2001). However, direct evidence for the involvement of this enzyme has yet to be generated. In pigs, only one SULT2B1 enzyme exists; however in humans this gene encodes two isoforms, SULT2B1a and SULT2B1b, which are generated by alternate splicing of the first exon (Her et al., 1998; Meloche and Falany, 2001; Kohjitani et al., 2006). These two isoforms have been shown to preferentially sulfonate pregnenolone and cholesterol, while DHEA was observed to be a poor substrate (Javitt et al., 2001; Fuda et al., 2002). Pigs share many similar anatomical and physiological characteristics with humans making them a valuable experimental model for multiple research applications (Patterson et al., 2008; Swindle et al., 2012). Consequently, similarities between human and porcine enzymes may further justify the increased use of pigs in research, particularly in 58 preclinical toxicological testing of pharmaceuticals, as many SULTs are known to conjugate drugs and in some cases, are required to activate them (Weinshilboum and Otterness, 1994; Swindle et al., 2012). The purpose of this study was to further characterize and compare the sulfoconjugation activity of human and porcine sulfotransferase enzymes towards androstenone and DHEA. 4.3 Materials and methods 4.3.1 Materials Human embryonic kidney cells (HEK293) were purchased from ATCC (Manassas, VA, USA). The pMK-RQ vector containing cDNA encoding porcine SULT2B1 was custom synthesized by Life Technologies Inc. (Burlington, ON, Canada). The pKK233-2 vector containing cDNA encoding for human SULT2A1 was kindly provided by Dr. Charles Falany (University of Alabama at Birmingham, Birmingham, AL, USA) while pCR3.1 vectors containing cDNA encoding for human SULT2B1a and human SULT2B1b were generously donated by Dr. Richard Weinshilboum (Mayo Clinic, Rochester, MN, USA). Radiolabelled [3H]-androstenone (10 Ci/mmol) was obtained from Moravek Biochemicals (Brea, CA, USA) and radiolabelled [1,2,6,73 H(N)]-DHEA (70.5 Ci/mmol) was purchased from Perkin-Elmer (Boston, MA, USA). Nonradioactive steroids were purchased from Steraloids Inc. (Newport, RI). Organic solvents of analytical grade were obtained from Fisher Scientific (Toronto, ON, Canada). All other chemicals used for the incubation of HEK293 cells were of analytical grade and were purchased from Sigma–Aldrich Canada, Ltd. (Oakville, ON, Canada) or Fisher Scientific, unless specified otherwise. 59 4.3.2 Plasmid Constructs The pSULT2A1 coding sequence was amplified from porcine liver cDNA and the pSULT2B1, hSULT2B1a, and hSULT2B1b coding sequences were amplified from their respective vectors by polymerase chain reaction (PCR) using platinum Taq DNA polymerase and gene-specific primers (Table 4.1). These primers were designed based on sequences available from the National Center for Biotechnology Information (NCBI). The PCR procedure consisted of heating at 94°C for 2 minutes, 34 cycles of 94°C for 30 seconds, 60°C for 30 seconds, and 68°C for 1 minute, and finally incubating at 68°C for 10 minutes. Amplified DNA fragments containing appropriate start codon and Kozak sequence (GCC) were gel-purified using a PureLink quick gel extraction kit (Invitrogen) and were subcloned into mammalian expression vector pcDNA3.1/V5-His-TOPO using a TA cloning kit (Invitrogen). Expression vectors for V5-His tagged proteins were generated in order to detect protein expression by Western blot using anti-V5-horseradish peroxidase (HRP) antibody (Billen and Squires, 2009). Sequences of all constructs were confirmed by DNA sequence analysis. Table 4.1 Gene-specific primers used for PCR of human and porcine SULTs Gene Forward Primer (5’→3’) Reverse Primer (5’→3’) Accession Number pSULT2A1 GCCATGACAGAAGAGGAGG TTGCCATGGGAACAGCTCTT NM 001037150.1 pSULT2B1 GCCATGGATGGGCCTGCCG CGGGTGGGGGACCTCG NM 001243697.1 hSULT2A1 GCCATGTCGGACGATTTCT TTCCCATGGGAACAGC NM 003167.3 hSULT2B1a GCCATGGCGTCTCCCCCAC TGAGGGTCGTGGGTGC NM 004605.2 hSULT2B1b GCCATGGACGGGCCCGCCG TGAGGGTCGTGGGTGC NM 177973.1 60 4.3.3 Cell Culture Transfection and Enzyme Expression HEK293 cells were routinely maintained as monolayer cultures in 75 cm2 cell culture flasks (Greiner Bio-One North America Inc., Monroe, NC, USA) and grown in complete growth medium (Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1% L-glutamine, 1% penicillin/streptomycin, 1% nonessential amino acids, 1% sodium pyruvate, and 1% geneticin) in a humidified atmosphere at 37ºC and 5% CO2 in air. Cells were plated in triplicate at 7 × 105 cells per well in 6-well cell culture plates (Greiner Bio-One) with 2 ml/well of complete growth medium and allowed 24 hours for adherence. Vectors expressing sulfotransferases were then transfected into cells using Nano Juice core transfection reagent (1:2, w/v of DNA:reagent) and Nano Juice transfection booster (1:3.2, w/v of DNA:reagent) (Novagen, Etobicoke, ON, Canada) with 0.08 µg pSULT2A1, 1.25 µg pSULT2B1, 1.25 µg hSULT2A1, 0.045 µg hSULT2B1a, and 0.045 µg hSULT2B1b. Vector amounts were varied in order to obtain similar levels of protein expression assessed by Western blotting without compromising activity. Empty pcDNA 3.1 vector was added, when appropriate, so that the total plasmid DNA level was 1.25 µg/well. Empty pcDNA 3.1 vector was also used to transfect control samples. After 48 hours, transfected cells were treated with radiolabelled [3H]-androstenone (20 µM, 31.2 µCi/µmol) and [1,2,6,7-3H(N)]-DHEA (20 µM, 15.5 µCi/µmol) dissolved in ethanol for 4, 8, 16, and 24 hours in a time-course study. Kinetic parameters were also obtained in three separate experiments by incubating cells for 24 hours with varying concentrations of androstenone or DHEA prepared by serial dilution. Media was subsequently removed and diluted 1:1 (v/v) with acetonitrile, centrifuged for 10 minutes at room temperature to precipitate protein, and analyzed by HPLC. Cells were harvested and lysed by sonication in 300 µl of radio61 immunoprecipitation assay (RIPA) buffer (0.5% sodium deoxycholate, 1% Nonnidet P40, and 0.1% SDS) with 1% protease inhibitor cocktail (EMD Millipore Corp., San Diego, CA, USA). Protein concentrations were determined using the Bradford (Bio-Rad) protein assay (Bradford, 1976). 4.3.4 HPLC Media samples diluted with acetonitrile were analyzed using reversed phase HPLC by injecting a 100 µl aliquot onto a Luna 5µ C18(2) HPLC column (250 × 4.60 mm) from Phenomenex (Torrance, CA, USA). Elution of radiolabelled steroids was monitored by a β-RAM model 2 isotope detector (IN/US Systems, Tampa, FL, USA). The HPLC mobile phase system for samples incubated with radiolabelled [3H]androstenone consisted of an isocratic flow with acetonitrile/water (33:67, v/v) for 8 minutes, a linear gradient from 33 – 60% acetonitrile for the next 17 minutes, an isocratic flow with 100% acetonitrile for the next 5 minutes, and an isocratic flow with acetonitrile/water (33:67, v/v) for the remaining 10 minutes. In this system, free steroids eluted between 36 and 38 minutes and sulfoconjugated steroids eluted starting at approximately 20 minutes. Conjugated steroids were further characterized by manually collecting entire peak fractions from the HPLC system followed by further processing as outlined below. The HPLC mobile phase gradient for samples incubated with labelled [1,2,6,7-3H(N)]-DHEA consisted of 50% acetonitrile for 15 minutes, 100% acetonitrile for the next 5 minutes, and 85% acetonitrile for the last five minutes. In this system, DHEA eluted at approximately 11 minutes and DHEAS at 2.5 minutes, which was confirmed using authentic steroid standards. Both gradients were delivered at a flow rate of 1.0 ml/minute. 62 4.3.5 Steroid Conjugate Analyses Collected fractions were dried using a Savant Speed Vac centrifugal vacuum concentrator and any conjugated steroids present in the fraction were deconjugated as previously described (Raeside et al., 1999), with modifications. Briefly, dried fractions were incubated overnight with 5 ml of trifluoroacetic acid/ethyl acetate (1/100, v/v) at 45ºC. This process chemically removes sulfonate groups from steroids, if present. To remove glucuronide groups, separate fractions were incubated overnight with 1.0 ml of 0.5 M sodium acetate buffer, pH 5.0, containing 2500 units/ml of β-glucuronidase (type B-1, from bovine liver) at 37º. Steroids treated with β-glucuronidase were extracted twice with 5 ml of diethyl ether and supernatants were pooled. Samples from both treatments were dried under nitrogen at 45ºC, reconstituted with 1.0 ml of 85% acetonitrile, filtered with GHP Acrodisc syringe filters (0.2 μm) (Pall Corp., Ville St. Laurent, Quebec, Canada), and analyzed by HPLC using the same gradient systems as above. 4.3.6 Western Blot Analysis Protein expression of HEK293 cells transfected with pSULT2A1, pSULT2B1, hSULT2A1, hSULT2B1a, and hSULT2B1b were analyzed by Western blot as previously described (Billen and Squires, 2009), with modifications. Briefly, 20 µg protein samples were separated on 12% sodium dodecyl sulfate – polyacrylamide gel by electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (EMD Millipore) by semi-dry transblot transfer. Blots were incubated overnight at 4ºC in 5% (w/v) dried skim milk in phosphate buffered saline (PBS) with 0.1% Tween 20 and then incubated with a 1:16000 dilution of primary anti-V5 antibody followed by a 1:8000 63 dilution of secondary goat-anti-mouse-HRP antibody in the 5% skim milk solution, as above. Protein bands were visualized and quantified by incubating with detection solution (50 µl of 68 mM p-coumaric acid in dimethyl sulfoxide (DMSO), 5 ml of 1.25 mM luminol in 0.1 M Tris, pH 8.5, and 15 µl of 3% hydrogen peroxide) for 1 minute and using a ChemiDoc MP imaging system (Bio-Rad) with Image Lab 5.0 software. Enzyme activity was normalized to the protein content of expressed sulfotransferase enzymes based on immunoblot band density. 4.3.7 Data Analysis Multiple amino acid sequence alignment analysis was carried out using MAFFT Version 7 software. Statistical analysis was carried out using a Student’s t-test using Microsoft Excel (2013) and the statistical significance level was defined as p < 0.05. Enzyme kinetics analysis was carried out using SigmaPlot 12.0 and Microsoft Excel with the Solver supplement. If no substrate inhibition was observed, data were fitted to the Michaelis-Menten equation (1) and the Hill equation (2): 𝑣= 𝑉𝑚𝑎𝑥 ⋅ [𝑆] 𝐾𝑚 + [𝑆] (1) 𝑣= 𝑉𝑚𝑎𝑥 ⋅ [𝑆]𝑛 𝐾𝑚 𝑛 + [𝑆]𝑛 (2) where v is the rate of substrate sulfoconjugation, Vmax is the apparent maximum rate of substrate sulfoconjugation, S is the substrate incubation concentration, Km is the substrate concentration at which the reaction rate is half of Vmax, and n describes binding cooperativity. The appropriate kinetic model was determined by examining EadieHofstee plots. Where a hook-shaped Eadie-Hofstee plot was observed, indicative of autoactivation, data were fitted to the Hill equation, and where a linear Eadie-Hofstee plot was observed, data were fitted to the Michaelis-Menten equation (Houston and 64 Kenworthy, 2000; Hutzler and Tracy, 2002). When substrate inhibition was observed, kinetic parameters were determined as previously described (Zhang et al., 1998; Geese and Raftogianis, 2001). Briefly, Vmax and Km were estimated at low concentrations, as above. The entire data set was then fitted to a partial substrate inhibition equation (Fromm, 1975): 𝑉 ⋅𝑆 1+𝑉 2 ⋅𝐾 𝑚𝑎𝑥 𝑖 𝑣 = 𝑉𝑚𝑎𝑥 ⋅ ( ) 𝐾𝑚 𝑆 1+ + 𝑆 𝐾𝑖 (3) where v, Vmax, S, and Km are defined as above, V2 is the estimated rate plateau that is reached when inhibition occurs, and Ki is the substrate concentration at which the Vmax is decreased by half. Vmax and Km were then determined over substrate concentrations much less than Ki, where substrate inhibition was negligible. Vmax/Km ratios, representing enzyme catalytic efficiencies, were also calculated. All data were expressed as mean ± standard error. 4.4 Results The amino acid sequence alignments of human and porcine SULTs are shown in Figure 4.1 and Figure 4.2. Both human and porcine proteins contain the classical PAPS binding domain that is nearly identical among SULTs (Dong et al., 2012; Falany and Rohn-Glowacki, 2013). They also contain the highly conserved ‘KTVE’ dimerization domain. The pSULT2A1 and hSULT2A1 cDNAs both contain open reading frames of 858 base pairs (bp) which encode for 285 amino acids. These proteins share an identity of 66% (Table 4.2) and a similarity of 81%. The pSULT2B1, hSULT2B1a, and hSULT2B1b cDNAs contain open reading frames of 1017, 1053, and 1098 bp which 65 hSULT2A1 MSDDFLWFEG IAFPTMGFRS ETLRKVRDEF VIRDEDVIIL TYPKSGTNWL pSULT2A1 -TEEEVR--- -F--KQILSP -M-QE--E-- TFKE---L-- -F-------M 50 50 hSULT2A1 AEILCLMHSK GDAKWIQSVP IWERSPWVES EIGYTALSET ESPRLFSSHL pSULT2A1 I--I--IL-- --T------- N-D----L-- IS--EN-KNK -G---I---- 100 100 hSULT2A1 PIQLFPKSFF SSKAKVIYLM RNPRDVLVSG YFFWKNMKFI KKPKSWEEYF pSULT2A1 -------A-- K----M--II -----II--- -----STNLV –R-E-L-Q-- 150 150 hSULT2A1 EWFCQGTVLY GSWFDHIHGW MPMREEKNFL LLSYEELKQD TGRTIEKICQ pSULT2A1 ---I--N-P- -------R-- L---DKE-V- I-------R- -RSAV----- 200 200 hSULT2A1 FLGKTLEPEE LNLILKNSSF QSMKENKMSN YSLLSVDYVV DKAQLLRKGV pSULT2A1 ----K----- -SSVVE---- -V----N--- F---KGLHLG -TGC-----T 250 250 hSULT2A1 SGDWKNHFTV AQAEDFDKLF QEKMADLPRE LFPWE pSULT2A1 P-----Y--- ----A----- --------Q- ----Q 285 285 Figure 4.1 Amino acid sequence alignment of SULT2A1 enzymes from human and pig species. The amino acid sequences were aligned using MAFFT with hSULT2A1 as reference. Only dissimilar residues are displayed. The conserved PAPS binding domain is doubly underlined, the conserved catalytic residue histidine is highlighted black, loop 3 which makes up the outer surface of both the PAPS and substrate binding pockets is singly underlined, and the conserved ‘KTVE’ dimerization domain is highlighted gray (adapted from Falany and Rohn-Glowacki, 2013). encode for 338, 350, and 365 amino acids, respectively. The pSULT2B1 protein shares identities of 72 and 76%, with hSULT2B1a and hSULT2B1b, respectively, and is 84% similar to both enzymes. Both of the human SULT2B1 isoforms contain an extended identical 50 amino acid carboxy-terminal sequence that is enriched in proline and serine residues and is not found in other human SULTs (Falany and Rohn-Glowacki, 2013). pSULT2B1 also has an extended carboxy-terminal sequence that is not found in pSULT2A1, although it contains 25 amino acid residues and is only 40% identical to hSULT2B1a and hSULT2B1b. The amino-termini of hSULT2B1a, hSULT2B1b, and pSULT2B1 are also extended and contain 8, 23, and 23 amino acid residues, respectively. 66 hSULT2B1a MASPPPFH-- ---------- ---SQKLPGE YFRYKGVPFP VGLYSLESIS 35 hSULT2B1b -DG-AEPQIP GLWDTYEDDI SEI------- ---------- ---------- 50 pSULT2B1 -DG-AEPRNQ AEWDPYEKNI SEI--N-S-- ------I--- --V--P---- 50 hSULT2B1a LAENTQDVRD DDIFIITYPK SGTTWMIEII CLILKEGDPS WIRSVPIWER 85 hSULT2B1b ---------- ---------- ---------- ---------- ---------- 100 pSULT2B1 I---AK*-Q- ---------- ---N------ S----D---- --Q-----K- 99 hSULT2B1a APWCETIVGA FSLPDQYSPR LMSSHLPIQI FTKAFFSSKA KVIYMGRNPR 135 hSULT2B1b ---------- ---------- ---------- ---------- ---------- 150 pSULT2B1 S----A-M-- ----N-P--- -----P---L ------N--- ----L----- 149 hSULT2B1a DVVVSLYHYS KIAGQLKDPG TPDQFLRDFL KGEVQFGSWF DHIKGWLRMK 185 hSULT2B1b ---------- ---------- ---------- ---------- ---------- 200 pSULT2B1 --L------- ---------- ------QN-- ---------- ------I--R 199 hSULT2B1a GKDNFLFITY EELQQDLQGS VERICGFLGR PLGKEALGSV VAHSTFSAMK 235 hSULT2B1b ---------- ---------- ---------- ---------- ---------- 250 pSULT2B1 --E------- -------HS- -Q-V-Q---- -------D-- ----A-N--- 249 hSULT2B1a ANTMSNYTLL PPSLLDHRRG AFLRKGVCGD WKNHFTVAQS EAFDRAYRKQ 285 hSULT2B1b ---------- ---------- ---------- ---------- ---------- 300 pSULT2B1 --A---F--- -T----Q--- ------I--- ---------- ----SV--E- 299 hSULT2B1a MRGMPTFPWD EDPEEDGSPD PEPSPEPEPK PSLEPNTSLE REPRPNSSPS 335 hSULT2B1b ---------- ---------- ---------- ---------- ---------- 350 pSULT2B1 ---L------ -T-*--AR-- -D---D**** ********** ********-- 326 hSULT2B1a PSPGQASETP HPRPS hSULT2B1b ---------- ----pSULT2B1 --------V- --*** 350 365 338 Figure 4.2 Amino acid sequence alignment of SULT2B1 enzymes from human and pig species. The amino acid sequences were aligned using MAFFT with hSULT2B1a as reference. Only dissimilar residues are displayed. The conserved PAPS binding domain is doubly underlined, the conserved catalytic residue histidine is highlighted black, loop 3 which makes up the outer surface of both the PAPS and substrate binding pockets is singly underlined, the conserved ‘KTVE’ dimerization domain is highlighted gray, and gaps are indicated by asterisks (*) (adapted from Falany and Rohn-Glowacki, 2013). 67 Table 4.2 Sequence identities of human and porcine SULT amino acid sequences pSULT2A1 pSULT2B1 hSULT2A1 pSULT2A1 ─ pSULT2B1 45 ─ hSULT2A1 66 41 ─ hSULT2B1a 41 72 39 hSULT2B1a ─ hSULT2B1b 39 76 37 94 Note. Values are presented as percent identity for each individual pair of SULTs Interestingly, the Ile20 and Ile23 residues found in the amino-terminal of hSULT2B1b, which are responsible for the high cholesterol sulfoconjugating activity of this enzyme (Fuda et al., 2002), are also conserved in the pSULT2B1 sequence. Vectors containing cDNA encoding SULT proteins were over-expressed in HEK cells in order to determine relative sulfoconjugation activities over time and to characterize and compare the kinetic properties between human and porcine SULTs. Although vector concentrations were varied in order to obtain similar levels of protein expression, hSULT2B1a and hSULT2B1b protein were still expressed approximately 3 to 9 times more when compared to the other SULTs (Figure 4.3). Therefore, protein expression was normalized to a standard sample via Western blot analysis. Characteristic HPLC chromatographs of steroid metabolites produced from incubations with androstenone and DHEA are shown in Figure 4.4. Unconjugated androstenone eluted at approximately 36.4 minutes which was confirmed by a comparison to authentic steroid standard. A sulfoconjugated fraction occurred at approximately 20.9 minutes which was identified by fraction collection and solvolysis treatment. Following this treatment, the collected fraction eluted from the HPLC column at approximately the same time as unconjugated androstenone. A third peak was 68 observed at approximately 2.8 minutes however there was no change in RT following either solvolysis or β-glucuronidase treatment. Since this peak was also seen in control samples, and since the production of this peak was not significantly different (p > 0.05) when compared to controls for all enzymes, it was thought to be an artifact rather than a product of androstenone metabolism. A fourth peak was also observed at approximately 4.7 minutes, however its appearance was not consistent. In addition, fraction collection and either solvolysis or β-glucuronidase treatment did not result in an RT change, suggesting that this peak may have been an artifact as well. DHEA had a retention time of approximately 11.2 minutes while DHEAS eluted at approximately 2.7 minutes. This was confirmed by HPLC analysis of authentic steroid standards. Time course data for all SULTs describing androstenone and DHEA sulfoconjugation are shown in Figure 4.5. The production of androstenone sulfonate increased over time for all enzymes with the exception of pSULT2B1. Conversion was less than 0.3% across all time points for this enzyme and was only significantly different (p < 0.05) when compared to control samples at 8h. Conversion of androstenone reached a maximum of 12.3% ± 1.8 after 24 hours by hSULT2A1 activity. Similar results were found for incubations with DHEA where the production of DHEAS increased over time for all enzymes, again with the exception of pSULT2B1. Conversion was less than 0.8% for cells transfected with pSULT2B1 expression vector across all time points but was significantly different (p < 0.05) when compared to control samples at 4h, 16h, and 24h incubation periods. The maximum conversion of DHEA was 9.8% ± 0.5 after 24 hours by hSULT2A1 activity. Since pSULT2B1 activity did not show increased sulfoconjugation production over time with either substrate, it was not included in enzyme kinetics analyses. In addition, initial rate enzyme kinetics were carried out using 69 an incubation of 24 hours, as the majority of enzymes had less than 10% conversion of substrate. Enzyme kinetics data obtained from incubations with androstenone as substrate were fit to either the Michaelis-Menten equation or the Hill equation (see Materials and Methods) (Figure 4.6 and Figure 4.7), while those obtained using DHEA as substrate showed substrate inhibition and were fit to an equation that describes partial substrate inhibition (Figure 4.8 and Figure 4.9). Corresponding kinetic parameters are shown in Table 4.3. Comparisons between human and porcine SULT2A1 showed substantially different substrate affinities for both androstenone and DHEA. However, these enzymes did show relatively similar sulfonation efficiencies for DHEA. Interestingly, the sulfonation efficiency for hSULT2A1 was approximately 5-fold higher than for pSULT2A1 when using androstenone as substrate. Vmax/Km ratios for hSULT2B1a and hSULT2B1b were significantly smaller than those of the other enzymes for both substrates suggesting that androstenone and DHEA are poor substrates for these human orthologs. Figure 4.3 Typical Western blot of human and porcine SULT proteins. Lanes are as follows: 1, protein marker; 2, pSULT2A1; 3, pSULT2B1; 4, hSULT2A1; 5, hSULT2B1a; 6, hSULT2B1b. 70 Figure 4.4 Characteristic HPLC profiles of conjugated and unconjugated steroids from HEK cells expressing SULT enzymes and incubated with radiolabelled [3H ]androstenone (A) and [1,2,6,7-3H(N)]-DHEA (B). RTs for reference standards of androstenone and DHEA were approximately 36-37 and 11-12 minutes. 71 A B Figure 4.5 Relative peak areas, as determined by HPLC analysis, of androstenone sulfonate (A) and DHEAS (B) production over time for human and porcine SULTs. Values are plotted as mean ± standard error for two independent experiments performed in triplicate (n = 2). Significant differences compared to the control (p < 0.05) were evaluated using a Student’s t-test and are denoted by (*). 72 A B Figure 4.6 The production of androstenone sulfonate by porcine SULT2A1 (A) and human SULT2A1 (B) with increasing androstenone concentrations. Values are plotted as mean ± standard error for three independent experiments performed in triplicate (n = 3). 73 A B Figure 4.7 The production of androstenone sulfonate by human SULT2B1a (A) and human SULT2B1b (B) with increasing androstenone concentrations. Values are plotted as mean ± standard error for three independent experiments performed in triplicate (n = 3). 74 A B Figure 4.8 The production of DHEAS by porcine SULT2A1 (A) and human SULT2A1 (B) with increasing androstenone concentrations. Values are plotted as mean ± standard error for three independent experiments performed in triplicate (n = 3). 75 A B Figure 4.9 The production of DHEAS by human SULT2B1a (A) and human SULT2B1b (B) with increasing androstenone concentrations. Values are plotted as mean ± standard error for three independent experiments performed in triplicate (n = 3). 76 Table 4.3 Kinetic constants for porcine and human SULTs Vmax (pmol/hr/mg protein) Km (µM) Vmax/Km Ki (µM) pSULT2A1 689.7 ± 92.2 74.1 ± 15.9 9.3 ND hSULT2A1 276.4 ± 52.5 5.8 ± 0.6 47.7 ND hSULT2B1a 22.5 ± 5.0 26.8 ± 13.2 0.8 ND hSULT2B1b 29.3 ± 6.5 46.9 ± 17.9 0.6 ND pSULT2A1 240.4 ± 8.2 3.3 ± 1.9 72.9 23 hSULT2A1 475.1 ± 15.3 9.4 ± 2.5 50.5 11 hSULT2B1a 18.8 ± 1.1 3.6 ± 3.0 5.2 29 hSULT2B1b 13.7 ± 0.8 2.4 ± 4.6 5.7 16 Androstenone DHEA Note. The kinetic constants reported in this table were derived from plots in Figure 4.6 to Figure 4.9. The data represent means ± standard errors for three independent experiments performed in triplicate (n = 3). ND, no detectable substrate inhibition. 4.5 Discussion In the present study, HEK cells were overexpressed with SULT proteins in order to determine which SULT plays a role in androstenone sulfoconjugation. The sulfoconjugation activities of porcine and human SULTs were also further characterized and compared using DHEA as a substrate. Previous studies have identified pSULT2A1 as a key enzyme involved in the sulfoconjugation of steroids (Sinclair and Squires, 2005; Sinclair et al., 2006), which is supported by results found in the present study. Porcine SULT2B1 was also suggested to be important, as hepatic expression was negatively correlated to androstenone concentration in adipose tissue (Moe et al., 2007a). However, results from the present study provide direct evidence suggesting that pSULT2B1 is not involved in the sulfoconjugation of androstenone or DHEA. Authentic steroid standards of DHEA and DHEAS were commercially available 77 and thus used to confirm and quantify sulfoconjugate production by HPLC. However, an authentic steroid standard of androstenone sulfonate is not yet commercially available. Therefore, fraction collection from HPLC and solvolysis treatment were used to identify sulfoconjugate metabolites. The sulfoconjugated metabolite peak that eluted at approximately 20.9 minutes in the current study was previously shown to include androstenol sulfonate and 3-keto-sulfoxy androstenone in incubations with porcine Leydig cells (Chapter 3). These metabolites were identified by solvolysis treatment as well as by LC-MS analysis. In the current study, LC-MS was not used for metabolite analysis so it is unclear whether the sulfoconjugated peak was only androstenol sulfonate, only 3-keto-sulfoxy androstenone, a mixture of both, or another form of sulfoconjugated androstenone. Previous studies have shown that expression and activity of 3α- and 3βhydroxysteroid dehydrogenase in HEK cells is minimal (Zhang et al., 2000; Mason et al., 2004; Udhane et al., 2013), and since androstenone is first metabolized by these enzymes to produce 3α-androstenol and 3β-androstenol prior to sulfoconjugation, the production of androstenol sulfonate in the current study is unlikely. However, more studies are needed to confirm this and to identify the sulfoconjugated compound observed. Reported calculations of Vmax and Km are usually difficult to compare due to differing assay conditions, source and purity of the enzyme, and kinetic assumptions made about substrate inhibition (Liu et al., 2006). However, since sulfonation reactions for both human and porcine SULTs were carried out under identical assay conditions, a direct and straightforward comparison can be made here. Both hSULT2B1a and hSULT2B1b isoforms were not efficient at sulfoconjugating androstenone or DHEA when compared to pSULT2A1 and hSULT2A1. Previous studies have shown that hSULT2A1 preferentially sulfonated DHEA but not cholesterol, whereas hSULT2B1b 78 preferentially sulfonated cholesterol, but not DHEA (Javitt et al., 2001; Fuda et al., 2002). In addition, hSULT2B1b, but not hSULT2A1, was selectively expressed in skin where cholesterol sulfonation plays an important role in skin barrier development (Javitt et al., 2001). This suggests that SULTs have selective physiological roles (Strott, 2002). Since the Ile20 and Ile23 residues present in the amino-terminal of hSULT2B1b were found to be essential for cholesterol sulfoconjugation (Fuda et al., 2002) and are conserved in pSULT2B1, and since the full-length cDNA sequence for this latter enzyme was also generated from skin (Uenishi et al., 2007), it is suggested that pSULT2B1 may have a specific physiological role and act in a similar manner as hSULT2B1b. However, the sulfoconjugation activities of pSULT2B1 and hSULT2B1b towards androstenone and DHEA were noticeably different. One explanation for the low activity of pSULT2B1 may be related to protein stability issues. In humans, the native form of hSULT2B1b was shown to be expressed in E. coli using different expression vectors but was not catalytically active following lysis of the cells for protein isolation (Meloche and Falany, 2001; Falany and Rohn-Glowacki, 2013). The addition of a histidine (His)-tag allowed for the recovery of the active enzyme; however removal of the His-tag resulted in an unstable enzyme that was inactivated by freeze-thawing (Meloche and Falany, 2001; Falany, 2005). Consequently, it was suggested that the added His-tag stabilized the active form of hSULT2B1b or allowed for more efficient enzyme folding (Falany et al., 2006; Falany and RohnGlowacki, 2013). Although vectors containing the V5-His-tag were used in the present study, the pSULT2B1 protein may not be stabilized by this tag due to species differences in amino acid sequences. Alternatively, the low activity of pSULT2B1 may be normal 79 and this enzyme may simply not sulfoconjugate androstenone or DHEA to any significant degree. Future studies should therefore examine other possible substrates for this enzyme. In the present study, hSULT2A1 had a significantly higher binding affinity for androstenone, but a lower Vmax, and was approximately 5-fold more efficient at sulfoconjugating this substrate when compared to pSULT2A1. Androstenone has largely been investigated in pigs in relation to boar taint; however much less is known about its role in humans. In plasma, androstenone concentrations have been reported to be twice as high in boars compared with humans (Smals and Weusten, 1991). Androstenone has also been found in the axillary region in humans (Austin and Ellis, 2003) and has been suggested to act as a chemosignal that mediates social behaviour and influences mood and physiology (reviewed by Havlicek et al., 2010). This is similar to the pheromonal role of androstenone in pigs (Andresen, 1976, 2006). Therefore, results from the current study warrant further investigation into the significance of androstenone sulfoconjugation in humans. Many SULTs are known to conjugate drugs, and in some cases are required to activate them (Weinshilboum and Otterness, 1994). Since pigs share many similar anatomical and physiological characteristics with humans, similarities between human and porcine enzymes may further justify the increased use of pigs in research, particularly in preclinical toxicological testing of pharmaceuticals (Swindle et al., 2012). However, care should be taken when interpreting data from these investigations, as orthologous SULT forms in pigs and humans may have substantially different sulfoconjugation activities. In the present study, although pSULT2A1 and hSULT2A1 had relatively similar sulfoconjugation efficiencies for DHEA, pSULT2A1 had an almost 3-fold higher 80 binding affinity for this substrate when compared to hSULT2A1. In addition, previous studies have shown pronounced differences in substrate specificities toward promutagens between human and mouse SULT1B1 enzymes as well as between human and rat SULT2A1 orthologs (reviewed by Glatt, 2000). Many sulfotransferases are often inhibited by their own substrates (Reed et al., 2010; Dong et al., 2012), which was demonstrated in the present study with DHEA but not androstenone. DHEA substrate inhibition has also been shown in previous studies with hSULT2A1, hSULT2B1a, and hSULT2B1b (Falany, 1997; Geese and Raftogianis, 2001; Chang et al., 2004; Liu et al., 2006). Two major potential mechanisms for substrate inhibition in SULTs have been proposed (reviewed by Wu, 2011; Dong et al., 2012). In the first, substrate inhibition may occur due to the presence of both an inhibitory binding site and an active primary binding site within the enzyme. When substrate concentration is high, a second DHEA molecule can bind to the inhibitory binding site and affect the sulfonate transfer efficiency (Figure 4.10) (Gamage et al., 2003; Berger et al., 2011). In the second mechanism, substrate inhibition may occur via formation of a ternary deadend enzyme complex [SULT•PAP•DHEA] which is non-productive and reaction inhibitory (Wu, 2011). In many cases, substrate inhibition is thought of as an abnormality SULT ∙PAPS Km SULT∙PAPS ∙DHEA 𝑉𝑚𝑎𝑥 SULT+DHEAS+PAP Ki ∙PAPS DHEA∙SULT∙DHEA V2 SULT+DHEA+DHEAS+PAP Figure 4.10 Kinetic model for partial substrate inhibition by DHEA 81 that results from using artificially high substrate concentrations in a laboratory setting (Reed et al., 2010); however several studies have shown that it may be a biologically relevant regulatory mechanism. The substrate inhibition of DHEA, in particular, has been suggested to play an important role in steroid homeostasis (Chang et al., 2004). In summary, this study used a transfected overexpression system in order to further characterize and compare human and porcine sulfotransferase enzymes. Direct evidence was provided, for the first time, showing that pSULT2B1 was not involved in androstenone metabolism. In addition, comparisons between human and porcine SULT2A1 showed substantially different substrate affinities for androstenone and DHEA. Future studies should focus on confirming the results observed in the present study regarding the sulfoconjugation activity of pSULT2B1 as well as on the biological role of substrate inhibition in both humans and pigs. 82 CHAPTER 5: GENERAL DISCUSSION AND CONCLUSIONS 5.1 Discussion and Future Considerations The purpose of this project was to investigate the metabolism of androstenone in the liver and testes, and to further characterize the enzymes involved in androstenone sulfoconjugation. It was expected that both porcine Leydig cells and hepatocytes would sulfoconjugate androstenone through the action of SULT2A1 and SULT2B1 enzymes. Results from primary cell incubation studies showed that the Leydig cells produced steroid sulfonates, but not glucuronides, whereas the hepatocytes produced androstenol glucuronide, but not sulfonates. HPLC analysis showed that the steroid sulfonates eluted as a group at approximately 20 minutes and analysis by LC-MS identified androstenol sulfonate and a sulfated form of androstenone as the metabolites making up this conjugated group. In addition, approximately 85.7% of androstenone was metabolized into these compounds after 8 hours of incubation with Leydig cells. The high proportion of sulfoconjugates observed in the current study are similar to that found in previous studies involving 16-androstene steroids, androgens, and estrogens in pigs (Saat et al., 1974; Raeside and Renaud, 1983; Sinclair and Squires, 2005; Sinclair et al., 2005a), and are a reflection of the high levels of sulfotransferase enzymes present in Leydig cells (Hobkirk et al., 1989; Raeside and Renaud, 1983). Of particular importance was the identification of a sulfoconjugated form of androstenone. LC-MS analysis in negative ionization mode showed that this metabolite had an m/z of 367.16 which was comparable to that reported in a similar study (Desnoyer, 2011). This author suggested a corresponding structure of 3-keto-sulfoxy83 androstneone. It has generally been thought that androstenone is not capable of undergoing sulfoconjugation as it does not possess any hydroxyl groups, however androstenone sulfonate has been found in the plasma of boars (Sinclair and Squires, 2005). The production of androstenone sulfonate was suggested to occur through prior enolisation of the 3-keto group (Sinclair et al., 2005a,b, 2006). This involves the removal of an alpha hydrogen atom from a carbon that is placed on an oxygen atom to form a hydroxyl group (Toullec, 1982), which may then be acted on by sulfotransferase enzymes. The resultant 3-enol sulfonate compound, however, would have an expected mass of approximately 352.5, which was not observed in the current study. Conversely, Desnoyer (2011) suggested that, following enolisation, sulfotransferase enzymes may act on either the C2 or C4 position to produce a 3-keto-sulfoxy androstenone metabolite with a mass similar to that observed in the current study. However, the sulfotransferase enzyme responsible for conjugating 16-androstene steroids is a hydroxysteroid SULT and thus only acts on hydroxyl groups (Strott, 2002; Sinclair and Squires, 2005; Sinclair et al., 2005b ). Since enolisation of androstenone would result in a hydroxyl group at the C3 position, and not the C2 or C4 positions, the production of 3-keto-4-sulfoxy androstenone may not be possible through this enolisation mechanism. Instead, the production of the 3keto-sulfoxy androstenone compound observed in the current study is suggested to occur via the addition of a hydroxyl group at a position other than C3. Hydroxylated forms of androstenone have been shown to be produced by porcine hepatocytes (Sinclair et al., 2005b). In addition, Austin and Ellis (2003) identified 6-hydroxy androstenone as a metabolite produced by axillary microflora. Although hydroxylation of other steroids has also been shown to occur at the C6 position (Zimniak et al., 1991; Miyata et al., 1994), 84 there are multiple sites where this reaction can occur due to the various enzymes that may be involved (Waxman, 1988; Martucci and Fishman, 1993; Gilep et al., 2011; Robic et al., 2014). Previously, identification of conjugated steroids was indirect and potentially inaccurate, as isolation and derivatization were required prior to analysis (Guazzo et al., 1996; Higashi and Shimada, 2004; Penning et al., 2010). However, in the present investigation, entire samples containing both conjugated and unconjugated steroids were successfully analyzed. Unfortunately, the identification of the sulfoconjugated metabolites observed in this study could not be confirmed since authentic reference standards are not yet commercially available. Future research should therefore focus on identifying the structure of androstenone sulfonate. This could be accomplished by analyzing samples using various spectroscopy techniques such as nuclear magnetic resonance (NMR) which determines molecular structures based on the magnetic properties of atomic nuclei. In addition, future research should also focus on identifying the enzymes involved in androstenone hydroxylation as this may affect the level of unconjugated steroid available to accumulate in adipose tissue and will consequently play a role in the development of boar taint. In addition to the sulfoconjugated metabolites, androstenol glucuronide was also produced, although this only occurred in incubations with hepatocytes and not with Leydig cells. Unfortunately, the amount of metabolite produced could not be quantified, as it was only observed by LC-MS and not by HPLC. Although the output from LC-MS provided an abundance value, this could not be compared with those of the sulfonated steroids since this value is dependent on the ionizability of a compound which changes with size and chemical nature. Therefore, these values were not reported for any of the 85 metabolites detected. However, the observance of androstenol glucuronide by LC-MS and not HPLC suggests that the levels of this compound may have been relatively low and below the limit of detection of the HPLC. This further suggests that the viability of hepatocytes used in this study may have been compromised, as similar studies have shown relatively high levels of 3α- and 3β- androstenol glucuronides produced by porcine hepatocytes (Sinclair et al., 2005b). The production of 16-androstene glucuronides by hepatocytes, but not Leydig cells, has been shown in previous studies (Sinclair et al., 2005a,b; Chen et al., 2015) and corresponds to the theory that glucuronidation serves primarily as a mechanism to facilitate metabolic clearance (Sinclair et al., 2005b). This is further supported by the high concentrations of androgen, estrogen, and androstene glucuronides found in the urine and bile, as well as the high levels of UGT enzymes found in the liver and kidney (Gower and Patterson, 1970; Sinclair et al., 2005b). Therefore, future research should focus on the metabolism of androstenone into glucuronide conjugates as well as the enzymes involved in these processes. Recently, one study investigated gene expression patterns involved in porcine hepatic androstenone metabolism in high and low androstenone boars from three different breeds (Sahadevan et al., 2015). Based on co-expression cluster analysis, these authors showed that UGT2B17, UGT2B18-like, and UGT2B31-like were co-expressed in one of the clusters from low androstenone boars across all three breeds. The enzyme or enzymes responsible for glucuronidating the androstenols have not been identified, although members from the UGT2B family, which are known to conjugate steroids (Mackenzie et al., 1992; Turgeon et al., 2001), would be the most likely candidates. In addition, it is not known whether androstenone itself is glucuronidated in the pig although 86 this is possible and would most likely occur through prior enolisation, as described above. In humans, UGT2B17 was shown to glucuronidate both the 3- and 17-hydroxy positions of C19 steroids (Beaulieu et al., 1996; Bélanger et al., 2003) while monkey UGT2B18 was shown to act on the hydroxyl group at the 3α position of C19 steroids (Beaulieu et al., 1998). If the enzyme responsible for androstenone/androstenol glucuronidation can be identified, and if enzyme expression and activity can be correlated to low androstenone concentrations in adipose tissue and plasma, this enzyme could potentially be used as a genetic marker to select for low boar taint boars. In the second part of this study, HEK cells were transfected with plasmids containing porcine and human SULT genes. The objective here was to further characterize the sulfoconjugation activity of porcine SULTs. Previously, pSULT2A1 was identified as a key enzyme involved in the metabolism of androstenone, as its activity and expression were negatively correlated with both fat and plasma androstenone concentrations in boars (Falany et al., 1989, 1994; Sinclair and Squires, 2005; Sinclair et al., 2006). Porcine SULT2B1 expression was also shown to be negatively correlated to fat androstenone concentration, although these results were achieved indirectly since goat anti-human SULT2B1 antibodies were used to identify the presence of pSULT2B1 via Western blot (Moe et al., 2007a,b). However, results from the current study suggested that pSULT2B1 was not directly involved with androstenone metabolism as this enzyme did not appear to conjugate androstenone to any significant degree. Porcine SULT2B1 did not appear to sulfoconjugate DHEA either. Comparisons with the human orthologs, hSULT2B1a and hSULT2B1b, showed that although these enzymes were capable of sulfoconjugating androstenone and DHEA, their efficiencies were relatively low. These 87 human isoforms possess a 50 amino acid carboxy-terminal extension that makes them structurally unique when compared to other human SULTs (Falany et al., 2006, 2013). In addition, the amino-termini of hSULT2B1a and hSULT2B1a are also extended and include 8 and 23 amino acids, respectively (Falany et al., 2013). Fuda et al. (2002) showed that the hSULT2B1b isoform preferentially sulfonates cholesterol, while hSULT2B1a sulfonates pregnenolone, but not cholesterol. These authors determined that isoleucines present at positions 21 and 23 in the extended amino-terminus of hSULT2B1b were crucial for cholesterol sulfoconjugation. Interestingly, the porcine SULT2B1 enzyme also contains an extended amino-terminus which is 65% similar to that of the human ortholog and includes the Ile21 and Ile23 residues. Therefore, like hSULT2B1b, the pSULT2B1 enzyme may preferentially sulfoconjugate cholesterol. Further, since cholesterol sulfonate can be metabolized to other sulfoconjugated steroids via the steroid-sulfonate synthesis pathway (Roberts et al., 1964; Gasparini et al., 1976; Falany et al., 1995), pSULT2B1 may still play an indirect role in the biosynthesis of androstenone and, consequently, in the development of boar taint. Future studies should therefore focus on determining which substrates are capable of being sulfoconjugated by pSULT2B1. In addition, hSULT2B1 isoforms have shown distinct tissue expression patterns compared to the hSULT2A1 enzyme (reviewed by Falany and Rohn-Glowacki, 2013). Generally, hSULT2A1 is expressed in tissues that do not express hSULT2B1b and vice versa. Therefore, studies comparing pSULT2A1 and pSULT2B1 tissue expression are warranted in order to further understand the function of these enzymes and role that they play in the development of boar taint. Finally, since pSULT2B1 expression was negatively correlated to androstenone concentration in adipose tissue (Moe et al., 88 2007a,b), future studies investigating potential polymorphisms within this enzyme may be useful for developing genetic markers for boar taint. 5.2 Conclusions This research has provided direct evidence, for the first time, showing that androstenone sulfoconjugation does occur in the boar. In addition, porcine SULT2B1 did not sulfoconjugate androstenone, although it may still be indirectly involved in boar taint. The results presented in the current investigation add to the further understanding of the pathways and genes involved in the development of boar taint, and emphasize the importance of conjugation in steroid metabolism. Although a new method for controlling boar taint was not discovered here, it is hoped that this research will lead to new lines of investigation and contribute to the identification of genetic markers, allowing for the genetic selection of low boar taint boars without negative consequences. 89 LITERATURE CITED Abbaszade, I. G., J. Arensburg, C. H. Park , J. Z. Kasa-Vubu, J. Orly, and A. H. Payne. 1997. Isolation of a new mouse 3β-hydroxysteroid dehydrogenase isoform, 3βHSD VI, expressed during early pregnancy. Endocrinology. 138(4):1392 – 1399. Adams, J. B. and D. McDonald. 1981. Enzymic synthesis of steroid sulphates XIV. Properties of human adrenal steroid alcohol sulphotransferase. BBA – Lipid Lipid Met. 664(3):460 – 468. Adlercreutz, H. and F. Martin. 1980. Biliary excretion and intestinal metabolism of progesterone and estrogens in man. J. Steroid Biochem. 13(2):231 – 244. Ahmad, N. and D. B. Gower. 1968. The biosynthesis of some androst-16-enes from C21 and C19 steroids in boar testicular and adrenal tissue. Biochem. J. 108(2):233 – 241. Albrecht, A. K. 2013. Review on the consequences of using ImprovacTM in modern pig production. Vet. Sci. Dev. 3(1):e1 – e5. Andersson, K., A. Schaub, K. Andersson, K. Lundström, S. Thomke, and I. Hansson. 1997. The effects of feeding system, lysine level and gilt contact on performance, skatole levels and economy of entire male pigs. Livest. Prod. Sci. 51(1-3):131 – 140. Andresen, O. 1976. Concentrations of fat and plasma 5α -androstenone and plasma testosterone in boars selected for rate of body weight gain and thickness of back fat during growth, sexual maturation and after mating. J. Reprod. Fertil. 48(1):51 – 9. Andresen, O. 2006. Boar taint related compounds: Androstenone/skatole/other substances. Acta Vet. Scand. 48(Suppl 1):S5 – S8. 90 Annor-Frempong, I. E., G. R. Nute, F. W. Whittington, and J. D. Wood. 1997. The problem of taint in pork: 1. Detection thresholds and odour profiles of androstenone and skatole in a model system. Meat Sci. 46(1):45 – 55. Austin, C. and J. Ellis. 2003. Microbial pathways leading to steroidal malodour in the axilla. J. Steroid Biochem. 87(1):105 – 110. Austin, C., L. Emberson, and P. Nicholls. 2004. Purification and characterization of pheromaxein, the porcine steroid-binding protein. Eur. J. Biochem. 271(13):2593 – 2606. Babol, J. and E. J. Squires. 1995. Quality of meat from entire male pigs. Food Res. Int. 28(3):201 – 212. Babol, J. and E. J. Squires. 1999. Liver metabolic activities of intact male pigs injected with skatole. Can. J. Anim. Sci. 79(4):549 – 552. Babol, J., E. J. Squires, and K. Lundström. 1998. Hepatic metabolism of skatole in pigs by cytochrome P4502E1. J. Anim. Sci. 76(3):822 – 828. Barton-Gade, P. 1987. Meat and fat quality in boars, castrates and gilts. Livest. Prod. Sci. 16(2):187 – 196. Bean, K. and J. D. Henion. 1997. Direct determination of anabolic steroid conjugates in human urine by combined high-performance liquid chromatography and tandem mass spectrometry. J. Chromatogr. B. 690(1):65 – 75. Beaulieu, M., E. Lévesque, D. W. Hum, and A. Bélanger. 1996. Isolation and characterization of a novel cDNA encoding a human UDPglucuronosyltransferase active on C19 steroids. J. Biol. Chem. 271(37):22855 – 22862. 91 Beaulieu, M., E. Lévesque, O. Barbier, D. Turgeon, G. Bélanger, D. W. Hum, A. . Bélanger. 1998. Isolation and characterization of a simian UDPglucuronosyltransferase UGT2B18 active on 3-hydroxyandrogens. J. Mol. Biol. 275(5):785 – 794. Bélanger, A., D. W. Hum, M. Beaulieu, E. Lévesque, C. Guillemette, A. Tchernof, G. Bélanger, D. Turgeon, and S. Dubois. 1998. Characterization and regulation of UDP-glucuronosyltransferases in steroid target tissues. J. Steroid Biochem. 65(1):301 – 310. Bélanger, A., G. Pelletier, F. Labrie, O. Barbier, and S. Chouinard. 2003. Inactivation of androgens by UDP-glucuronosyltransferase enzymes in humans. Trends Endocrin. Met. 14(10):473 – 479. Berger, I., C. Guttman, D. Amar, R. Zarivach, and A. Aharoni. 2011. The molecular basis for the broad substrate specificity of human sulfotransferase 1A1. Plos One. 6(11):e26794 – e26803. Berkoz, B., E. P. Chavez, and C. Djerassi. 1962. Steroids. Part CLXXII. Factors controlling the direction of enol acetylation of 3-oxo-steroids. J. Chem. Soc. 1323 – 1329. Billen, M. J. and E. J. Squires. 2009. The role of porcine cytochrome b5A and cytochrome b5B in the regulation of cytochrome P45017A1 activities. J. Steroid Biochem. 113(1):98 – 104. Blanchard, R. L., R. R. Freimuth, J. Buck, R. M. Weinshilboum, and M. W. H. Coughtrie. 2004. A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily. Pharmacogenetics. 14(3):199 – 211. Bonneau, M. 1982. Compounds responsible for boar taint, with special emphasis on androstenone: A review. Livest. Prod. Sci. 9(6):687 – 705. 92 Bonneau, M. 1998. Use of entire males for pig meat in the European Union. Meat Sci. 49:S257 – S272. Booth, W. D. 1975. Changes with age in the occurrence of C19 steroids in the testis and submaxillary gland of the boar. J. Reprod. Fertil. 42(3):459 – 472. Booth, W. D. 1980a. A study of some major testicular steroids in the pig in relation to their effect on the development of male characteristics in the prepubertally castrated boar. J. Reprod. Fertil. 59(1):155 – 62. Booth, W. D. 1980b. Endocrine and exocrine factors in the reproductive behaviour of the pig. Sym. Zool. S. 45:298 – 311. Booth, W. D. 1987. Factors affecting the pheromone composition of voided boar saliva. J. Reprod. Fertil. 81(2):427 – 31. Booth, W. D. and K. I. von Glos. 1991. Pheromaxein, the pheromonal steroid-binding protein, is a major protein synthesized in porcine submaxillary salivary glands. J. Endocrinol. 128(2):205 – 212. Bowlby, M. R. 1993. Pregnenolone sulfate potentiation of N-methyl-D-aspartate receptor channels in hippocampal neurons. Mol. Pharmacol. 43(5):813 – 819. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248 – 254. Bradlow, H. L. 1970. The hydrolysis of steroid conjugates. In: S. Bernstein and S. Solomon, eds., Chemical and biological aspects of steroid conjugation. SpringerVerlag, New York, NY. p. 131 – 181. Bremner, E. A., J. D. Mainland, R. M. Khan, and N. Sobel. 2003. The prevalence of androstenone anosmia. Chem. Senses. 28(5):423 – 432. 93 Brooks, R. I. and A. M. Pearson. 1986. Steroid hormone pathways in the pig, with special emphasis on boar odor: A review. J. Anim. Sci. 62(3):632 – 645. Brophy, P. J. and D. B. Gower. 1972. 16-unsaturated C19 3-oxo steroids as metabolic intermediates in boar testis. Biochem. J. 128(4):945 – 52. Buhr, B. L., K. Zering, and D. DiPietre. 2013. A comprehensive, full chain and US meat sector economic analysis of the adoption of Improvest® by the US pork industry. Am. Assoc. Swine Vet. 189 – 194. Burchell, B. and M. W. H. Coughtrie. 1989. UDP-glucuronosyltransferases. Pharmacol. Ther. 43(2):261 – 289. Chang, H. J., R. Shi, P. Rehse, and S. X. Lin. 2004. Identifying androsterone (ADT) as a cognate substrate for human dehydroepiandrosterone sulfotransferase (DHEA-ST) important for steroid homeostasis - structure of the enzyme-ADT complex. J. Biol. Chem. 279(4):2689 – 2696. Chatman, K., T. Hollenbeck, L. Hagey, M. Vallee, R. Purdy, F. Weiss, G. Siuzdak. 1999. Nanoelectrospray mass spectrometry and precursor ion monitoring for quantitative steroid analysis and attomole sensitivity. Anal. Chem. 71(13):2358 – 2363. Chen, G., E. Bourneuf, S. Marklund, G. Zamaratskaia, A. Madej, and K. Lundström. 2007a. Gene expression of 3β-hydroxysteroid dehydrogenase and 17βhydroxysteroid dehydrogenase in relation to androstenone, testosterone, and estrone sulphate in gonadally intact male and castrated pigs. J. Anim. Sci. 85(10):2457 – 2463. 94 Chen, G., G. Zamaratskaia, H. K. Andersson, and K. Lundström. 2007b. Effects of raw potato starch and live weight on fat and plasma skatole, indole and androstenone levels measured by different methods in entire male pigs. Food Chem. 101(2):439 – 448. Chen, G., Y. Bai, L. Ren, D. Zhu, Y. Li, M. Fang, H. Al-Kateb, O. Doran. 2015. Metabolism of androstenone, 17β-estradiol and dihydrotestosterone in primary cultured pig hepatocytes and the role of 3β-hydroxysteroid dehydrogenase in this process. Plos One. 10(1):e113194 – e113200. Clarke, D. J. and B. Burchell. 1994. The uridine diphosphate glucuronsyltransferase multigene family: Function and regulation. In: F.C. Kauffman, ed., Conjugationdeconjugation reactions in drug metabolism and toxicity. Springer-Verlag, Germany. p. 3 – 43. Claus, R., U. Weiler, and A. Herzog. 1994. Physiological aspects of androstenone and skatole formation in the boar – a review with experimental data. Meat Sci. 38(2):289 – 305. Cook, B., R. H. F. Hunter, and A. S. L. Kelly. 1977. Steroid-binding proteins in follicular fluid and peripheral plasma from pigs, cows and sheep. J. Reprod. Fertil. 51(1):65 – 71. Cooke, G. M. and D. B. Gower. 1977. The submicrosomal distribution in rat and boar testis of some enzymes involved in androgen and 16-androstene biosynthesis. BBA – Gen. Subjects. 498(1):265 – 271. Corpéchot, C., P. Robel, M. Axelson, J. Sjovall, and E. E. Baulieu. 1981. Characterization and measurement of dehydroepiandrosterone sulfate in rat brain. Proc. Natl. Acad. Sci. USA. 78(8):4704 – 4707. 95 Cronin, G. M., F. R. Dunshea, K. L. Butler, I. McCauley, J. L. Barnett, and P. H. Hemsworth. 2003. The effects of immuno- and surgical-castration on the behaviour and consequently growth of group-housed, male finisher pigs. Appl. Anim. Behav. Sci. 81(2):111 – 126. Davis, S. M. and E. J. Squires. 1999. Association of cytochrome b5 with 16-androstene steroid synthesis in the testis and accumulation in the fat of male pigs. J. Anim. Sci. 77(5):1230 – 1235. Desmoulin, B., M. Bonneau, A. Frouin, and J. P. Bidard. 1982. Consumer testing of pork and processed meat from boars: The influence of fat androstenone level. Livest. Prod. Sci. 9(6):707 – 715. Desnoyer, J. E. 2011. The formation of androstenone conjugates in testis tissue from the mature boar. MSc Thesis. University of Guelph, Guelph, Ontario. Dong, D., R. Ako, and B. Wu. 2012. Crystal structures of human sulfotransferases: Insights into the mechanisms of action and substrate selectivity. Expert Opin. Drug Met. 8(6):635 – 646. Doran, E., F. M. Whittington, J. D. Wood, and J. D. McGivan. 2004. Characterisation of androstenone metabolism in pig liver microsomes. Chem. Biol. Interact. 147(2):141 – 149. Drochner, W. 1993. Digestion of carbohydrates in the pig. Archiv. Tierernahr. 43(2):95 – 116. Dunshea, F. R., C. Colantoni, K. Howard, I. Mccauley, P. Jackson, K. A. Long, S. Lopaticki, E. A. Nugent, J. A. Simons, J. Walker, and D. P. Hennessy. 2001. Vaccination of boars with a GnRH vaccine (Improvac) eliminates boar taint and increases growth performance. J. Anim. Sci. 79(10):2524 – 2535. 96 Dutton, G. J. 1978. Developmental aspects of drug conjugation, with special reference to glucuronidation. Annu. Rev. Pharmacol. Toxicol. 18:17 – 35. Ebner, T. and B. Burchell. 1993. Substrate specificities of two stably expressed human liver UDP-glucuronosyltransferases of the UGT1 gene family. Drug Metab. Dispos. 21(1):50 – 55. EC Declaration. 2010. European declaration on alternatives to surgical castration of pigs. http://ec.europa.eu/food/animal/welfare/farm/initiatives_en.htm (Accessed 18 November 2014). Einarsson, S. 2006. Vaccination against GnRH: Pros and cons. Acta Vet. Scand. 48(Suppl 1):S10 – S13. Eriksson, H. 1971. Absorption and enterohepatic circulation of neutral steroids in the rat. Eur. J. Endocrinol. 19(3):416 – 423. Falany, C. and K. J. Rohn-Glowacki. 2013. SULT2B1: Unique properties and characteristics of a hydroxysteroid sulfotransferase family. Drug Metab. Rev. 45(4):388 – 400. Falany, C. N. 1997. Enzymology of human cytosolic sulfotransferases. FASEB J. 11(4):206 – 216. Falany, C. N. 2005. Human cytosolic sulfotransferases. Properties, physiological functions, and toxicology. In: L. H. Lash, ed. Methods in pharmacology and toxicology, drug metabolism and transport: molecular methods and mechanisms. Humana Press Inc., Totowa, NJ. p. 341 – 378. Falany, C. N., D. He, N. Dumas, A. R. Frost, and J. L. Falany. 2006. Human cytosolic sulfotransferase 2B1: Isoform expression, tissue specificity and subcellular localization. J. Steroid Biochem. 102(1-5):214 – 221. 97 Falany, C. N., J. Wheeler, S. O. Tae, and J. L. Falany. 1994. Steroid sulfation by expressed human cytosolic sulfotransferases. J. Steroid Biochem. 48(4):369 – 375. Falany, C. N., K. A. Comer, T. P. Dooley, and H. Glatt. 1995. Human dehydroepiandrosterone sulfotransferase. Purification, molecular cloning, and characterization. Ann. NY. Acad. Sci. 774:59 – 72. Falany, C. N., M. E. Vazquez, and J. M. Kalb. 1989. Purification and characterization of human liver dehydroepiandrosterone sulphotransferase. Biochem. J. 260(3):641 – 646. Fietz, D., K. Bakhaus, B. Wapelhorst, G. Grosser, S. Gunther, J. Alber, B. Doring, S. Kliesch, W. Weidner, C. E. Galuska, M. F. Hartmann, S. A. Wudy, M. Bergmann, and J. Geyer. 2013. Membrane transporters for sulfated steroids in the human testis-cellular localization, expression pattern and functional analysis. Plos One. 8(5):e62638 – e62651. Fink-Gremmels, J. and H. Malekinejad. 2007. Clinical effects and biochemical mechanisms associated with exposure to the mycoestrogen zearalenone. Anim. Feed Sci. Technol. 137(3–4):326 – 341. Font-i-Furnols, M. 2012. Consumer studies on sensory acceptability of boar taint: A review. Meat Sci. 92(4):319 – 329. Font-i-Furnols, M., M. Gispert, A. Diestre, and M. A. Oliver. 2003. Acceptability of boar meat by consumers depending on their age, gender, culinary habits, and sensitivity and appreciation of androstenone odour. Meat Sci. 64(4):433 – 440. Fromm, H. J. 1975. Initial rate enzyme kinetics. Springer-Verlag, New York, NY. 98 Fuda, H., Y. C. Lee, C. Shimizu, N. B. Javitt, and C. A. Strott. 2002. Mutational analysis of human hydroxysteroid sulfotransferase SULT2B1 isoforms reveals that exon 1B of the SULT2B1 gene produces cholesterol sulfotransferase, whereas exon 1A yields pregnenolone sulfotransferase. J. Biol. Chem. 277(39):36161 – 36166. Gamage, N. U., R. G. Duggleby, A. C. Barnett, M. Tresillian, C. F. Latham, N. E. Liyou, M. E. Mcmanus, J. L. Martin. 2003. Structure of a human carcinogen-converting enzyme, SULT1A1. Structural and kinetic implications of substrate inhibition. J. Biol. Chem. 278(9):7655 – 7662. García-Regueiro, J. A. and I. Diaz. 1989. Evaluation of the contribution of skatole, indole, androstenone and androstenols to boar-taint in back fat of pigs by HPLC and capillary gas chromatography (CGC). Meat Sci. 25(4):307 – 316. Gardi, R., P. P. Castelli, and A. Ercoli. 1962. Anomalous enolization of 3-keto-5αsteroids. Tetrahedron Lett. 3(11):497 – 500. Gasparini, F. J., R. B. Hochberg, and S. Lieberman. 1976. Biosynthesis of steroid sulfates by the boar testes. Biochemistry-US. 15(18):3969 – 3975. Geese, W. and R. B. Raftogianis. 2001. Biochemical characterization and tissue distribution of human SULT2B1. Biochem. Biophys. Res. Commun. 288(1):280 – 289. Geyer, J., B. Döring, K. Meerkamp, B. Ugele, N. Bakhiya, C. F. Fernandes, J. R. Godoy, H. Glatt, and E. Petzinger. 2007. Cloning and functional characterization of human sodium-dependent organic anion transporter (SLC10A6). J. Biol. Chem. 282(27):19728 – 19741. Geyer, J., T. Wilke, and E. Petzinger. 2006. The solute carrier family SLC10: More than a family of bile acid transporters regarding function and phylogenetic relationships. N-S. Arch. Pharmacol. 376(6):413 – 431. 99 Giersing, M., J. Ladewig, and B. Forkman. 2006. Animal welfare aspects of preventing boar taint. Acta Vet. Scand. 48(Suppl 1):S3 – S5. Gilep, A. A., T. A. Sushko, and S. A. Usanov. 2011. At the crossroads of steroid hormone biosynthesis: The role, substrate specificity and evolutionary development of CYP17. BBA – Proteins Proteom. 1814(1):200 – 209. Girard, C., O. Barbier, G. Veilleux, M. El-Alfy, and A. Bélanger. 2003. Human uridine diphosphate-glucuronosyltransferase UGT2B7 conjugates mineralocorticoid and glucocorticoid metabolites. Endocrinology. 144(6):2659 – 2668. Glatt, H. 2000. Sulfotransferases in the bioactivation of xenobiotics. Chem. Biol. Interact. 129(1):141 – 170. Glatt, H. and W. Meinl. 2004. Pharmacogenetics of soluble sulfotransferases (SULTs). N-S. Arch. Pharmacol. 369(1):55 – 68. Gosland, M., C. Tsuboi, T. Hoffman, S. Goodin, and M. Vore. 1993. 17β-estradiol glucuronide: An inducer of cholestasis and a physiological substrate for the multidrug resistance transporter. Cancer Res. 53(22):5382 – 5385. Gouveia, M. J., P. J. Brindley, L. L. Santos, J. M. Correia da Costa, P. Gomes, and N. Vale. 2013. Mass spectrometry techniques in the survey of steroid metabolites as potential disease biomarkers: A review. Metabolism. 62(9):1206 – 1217. Gower, D. B. 1972. 16-unsaturated C19 steroids. A review of their chemistry, biochemistry and possible physiological role. J. Steroid Biochem. 3(1):45 – 103. Gower, D. B. and R. L. Patterson. 1970. Identification of C19-16-unsaturated steroids in boar urine and spermatic vein plasma by combined gas chromatography and mass spectrometry. J. Endocrinol. 47:365 – 368. 100 Gray, M. and E. J. Squires. 2013. Effects of nuclear receptor transactivation on steroid hormone synthesis and gene expression in porcine Leydig cells. J. Steroid Biochem. 133:93 – 100. Griffiths, N. M., and R. L. S. Patterson. 1970. Human olfactory responses to 5α-androst16-en-3-one – principal component of boar taint. J. Sci. Food Agric. 21(1):4 – 6. Groh, H., K. Schade, and C. Hörhold‐schubert. 1993. Steroid metabolism with intestinal microorganisms. J. Basic Microbiol. 33(1):59 – 72. Guazzo, E. P., P. J. Kirkpatrick, I. M. Goodyer, H. M. Shiers, and J. Herbert. 1996. Cortisol, dehydroepiandrosterone (DHEA), and DHEA sulfate in the cerebrospinal fluid of man: Relation to blood levels and the effects of age. J. Clin. Endocrinol. Metab. 81(11):3951 – 3960. Guillemette, C., D. W. Hum, and A. Bélanger. 1996. Evidence for a role of glucuronosyltransferase in the regulation of androgen action in the human prostatic cancer cell line LNCaP. J. Steroid Biochem. 57(3–4):225 – 231. Gunn, M. G., P. Allen, M. Bonneau, D. V. Byrne, S. Cinotti, B. Fredriksen, L. L. Hansen, A. H. Karlsson, M. G. Linder, K. Lundström, D. B. Morton, A. Prunier, J. Squires, F. Tuyttes, A. V. Calvo, E. H. von Vorell, and J. Wood. 2004. Welfare aspects of the castration of piglets. Scientific report on the scientific panel for animal health and welfare on a request from the commission related to welfare aspects of the castration of piglets. The EFSA Journal. 91:1 – 18. Hagenbuch, B. and P. J. Meier. 2003. The superfamily of organic anion transporting polypeptides. BBA-Biomembranes. 1609(1):1 – 18. Harteneck, C. 2013. Pregnenolone sulfate: From steroid metabolite to TRP channel ligand. Molecules. 18(10):12012 – 12028. 101 Havlicek, J., A. K. Murray, T. K. Saxton, and S. C. Roberts. 2010. Current issues in the study of androstenes in human chemosignalling. In: G. Litwack, ed., Vitamins and hormones. Academic Press, London. p. 47 – 81. Hay, M., A. Vulin, S. Génin, P. Sales, and A. Prunier. 2003. Assessment of pain induced by castration in piglets: Behavioral and physiological responses over the subsequent 5 days. Appl. Anim. Behav. Sci. 82(3):201 – 218. Hébert, P. and G. M. Cooke. 1992. Kinetic evidence for separate 3β-hydroxysteroid dehydrogenase-isomerases in androgen and 16-androstene biosynthetic pathways in the pig testis. J. Steroid Biochem. 42(8):901 – 910. Her, C., T. C. Wood, E. E. Eichler, H. W. Mohrenweiser, L. S. Ramagli, M. J. Siciliano, and R. M. Weinshilboum. 1998. Human hydroxysteroid sulfotransferase SULT2B1: Two enzymes encoded by a single chromosome 19 gene. Genomics. 53(3):284 – 295. Higashi, T. and K. Shimada. 2004. Derivatization of neutral steroids to enhance their detection characteristics in liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 378(4):875 – 882. Hobkirk, R. 1985. Steroid sulfotransferases and steroid sulfate sulfatases: Characteristics and biological roles. Can. J. Biochem. Cell B. 63(11):1127 – 1144. Hobkirk, R. 1993. Steroid sulfation: Current concepts. Trends Endocrin. Met. 4(2):69 – 74. Hobkirk, R., R. Renaud, and J. I. Raeside. 1989. Partial characterization of steroid sulfohydrolase and steroid sulfotransferase activities in purified porcine Leydig cells. J. Steroid Biochem. 32(3):387 – 392. 102 Hochberg, R. B., S. Ladany, M. Welch, and S. Lieberman. 1974. Cholesterol and cholesterol sulfate as substrates for the adrenal side-chain cleavage enzyme. Biochemistry-US. 13(9):1938 – 1945. Houston, J. B. and K. E. Kenworthy. 2000. In vitro-in vivo scaling of CYP kinetic data not consistent with the classical michaelis-menten model. Drug Metab. Dispos. 28(3):246 – 254. Hu, G. X., B. H. Zhao, Y. H. Chu, H. Y. Zhou, B. T. Akingbemi, Z. Q. Zheng, and R. S. Ge. 2010. Effects of genistein and equol on human and rat testicular 3βhydroxysteroid dehydrogenase and 17β-hydroxysteroid dehydrogenase 3 activities. Asian J. Androl. 4:519 – 526. Huber, L., E. J. Squires, and C. F. M. de Lang. 2013. Dynamics of nitrogen retention in entire male pigs immunized against gonadotropin-releasing hormone. J. Anim. Sci. 91(10):4817 – 4825. Hughes, P. E. 1982. Factors affecting the natural attainment of puberty in the gilt. In: D. J. A. Cole and G. R. Foxcroft, eds., Control of pig reproduction. Butterworth Scientific, London. p. 117 – 138. Hum, D. W., A. Bélanger, E. Lévesque, O. Barbier, M. Beaulieu, C. Albert, M. Vallee, C. Guillemette, A. Tchernof, D. Turgeon, and S. Dubois. 1999. Characterization of UDP-glucuronosyltransferases active on steroid hormones. J. Steroid Biochem. 69(1):413 – 423. Hutzler, J. M. and T. S. Tracy. 2002. Atypical kinetic profiles in drug metabolism reactions. Drug Metab. Dispos. 30(4):355 – 362. 103 Ibrahim, A. R. S., A. M. Galal, M. S. Ahmed, and G. S. Mossa. 2003. O-Demethylation and sulfation of 7-methoxylated flavanones by Cunninghamella elegans. Chem. Pharm. Bull. 51(2):203 – 206. Igaz, P., E. Pap, A. Patócs, A. Falus, Z. Tulassay, and K. Rácz. 2002. Genomics of steroid hormones: In silico analysis of nucleotide sequence variants (polymorphisms) of the enzymes involved in the biosynthesis and metabolism of steroid hormones. J. Steroid Biochem. 82(4):359 – 367. Jaros, P., E. Burgi, K. D. C. Stark, R. Claus, D. Hennessy, and R. Thun. 2005. Effect of active immunization against GnRH on androstenone concentration, growth performance and carcass quality in intact male pigs. Livest. Prod. Sci. 92(1):31 – 38. Javitt, N. B., Y. C. Lee, C. Shimizu, H. Fuda, and C. A. Strott. 2001. Cholesterol and hydroxycholesterol sulfotransferases: Identification, distinction from dehydroepiandrosterone sulfotransferase, and differential tissue expression. Endocrinology. 142(7):2978 – 2984. Jedlitschky, G., A. J. Cassidy, M. Sales, N. Pratt, and B. Burchell. 1999. Cloning and characterization of a novel human olfactory UDP-glucuronosyltransferase. Biochem. J. 340:837 – 843. Jen, K. Y. 2009. An investigation of the efficacy of non-nutritive adsorbtion agents on decreasing androstenone levels in boars. MSc. Thesis. University of Guelph, Guelph, Ontario. Jensen, M. T., R. P. Cox, and B. B. Jensen. 1995. Microbial production of skatole in the hind gut of pigs given different diets and its relation to skatole deposition in backfat. Animal Science. 61(2):293 – 304. 104 Johnson, L. A. 1991. Sex preselection in swine: Altered sex ratios in offspring following surgical insemination of flow sorted x-and y-bearing sperm. Reprod. Domest. Anim. 26(6):309 – 314. Johnson, L. A. 2000. Sexing mammalian sperm for production of offspring: The state-ofthe-art. Anim. Reprod. Sci. 60:93 – 107. Johnson, L. A., D. Rath, J. M. Vazquez, W. M. C. Maxwell, and J. R. Dobrinsky. 2005. Preselection of sex of offspring in swine for production: Current status of the process and its application. Theriogenology. 63(2):615 – 624. Jonsson, P. and Ø. Andresen. 1979. Experience during two generation of within lines boar performance testing, using 5α-androst-16-ene-3-one (5α-androstenone) and an olfactory judgement of boar taint. Genet. Sel. Evol. 11(3):241 – 250. Jörnvall, H., B. Persson, M. Krook, S. Atrian, R. Gonzàlez-Duarte, J. Jeffery, and D. Ghosh. 1995. Short-chain dehydrogenases/reductases (SDR). Biochemistry-US. 34(18):6003 – 6013. Katkov, T. and D. B. Gower. 1970. The biosynthesis of androst-16-enes in boar testis tissue. Biochem. J. 117(3):533 – 538. Katkov, T., W. D. Booth, and D. B. Gower. 1972. The metabolism of 16-androstenes in boar salivary glands. BBA-Lipid Lipid Met. 270(4):546 – 556. Keller, A., H. Zhuang, Q. Chi, L. B. Vosshall, and H. Matsunami. 2007. Genetic variation in a human odorant receptor alters odour perception. Nature. 449(7161):468 – 472. Kershaw, E. E. and J. S. Flier. 2004. Adipose tissue as an endocrine organ. J. Clin. Endocr. Metab. 89(6):2548 – 2556. 105 Kirkwood, R. N., P. E. Hughes, and W. D. Booth. 1983. The influence of boar-related odours on puberty attainment in gilts. Anim. Sci. 36(1):131 – 136. Kohjitani, A., H. Fuda, O. Hanyu, and C. A. Strott. 2006. Cloning, characterization and tissue expression of rat SULT2B1a and SULT2B1b steroid/sterol sulfotransferase isoforms: Divergence of the rat SULT2B1 gene structure from orthologous human and mouse genes. Gene. 367:66 – 73. Krozowski, Z. 1994. The short-chain alcohol dehydrogenase superfamily: Variations on a common theme. J. Steroid Biochem. 51(3):125 – 130. Kullak-Ublick, G. A., M. G. Ismair, B. Stieger, L. Landmann, R. Huber, F. Pizzagalli, K. Fattinger, P. J. Meier, and B. Hagenbuch. 2001. Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gastroenterology. 120(2):525 – 533. Kwan, T. K., C. Orengo, and D. B. Gower. 1985. Biosynthesis of androgens and pheromonal steroids in neonatal porcine testicular preparations. FEBS Lett. 183(2):359 – 364. Lazard, D., K. Zupko, Y. Poria, P. Net, J. Lazarovits, S. Horn, M. Khen, and D. Lancet. 1991. Odorant signal termination by olfactory UDP glucuronosyl transferase. Nature. 349(6312):790 – 793. Lebeau, M. C., A. Alberga, and E. E. Baulieu. 1964. Adrenal biosynthesis of dehydroisoandrosterone sulfate. Biochem. Bioph. Res. Co. 17(5):570 – 572. Lerch, M. L., M. K. Harper, and D. J. Faulkner. 2003. Brominated polyacetylenes from the Philippines sponge Diplastrella sp. J. Nat. Prod. 66:667 – 670. 106 Leung, M. C. K., K. L. Bowley, and E. J. Squires. 2010. Examination of testicular gene expression patterns in Yorkshire pigs with high and low levels of boar taint. Anim. Biotechnol. 21(2):77 – 87. Lin, Z., Y. Lou, J. Peacock, and E. J. Squires. 2005. A novel polymorphism in the 5 ' untranslated region of the porcine cytochrome b5 (CYB5) gene is associated with decreased fat androstenone level. Mammalian Genome. 16(5):367 – 373. Liu, Y., T. I. Apak, H. J. Lehmler, L. W. Robertson, and M. W. Duffel. 2006. Hydroxylated polychlorinated biphenyls are substrates and inhibitors of human hydroxysteroid sulfotransferase SULT2A1. Chem. Res. Toxicol. 19(11):1420 – 1425. Lundström, K. and G. Zamaratskaia. 2006. Moving towards taint-free pork – alternatives to surgical castration. Acta Vet. Scand. 48(Suppl 1):S1 – S5. Lundström, K., B. Malmfors, G. Malmfors, S. Stern, H. Petersson, A. B. Mortensen, and S. E. Sørensen. 1988. Skatole, androstenone and taint in boars fed two different diets. Livest. Prod. Sci. 18(1):55 – 67. Macdonald, I. A., V. D. Bokkenheuser, J. Winter, A. M. McLernon, and E. H. Mosbach. 1983. Degradation of steroids in the human gut. J. Lipid Res. 24(6):675 – 700. Mackenzie, P. I., I. S. Owens, B. Burchell, K. W. Bock, A. Bairoch, A. Bélanger, S. F. Gigleux, M. Green, D. W. Hum, T. Iyanagi, D. Lancet, P. Louisot, J. Magadalou, C. J. Roy, J. K. Ritter, T. R. Tephly, H. Schachter, T. Tephly, K. F. Tipton, and D. W. Nebert. 1997. The UDP glycosyltransferase gene superfamily: Recommended nomenclature update based on evolutionary divergence. Pharmacogenet. Genom. 7(4):255 – 269. 107 MacKenzie, P., A. Rogers, D. J. Elliot, N. Chau, J. Hulin, J. O. Miners, and R. Meech. 2011. The novel UDP glycosyltransferase 3A2: Cloning, catalytic properties, and tissue distribution. Mol. Pharmacol. 79(3):472 – 478. Mackenzie, P., A. Rogers, J. Treloar, B. R. Jorgensen, J. O. Miners, and R. Meech. 2008. Identification of UDP glycosyltransferase 3A1 as a UDP Nacetylglucosaminyltransferase. J. Biol. Chem. 283(52):36205 – 36210. Mackenzie, P., K. W. Bock, B. Burchell, C. Guillemette, S. Ikushiro, T. Iyanagi, J. O. Miners, I. S. Owens, and D. W. Nebert. 2005. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet. Genom. 15(10):677 – 685. Mackenzie, P., L. Rodbourne, and S. Stranks. 1992. Steroid UDP glucuronosyltransferases. J. Steroid Biochem. 43(8):1099 – 1105. Makin, H. L. J. and D. J. H. Trafford. 1972. The chemistry of the steroids. Clin. Endocrinol. Metab. 1(2):333 – 360. Marchese, S., D. Pes, A. Scaloni, V. Carbone, and P. Pelosi. 1998. Lipocalins of boar salivary glands binding odours and pheromones. Eur. J. Biochem. 252(3):563 – 568. Martinez, E. A., J. M. Vazquez, J. Roca, X. Lucas, M. A. Gil, I. Parrilla, J. L. Vazquez, and B. N. Day. 2002. Minimum number of spermatozoa required for normal fertility after deep intrauterine insemination in non-sedated sows. Reproduction. 123(1):163 – 170. Martinez, E. A., J. M. Vazquez, J. Roca, X. Lucas, M. A. Gil, I. Parrilla, J. L. Vazquez, and B. N. Day. 2001. Successful non-surgical deep intrauterine insemination with small numbers of spermatozoa in sows. Reproduction. 122(2):289 – 296. 108 Martucci, C. P. and J. Fishman. 1993. P450 enzymes of estrogen metabolism. Pharmacol. Ther. 57(2):237 – 257. Marx, G., T. Horn, J. Thielebein, B. Knubel, and E. von Borell. 2003. Analysis of painrelated vocalization in young pigs. J. Sound Vibrat. 266(3):687 – 698. Mason, J. I., B. E. C. Howe, A. F. Howie, S. D. Morley, M. R. Nicol, and A. H. Payne. 2004. Promiscuous 3β-hydroxysteroid dehydrogenases: testosterone 17βhydroxysteroid dehydrogenase activities of mouse type I and VI 3βhydroxysteroid dehydrogenases. Endocr. Res. 30(4):709 – 714. Mason, J. I., D. Naville, B. W. Evans, and J. L. Thomas. 1998. Functional activity of 3βhydroxysteroid dehydrogenase/isomerase. Endocr. Res. 24(3-4):549 – 557. Mattioli, M., G. Galeati, F. Conte, and E. Seren. 1986. Effect of 5α-androst-16-en-3-one on oxytocin release in oestrous sows. Theriogenology. 25(3):399 – 403. McBride, M. W., A. J. McVie, S. M. Burridge, B. Brintnell, N. Craig, A. M. Wallace, R. H. Wilson, J. Varley, and R. G. Sutcliffe. 1999. Cloning, expression, and physical mapping of the 3β-hydroxysteroid dehydrogenase gene cluster (HSD3BP1– HSD3BP5) in human. Genomics. 61(3):277 – 284. McGlone, J. J., S. E. Curtis, and E. M. Banks. 1987. Evidence for aggression-modulating pheromones in prepubertal pigs. Behav. Neural Biol. 47:27 – 39. Meadus, W. J., J. I. Mason, and E. J. Squires. 1993. Cytochrome P450c17 from porcine and bovine adrenal catalyses the formation of 5,16-androstadien-3β-ol from pregnenolone in the presence of cytochrome b5. J. Steroid Biochem. 46(5):565 – 572. Meech, R. and P. I. Mackenzie. 2010. UGT3A: Novel UDP-glycosyltransferases of the UGT superfamily. Drug Metab. Rev. 42(1):45 – 54. 109 Meech, R., N. Mubarokah, A. Shivasami, A. Rogers, P. C. Nair, D. G. Hu, R. A. Mckinnon, and P. I. Mackenzie. 2015. A novel function for UDP glycosyltransferase 8: Galactosidation of bile acids. Mol. Pharmacol. 87(3):442 – 450. Meier, P. J., U. Eckhardt, A. Schroeder, B. Hagenbuch, and B. Stieger. 1997. Substrate specificity of sinusoidal bile acid and organic anion uptake systems in rat and human liver. Hepatology. 26(6):1667 – 1677. Meloche, C. and C. N. Falany. 2001. Expression and characterization of the human 3βhydroxysteroid sulfotransferases (SULT2B1a and SULT2B1b). J. Steroid Biochem. 77(4):261 – 269. Metz, C., K. Hohl, S. Waidelich, W. Drochner, and R. Claus. 2002. Active immunization of boars against GnRH at an early age: Consequences for testicular function, boar taint accumulation and N-retention. Livest. Prod. Sci. 74(2):147 – 157. Miettinen, T. A. and E. Leskinen. 1970. Glucuronic acid pathway. In: W. H. Fishman, ed., Metabolic conjugation and metabolic hydrolysis. Academic Press,Inc., New York, NY. p. 157 – 237. Miura, R., K. Shiota, K. Noda, S. Yagi, T. Ogawa, and M. Takahashi. 1994. Molecular cloning of cDNA for rat ovarian 20α-hydroxysteroid dehydrogenase (HSD1). Biochem. J. 299(Pt 2):561 – 567. Miyata, M., K. Nagata, M. Shimada, Y. Yamazoe, and R. Kato. 1994. Structure of a gene and cDNA of a major constitutive form of testosterone 6βhydroxylase (P450/6βA) encoding CYP3A2: Comparison of the cDNA with P450PCN2. Arch. Biochem. Biophys. 314(2):351 – 359. 110 Moe, M., E. Grindflek, and O. Doran. 2007a. Expression of 3β-hydroxysteroid dehydrogenase, cytochrome P450-c17, and sulfotransferase 2B1 proteins in liver and testis of pigs of two breeds: Relationship with adipose tissue androstenone concentration. J. Anim. Sci. 85(11):2924 – 2931. Moe, M., S. Lien, T. Aasmundstad, T. H. E. Meuwissen, M. H. S. Hansen, C. Bendixen, and E. Grindflek. 2009. Association between SNPs within candidate genes and compounds related to boar taint and reproduction. BMC Genetics. 10:32 – 45. Moe, M., T. Meuwissen, S. Lien, C. Bendixen, X. Wang, L. N. Conley, I. Berget, H. Tajet, and E. Grindflek. 2007b. Gene expression profiles in testis of pigs with extreme high and low levels of androstenone. BMC Genomics. 8:405 – 420. Neunzig, J., A. Sanchez-Guijo, A. Mosa, M. F. Hartmann, J. Geyer, S. A. Wudy, and R. Bernhardt. 2014. A steroidogenic pathway for sulfonated steroids: The metabolism of pregnenolone sulfate. J. Steroid Biochem. 144:324 – 333. Nicolau-Solano, S. I. and O. Doran. 2008. Effect of testosterone, estrone sulphate and androstenone on 3β-hydroxysteroid dehydrogenase protein expression in primary cultured hepatocytes. Livest. Sci. 114(2):202 – 210. Nicolau-Solano, S. I., J. D. McGivan, F. M. Whittington, G. J. Nieuwhof, J. D. Wood, and O. Doran. 2006. Relationship between the expression of hepatic but not testicular 3β-hydroxysteroid dehydrogenase with androstenone deposition in pig adipose tissue. J. Anim. Sci. 84(10):2809 – 2817. Park-Chung, M., A. Malayev, R. H. Purdy, T. T. Gibbs, and D. H. Farb. 1999. Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 830(1):72 – 87. 111 Parkinson, A. 2001. Biotransformation of xenobiotics. In: C. D. Klaassen, ed., Casarett and Doull’s toxicology: The basic science of poisons. McGraw-Hill, New York, NY. p. 133 – 224. Patterson, J. K., X. G. Lei, and D. D. Miller. 2008. The pig as an experimental model for elucidating the mechanisms governing dietary influence on mineral absorption. Exp. Biol. Med. 233(6):651 – 664. Patterson, R. L. S. 1968. 5α-androst-16-ene-3-one: Compound responsible for taint in boar fat. J. Sci. Food Agric. 19(1):31 – 38. Pawlowski, J. E., M. Huizinga, and T. M. Penning. 1991. Cloning and sequencing of the cDNA for rat liver 3α-hydroxysteroid/dihydrodiol dehydrogenase. J. Biol. Chem. 266(14):8820 – 8825. Payne, A. H. and G. I. Youngblood. 1995. Regulation of expression of steroidogenic enzymes in Leydig cells. Biol. Reprod. 52:217 – 225. Pearce, G. P., P. E. Hughes, and W. D. Booth. 1988. The involvement of boar submaxillary salivary gland secretions in boar-induced precocious puberty attainment in the gilt. Anim. Reprod. Sci. 16:125 – 134. Penning, T. M. 1997. Molecular endocrinology of hydroxysteroid dehydrogenases. Endocr. Rev. 18(3):281 – 305. Penning, T. M. 2003. Hydroxysteroid dehydrogenases and pre-receptor regulation of steroid hormone action. Hum. Reprod. Update. 9(3):193 – 205. Penning, T. M., M. J. Bennett, S. Smith-Hoog, B. P. Schlegel, J. M. Jez, and M. Lewis. 1997. Structure and function of 3α-hydroxysteroid dehydrogenase. Steroids. 62(1):101 – 111. 112 Penning, T. M., S. H. Lee, Y. Jin, A. Gutierrez, and I. A. Blair. 2010. Liquid chromatography–mass spectrometry (LC–MS) of steroid hormone metabolites and its applications. J. Steroid Biochem. 121:546 – 555. Perry, G. C., R. L. S. Patterson, H. J. H. MacFie, and C. G. Stinson. 1980. Pig courtship behaviour: Pheromonal property of androstene steroids in male submaxillary secretion. Anim. Sci. 31(2):191 – 199. Petzinger, E. and J. Geyer. 2006. Drug transporters in pharmacokinetics. N-S Arch. Pharmacol. 372(6):465 – 475. Prunier, A., M. Bonneau, E. H. von Borell, S. Cinotti, M. Gunn, B. Fredriksen, M. Giersing, D. B. Morton, F. A. M. Tuyttens, and A. Velarde. 2006. A review of the welfare consequences of surgical castration in piglets and the evaluation of nonsurgical methods. Anim. Welfare. 15(3):277 – 289. Raeside, J. I. and R. L. Renaud. 1983. Estrogen and androgen production by purified Leydig cells of mature boars. Biol. Reprod. 28(3):727 – 733. Raeside, J., H. Christie, and R. Renaud. 1999. Metabolism of oestrone and oestradiol-17β to conjugated steroids by the accessory sex glands of the male pig. J. Endocrinol. 163(1):49 – 53. Rasmussen, M. K., B. Ekstrand, and G. Zamaratskaia. 2013. Regulation of 3βhydroxysteroid dehydrogenase/Δ5-Δ4 isomerase: A review. Int. J. Mol. Sci. 14(9):17926 – 17942. Rasmussen, M. K., C. Brunius, B. Ekstrand, and G. Zamaratskaia. 2012a. Expression of hepatic 3β-hydroxysteroid dehydrogenase and sulfotransferase 2A1 in entire and castrated male pigs. Mol. Biol. Rep. 39(8):7927 – 7932. 113 Rasmussen, M. K., C. Brunius, G. Zamaratskaia, and B. Ekstrand. 2012b. Feeding dried chicory root to pigs decrease androstenone accumulation in fat by increasing hepatic 3β hydroxysteroid dehydrogenase expression. J. Steroid Biochem. 130(12):90 – 95. Reed, M. C., A. Lieb, and H. F. Nijhout. 2010. The biological significance of substrate inhibition: A mechanism with diverse functions. Bioessays. 32(5):422 – 429. Rittmaster, R. S. 1993. Androgen conjugates: Physiology and clinical significance. Endocr. Rev. 14(1):121 – 132. Rizea Savu, S., L. Silvestro, A. Haag, and F. Sörgel. 1996. A confirmatory HPLCMS/MS method for ten synthetic corticosteroids in bovine urines. J. Mass Spectrom. 31(12):1351 – 1363. Robbins, P. W. and F. Lipmann. 1956. Identification of enzymatically active sulfate as adenosine-3'-phosphate-5'-phosphosulfate. J. Am. Chem. Soc. 78(11):2652 – 2653. Roberts, K. D. and S. Lieberman. 1970. The biochemistry of the 3β-hydroxy-∆5-steroid sulfates. In: S. Bernstein and S. Solomon, eds., Chemical and biological aspects of steroid conjugation. Springer-Verlag, New York, NY. p. 291 – 320. Roberts, K. D., L. Bandi, H. I. Calvin, W. D. Drucker, and S. Lieberman. 1964. Evidence that steroid sulfates serve as biosynthetic intermediates. IV. Conversion of cholesterol sulfate in vivo to urinary C19 and C21 steroidal sulfates. Biochemistry-US. 3(12):1983 – 1988. Robic, A., C. Larzul, and M. Bonneau. 2008. Genetic and metabolic aspects of androstenone and skatole deposition in pig adipose tissue: A review. Genet. Sel. Evol. 40(1):129 – 143. 114 Robic, A., T. Faraut, and A. Prunier. 2014. Pathways and genes involved in steroid hormone metabolism in male pigs: A review and update. J. Steroid Biochem. 140:44 – 55. Rose, K., G. Stapleton, K. Dott, M. P. Kieny, R. Best, M. Schwarz, D. W. Russell, I. Bjorkhem, J. Seckl, R. Lathe. 1997. Cyp7b, a novel brain cytochrome P450, catalyzes the synthesis of neurosteroids 7α -hydroxy dehydroepiandrosterone and 7α -hydroxy pregnenolone. Proc. Natl. Acad. Sci. USA. 94(10):4925 – 4930. Roy, A. B. 1970. Enzymological aspects of steroid conjugation. In: A. Bernstein and S. Solomon, eds., Chemical and biological aspects of steroid conjugation. SpringerVerlag, New York, NY. p. 74 – 130. Ruder, H. J., L. Loriaux, and M. B. Lipsett. 1972. Estrone sulfate: Production rate and metabolism in man. J. Clin. Invest. 51(4):1020 – 1033. Ruokonen, A. 1978. Steroid metabolism in testis tissue: The metabolism of pregnenolone, pregnenolone sulfate, dehydroepiandrosterone and dehydroepiandrosterone sulfate in human and boar testes in vitro. J. Steroid Biochem. 9(10):939 – 946. Ruokonen, A. and R. Vihko. 1974a. Steroid metabolism in testis tissue: Concentrations of unconjugated and sulfated neutral steroids in boar testis. J. Steroid Biochem. 5(1):33 – 38. Ruokonen, A. and Vihko. 1974b. Steroid metabolism in human and boar testis tissue. Steroid concentrations and the position of the sulfate group in steroid sulfates. Steroids. 23(1):1 – 16. Saat, Y. A., D. B. Gower, F. A. Harrison, and R. B. Heap. 1972. Studies on the biosynthesis in vivo and excretion of 16-unsaturated C19 steroids in the boar. Biochem. J. 129(3):657 – 663. 115 Saat, Y. A., D. B. Gower, F. A. Harrison, and R. B. Heap. 1974. Studies on the metabolism of 5α-androst-16-en-3-one in boar tests in vivo. Biochem. J. 144(2):347 – 352. Schaber, G., G. Wiatr, H. Wachsmuth, M. Dachtler, K. Albert, I. Gaertner, and U. Breyer-Pfaff. 2001. Isolation and identification of clozapine metabolites in patient urine. Drug Metab. Dispos. 29(6):923 – 931. Sellier, P. and M. Bonneau. 1988. Genetic relationships between fat androstenone level in males and development of male and female genital tract in pigs. J. Anim. Breed. Genet. 105(1-6):11 – 20. Sellier, P., P. Le Roy, M. N. Fouilloux, J. Gruand, and M. Bonneau. 2000. Responses to restricted index selection and genetic parameters for fat androstenone level and sexual maturity status of young boars. Livest. Prod. Sci. 63(3):265 – 274. Senafi, S. B., D. J. Clarke, and B. Burchell. 1994. Investigation of the substrate specificity of a cloned expressed human bilirubin UDP-glucuronosyltransferase: UDP-sugar specificity and involvement in steroid and xenobiotic glucuronidation. Biochem. J. 303:233 – 240. Shadevan, S., E. Tholen, C. Große-Brinkhaus, K. Schellander, D. Tesfaye, M. HofmannApitius, M. U. Cinar, A. Gunawan, M. Hölker, and C. Neuhoff. 2015. Identification of gene co-expression clusters in liver tissues from multiple porcine populations with high and low backfat androstenone phenotype. BMC Genetics. 16:21 – 38. Shihan, M., U. Kirch, and G. Scheiner-Bobis. 2013. Dehydroepiandrosterone sulfate mediates activation of transcription factors CREB and ATF-1 via a Gα11-coupled receptor in the spermatogenic cell line GC-2. BBA-Mol. Cell Res. 1833(12):3064 – 3075. 116 Shimizu, K. 1979. Metabolism of [17-2H] pregnenolone into 5-[17β-2H, 17α-18O] androstene-3β,17α-diol and other products by incubation with the microsomal fraction of boar testis under 18O2 atmosphere. BBA-Lipid Lipid Met. 575(1):37 – 45. Shipkova, M. and E. Wieland. 2005. Glucuronidation in therapeutic drug monitoring. Clin. Chim. Acta. 358(1–2):2 – 23. Sim, S. M., S. Huijghebaert, D. J. Back, and H. J. Eyssen. 1983. Gastrointestinal absorption of estrone sulfate in germfree and conventional rats. J. Steroid Biochem. 18(4):499 – 503. Sim, S.M. and D. J. Back. 1985. Intestinal absorption of oestrone, oestrone glucuronide and oestrone sulphate in the rat in situ – i. Importance of hydrolytic enzymes on conjugate absorption. J. Steroid Biochem. 22(6):781 – 788. Simard, J., J. Couet, F. Durocher, Y. Labrie, R. Sanchez, N. Breton, C. Turgeon, and F. Labrie. 1993. Structure and tissue-specific expression of a novel member of the rat 3β-hydroxysteroid dehydrogenase/delta 5-delta 4 isomerase (3β-HSD) family. The exclusive 3β-HSD gene expression in the skin. J. Biol. Chem. 268(26):19659 – 19668. Sinclair, P. A. 2004. 16-androstene steroid metabolism and its impact on the development of boar taint. PhD Thesis. University of Guelph, Guelph, Ontario. Sinclair, P. A. and E. J. Squires. 2005. Testicular sulfoconjugation of the 16-androstene steroids by hydroxysteroid sulfotransferase: Its effect on the concentrations of 5αandrostenone in plasma and fat of the mature domestic boar. J. Anim. Sci. 83(2):358 – 365. 117 Sinclair, P. A., E. J. Squires, and J. I. Raeside. 2001. Early postnatal plasma concentrations of testicular steroid hormones, pubertal development, and carcass leanness as potential indicators of boar taint in market weight intact male pigs. J. Anim. Sci. 79(7):1868 – 1876. Sinclair, P. A., E. J. Squires, J. I. Raeside, and R. Renaud. 2005a. Synthesis of free and sulphoconjugated 16-androstene steroids by the Leydig cells of the mature domestic boar. J. Steroid Biochem. 96(2):217 – 228. Sinclair, P. A., S. Hancock, W. J. Gilmore, and E. J. Squires. 2005b. Metabolism of the 16-androstene steroids in primary cultured porcine hepatocytes. J. Steroid Biochem. 96(1):79 – 87. Sinclair, P. A., W. J. Gilmore, Z. Lin, Y. Lou, and E. J. Squires. 2006. Molecular cloning and regulation of porcine SULT2A1: Relationship between SULT2A1 expression and sulfoconjugation of androstenone. J. Mol. Endocrinol. 36(2):301 – 311. Smals, A. G. H. and J. J. A. M. Weusten. 1991. 16-ene-steroids in the human testis. J. Steroid Biochem. 40(4-6):587 – 592. Soldin, S. J. and O. P. Soldin. 2009. Steroid hormone analysis by tandem mass spectrometry. Clin Chem. 55(6):1061 – 1066. Sprong, H., B. Kruithof, R. Leijendekker, J. W. Slot, G. Van Meer, and P. Van Der Sluijs. 1998. UDP-galactose: Ceramide galactosyltransferase is a class I integral membrane protein of the endoplasmic reticulum. J. Biol. Chem. 273(40):25880 – 25888. Squires, E. J. 2006. Possibilities for selection against boar taint. Acta Vet. Scand. 48(Suppl 1):S8 – S11. Squires, E. J. 2010. Applied animal endocrinology. 2nd ed. CABI. Cambridge, MA. 118 Squires, J., M. Jafarikia, F. Schenkel, S. Wyss, F. Fortin, W. Van Berkel, R. de Wolde, and B. Sullivan. 2014. Potential application of genomics to reduce boar taint levels in three Canadian swine breeds. Proc. 10th World Congr. Genet. Appl. Livest. Prod., Vancouver, BC, Canada. Stanczyk, F. Z. and N. J. Clarke. 2010. Advantages and challenges of mass spectrometry assays for steroid hormones. J. Steroid Biochem. 121(3-5):491 – 495. Stinson, C. G. and R. L. Patterson. 1972. C19-Δ16 steroids in boar sweat glands. Brit. Vet. J. 128(9):41 – 43. Strott, C. A. 1996. Steroid sulfotransferases. Endocr. Rev. 17(6):670 – 697. Strott, C. A. 2002. Sulfonation and molecular action. Endocr. Rev. 23(5):703 – 732. Strott, C. A. and Y. Higashi. 2003. Cholesterol sulfate in human physiology: What's it all about? J. Lipid Res. 44(7):1268 – 1278. Swindle, M. M., A. Makin, A. J. Herron, F. J. Clubb, and K. S. Frazier. 2012. Swine as models in biomedical research and toxicology testing. Vet Pathol. 49:344 – 356. Tajet, H., O. Andresen, and T. H. E. Meuwissen. 2006. Estimation of genetic parameters of boar taint; skatole and androstenone and their correlations with sexual maturation. Acta Vet. Scand. 48:S9 – S12. Thomae, B. A., B. W. Eckloff, R. R. Freimuth, E. D. Wieben, and R. M. Weinshilboum. 2002. Human sulfotransferase SULT2A1 pharmacogenetics: Genotype-tophenotype studies. Pharmacogenomics J. 2(1):48 – 56. Thun, R., F. Gajewski, and F. Janett. 2006. Castration in male pigs: Techniques and animal welfare issues. J. Physiol. Pharmacol. 57(Suppl 8):189 – 194. 119 Toullec, J. 1982. Enolisation of simple carbonyl compounds and related reactions. In: V. Gold and D. Bethell, eds., Advances in physical organic chemistry. Volume 18. Academic Press Inc., New York, NY. p. 1 – 71. Tuckey, R. 1990. Side-chain cleavage of cholesterol sulfate by ovarian mitochondria. J. Steroid Biochem. 37(1):121 – 127. Turgeon, D., J. S. Carrier, E. Lévesque, D. W. Hum, and A. Bélanger. 2001. Relative enzymatic activity, protein stability, and tissue distribution of human steroidmetabolizing UGT2B subfamily members. Endocrinology. 142(2):778 – 787. Udhane, S., P. Kempna, G. Hofer, P. E. Mullis, and C. E. Flück. 2013. Differential regulation of human 3β-hydroxysteroid dehydrogenase type 2 for steroid hormone biosynthesis by starvation and cyclic amp stimulation: studies in the human adrenal NCI-H295R cell model. PLoS One. 8(7):e68691 – e68705. Uenishi, H., T. Eguchi-Ogawa, H. Shinkai, N. Okumura, K. Suzuki, D. Toki, N. Hamasima, and T. Awata. 2007. PEDE (pig EST data explorer) has been expanded into pig expression data explorer, including 10147 porcine full-length cDNA sequences. Nucleic Acids Res. 35(Database Issue):D650 – D653. Van Eldere, J., J. Mertens, and H. Eyssen. 1990. Influence of intestinal bacterial desulfation on the enterohepatic circulation of dehydroepiandrosterone sulfate. J. Steroid Biochem. 36(5):451 – 456. Van Oeckel, M. J., N. Warnants, M. De Paepe, M. Casteels, and C. V. Boucqué. 1998. Effect of fibre-rich diets on the backfat skatole content of entire male pigs. Livest. Prod. Sci. 56(2):173 – 180. Vazquez, J. M., J. Roca, M. A. Gil, C. Cuello, I. Parrilla, I. Caballero, J. L. Vazquez, and E. A. Martinez. 2008. Low-dose insemination in pigs: Problems and possibilities. Reprod. Domest. Anim. 43:347 – 354. 120 Von Teichman, A., H. Joerg, P. Werner, B. Brenig, and G. Stranzinger. 2001. cDNA cloning and physical mapping of porcine 3β-hydroxysteroid dehydrogenase/Δ5Δ4 isomerase. Anim. Genet. 32(5):298 – 302. Wang, J. F., Y. H. Zhu, D. F. Li, M. Wang, and B. B. Jensen. 2004. Effect of type and level of dietary fibre and starch on ileal and faecal microbial activity and shortchain fatty acid concentrations in growing pigs. Anim. Sci. 78:109 – 117. Warriss, P. and S. N. Brown. 1985. The physiological responses to fighting in pigs and the consequences for meat quality. J. Sci. Food Agric. 36(2):87 – 92. Waxman, D. 1988. Interactions of hepatic cytochromes P-450 with steroid hormones: Regioselectivity and stereospecificity of steroid metabolism and hormonal regulation of rat P-450 enzyme expression. Biochem. Pharmacol. 37(1):71 – 84. Weiler, U., K. Fischer, H. Kemmer, A. Dobrowolski, and R. Claus. 1997. Influence of androstenone sensitivity on consumer reactions to boar meat. In: M. Bonneau, K. Lundström, and B. Malmfors, eds., Boar taint in entire male pigs. EAAP Publication No. 92. Wageningen Pers, Wageningen, Neatherlands. p. 147 – 151. Weiler, U., M. Font i Furnols, K. Fischer, H. Kemmer, M. A. Oliver, M. Gispert, A. Dobrowolski, and R. Claus. 2000. Influence of differences in sensitivity of Spanish and German consumers to perceive androstenone on the acceptance of boar meat differing in skatole and androstenone concentrations. Meat Sci. 54(3):297 – 304. Weinshilboum, R. and D. Otterness. 1994. Sulfotransferase enzymes. In: F. C. Kauffman, ed., Conjugation-deconjugation reactions in drug metabolism and toxicity. 112th ed. Springer-Verlag, Germany. p. 45 – 78. 121 Weinshilboum, R. M., D. M. Otterness, I. A. Aksoy, T. C. Wood, C. Her, and R. B. Raftogianis. 1997. Sulfation and sulfotransferases 1: Sulfotransferase molecular biology: CDNAs and genes. FASEB Journal. 11(1):3 – 14. Whittington, F. M., G. R. Nute, S. I. Hughes, J. D. McGivan, I. J. Lean, J. D. Wood, and E. Doran. 2004. Relationships between skatole and androstenone accumulation, and cytochrome P4502E1 expression in meishan × large white pigs. Meat Sci. 67(4):569 – 576. Willeke, H., R. Claus, E. Müller, F. Pirchner, and H. Karg. 1987. Selection for high and low level of 5α‐androst‐16‐en‐3‐one in boars. I. Direct and correlated response of endorcinological traits. J. Anim. Breed Genet. 104(1-5):64 – 73. Willeke, H., R. Claus, F. Pirchner, and W. Alsing. 1980. A selection experiment against 5α-androst-16-en-3-one, the boar taint steroid, in adipose tissue of boars. Z. Tierz. Zuchtungsbio. 97(1-4):86 – 94. Wood, J. D., P. J. Buxton, F. W. Whittington, and M. Enser. 1986. The chemical composition of fat tissues in the pig: Effects of castration and feeding treatment. Livest. Prod. Sci. 15(1):73 – 82. Wu, B. 2011. Substrate inhibition kinetics in drug metabolism reactions. Drug Metab. Rev. 43(4):440 – 456. Wysocki, C. J. and K. Beauchamp. 1984. Ability to smell androstenone is genetically determined. Proc. Natl. Acad. Sci. USA. 81(15):4899 – 4902. Xue, J., G. D. Dial, E. E. Holton, Z. Vickers, E. J. Squires, Y. Lou, D. Godbout, and N. Morel. 1996. Breed differences in boar taint: Relationship between tissue levels of boar taint compounds and sensory analysis of taint. J. Anim. Sci. 74(9):2170 – 2177. 122 Yi, L., J. Dratter, C. Wang, J. A. Tunge, and H. Desaire. 2006. Identification of sulfation sites of metabolites and prediction of the compounds’ biological effects. Anal. Bioanal. Chem. 386:666 – 674. Zamaratskaia, G. and E. J. Squires. 2009. Biochemical, nutritional and genetic effects on boar taint in entire male pigs. Animal. 3(11):1508 – 1521. Zamaratskaia, G., E. Dahl, A. Madej, E. J. Squires, and O. Andresen. 2008a. Studies on 5α-androst-16-en-3-one binding to porcine serum, plasma and testicular cytosolic fraction and to human serum. J. Steroid Biochem. 111(1-2):24 – 28. Zamaratskaia, G., H. K. Andersson, G. Chen, K. Andersson, A. Madej, and K. Lundström. 2008b. Effect of a gonadotropin-releasing hormone vaccine (ImprovacTM) on steroid hormones, boar taint compounds and performance in entire male pigs. Reprod. Domest. Anim. 43(3):351 – 359. Zamaratskaia, G., J. Babol, H. K. Andersson, K. Andersson, and K. Lundström. 2005. Effect of live weight and dietary supplement of raw potato starch on the levels of skatole, androstenone, testosterone and oestrone sulphate in entire male pigs. Livest. Prod. Sci. 93(3):235 – 243. Zamaratskaia, G., W. J. Gilmore, K. Lundström, and E. J. Squires. 2007. Effect of testicular steroids on catalytic activities of cytochrome P450 enzymes in porcine liver microsomes. Food Chem. Toxicol. 45(4):676 – 681. Zhang, H., O. Varlamova, F. M. Vargas, C. N. Falany, T. S. Leyh, and O. Varmalova. 1998. Sulfuryl transfer: The catalytic mechanism of human estrogen sulfotransferase. J. Biol. Chem. 273(18):10888 – 10892. Zhang, Y., I. Dufort, P. Rheault, and V. Luu-The. 2000. Characterization of a human 20α-hydroxysteroid dehydrogenase. J. Mol. Endocrinol. 25:221 – 228. 123 Zimniak, P., E. J. Holsztynska, A. Radominska, M. Iscan, R. Lester, and D. J. Waxman. 1991. Distinct forms of cytochrome P-450 are responsible for 6β-hydroxylation of bile acids and of neutral steroids. Biochem. J. 275(Pt 1):105 – 111. 124






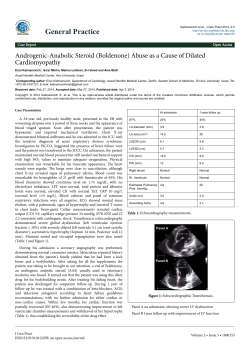


![” ⊙ Prohibited Substances [1] Anabolic-Androgenic Steroids](http://cdn1.abcdocz.com/store/data/000006330_2-6488874179c020189ebf4ca4aba01440-250x500.png)
© Copyright 2026