
Generation and Application of Recombinant Antibody Fragments for Prostate Cancer Detection
Generation and Application of Recombinant Antibody Fragments for Prostate Cancer Detection A thesis submitted for the degree of Ph.D. By Conor Hayes B.Sc. (Hons) Based on research carried out at School of Biotechnology, Dublin City University, Dublin 9, Ireland. Under the supervision of Professor Richard O’Kennedy. Declaration I hereby certify that this material, which I now submit for assessment on the programme of study leading to the award of Doctor of Philosophy is entirely my own work, that I have exercised reasonable care to ensure that the work is original, and does not to the best of my knowledge breach any law of copyright, and has not been taken from the work of others save and to the extent that such work has been cited and acknowledged within the text of my work. Signed: _______________ ID No.: Date: _________________ ii Acknowledgements iii Table of contents Declaration ........................................................................................................................ ii Acknowledgements ..........................................................................................................iii Abbreviations .................................................................................................................... x Units ...............................................................................................................................xiii Publications and Poster Presentations ............................................................................ xiv 1.0 Abstract .................................................................................................................... xvi Chapter 1 ......................................................................................................................... 1 Introduction ...................................................................................................................... 1 1.1 Section overview ......................................................................................................... 2 1.2 History of PCa ............................................................................................................. 2 1.2.1 Background to PCa .................................................................................................. 3 1.2.2 Environmental factors .............................................................................................. 4 1.2.3 PSA ........................................................................................................................... 5 1.2.4 PSA and its isoforms .............................................................................................. 10 1.2.5 Other potential markers of PCa ............................................................................. 14 1.3 Introduction to antibodies and immunity .................................................................. 16 1.3.1 Antibodies and their structure................................................................................ 16 1.3.2 Antibody classes ..................................................................................................... 17 1.3.3 Innate and acquired Immunity ............................................................................... 18 1.4 Antibody Production ................................................................................................. 20 1.4.1 Hybridoma technology ........................................................................................... 20 1.4.2 Recombinant antibodies ......................................................................................... 21 1.4.3 Phage display ......................................................................................................... 22 1.5 Immunoassays for PCa detection .............................................................................. 28 1.5.1 Current immunoassays for PSA detection ............................................................. 29 1.5.2 History of PSA immunoassay development for clinical use ................................... 29 1.6 PSA assays for forensic applications ........................................................................ 32 1.7 Novel immunoassay formats reported in the literature ............................................. 34 1.8 Biosensor devices ...................................................................................................... 36 1.9 Thesis Outline ........................................................................................................... 41 Chapter 2 ....................................................................................................................... 42 Materials & Methods...................................................................................................... 42 iv 2.1 Materials & Methods................................................................................................. 43 2.1.1 Materials ................................................................................................................ 43 2.1.2 Culture media formulation ..................................................................................... 44 2.1.3 Buffer compositions................................................................................................ 44 2.1.4 Equipment list ........................................................................................................ 46 2.1.5 Bacterial strains used ............................................................................................. 47 2.2 Production of murine recombinant antibody fragments............................................ 48 2.2.1 Preparation of bacterial stocks .............................................................................. 48 2.2.2 Immunisation of Balb/c mice with fPSA and antibody titre determination ............ 48 2.2.3 Extraction and isolation of total RNA from the spleen of an immunised Balb/c mouse ................................................................................................................... 49 2.2.4 Reverse transcription of total RNA to cDNA ......................................................... 49 2.2.5 PCR Primers .......................................................................................................... 50 2.2.6 Components and PCR conditions for antibody variable domain genes amplification using Krebber-specific primers ................................................................ 52 2.2.7 Purification of VH and VL variable gene fragments using the Promega gel clean up Kit ..................................................................................................................... 53 2.2.8 Splice by Overlap extension PCR (SOE-PCR)....................................................... 54 2.2.9 pAK 100 vector purification using Wizard miniprep DNA purification system .... 61 2.2.10 SOE-PCR restriction using SfiI enzyme and ligation into pAK 100 vector for phage display analysis and ethanol precipitation ..................................................... 62 2.2.11 Preparation of electrocompetent E. coli cells ...................................................... 63 2.2.12 Electro-transformation of XL-1 blue E. coli cells with an scFv-containing plasmid ............................................................................................................................ 64 2.2.13 Rescue of scFv-displaying phages and precipitation of phage-displayed fragments ......................................................................................................................... 65 2.2.14 Phage precipitation using PEG/NaCl .................................................................. 65 2.2.15 Selection of antigen-binding phage scFv fragments by biopanning using immobilised antigens....................................................................................................... 66 2.2.16 Polyclonal and monoclonal phage ELISA and scFv check. ................................. 68 2.2.17 Inhibition phage ELISA ........................................................................................ 70 2.2.18 ScAb expression vectors (pMoPAC 16 & 53), plasmid preparation and gel purification ...................................................................................................................... 71 v 2.2.19 Vector and SOE-PCR product digestion .............................................................. 71 2.2.20 Sub-cloning the restricted scFv fragment into the restricted vector fragments for scAb expression ........................................................................................ 73 2.2.21 Soluble expression of scAb fragments in pMoPAC 16 and pMoPAC 53 vectors ............................................................................................................................. 74 2.3 Generation of chicken-derived anti-fPSA recombinant antibody library ................. 75 2.3.1 Removal of spleens from chickens immunised with fPSA, RNA extraction and cDNA synthesis ............................................................................................................... 75 2.3.2 PCR Primers .......................................................................................................... 75 2.3.3 Amplification of VH and VL chain genes for Fab library generation ..................... 77 2.3.4 Transformation into XL-1 Blue E. coli cells .......................................................... 80 2.4 Murine anti-fPSA scAb analysis using the Biacore 3000 instrument ....................... 82 2.4.1 Mouse monoclonal anti-human kappa light chain purification ............................. 82 2.4.2 Biacore maintenance .............................................................................................. 83 2.4.3 Preconditioning of the CM5 dextran sensor chip .................................................. 83 2.4.4 Preconcentration studies........................................................................................ 84 2.4.5 Immobilisation conditions ...................................................................................... 84 2.4.6 High throughput analysis of murine anti-fPSA scAbs using Biacore 3000 ........... 85 2.4.7 Kinetic analysis of selected clones using Biacore 3000 instrument ...................... 86 2.5 Murine anti-fPSA scAb clone sequencing ................................................................ 86 2.6 Selection of most sensitive murine anti-fPSA scAb by ELISA ................................ 86 2.7 Large-scale expression of a murine anti-fPSA scAb and subsequent purification using immobilised metal affinity chromatography (IMAC) ....................... 87 2.8 Induced scAb cell expression profile ........................................................................ 88 2.9 Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) .......... 88 2.10 Western blotting ...................................................................................................... 89 2.11 Avian clone analysis on Biacore A100 ................................................................... 89 2.12 Epitope Mapping of murine and avian anti-fPSA recombinant antibody fragments using the Biacore 3000 instrument ................................................................. 90 2.13 Biacore subtractive inhibition assay for the detection of fPSA using the purified murine anti-fPSA scAb ..................................................................................... 91 2.14 Murine anti-fPSA scAb inhibition ELISA .............................................................. 92 vi 2.15 Biacore sandwich assay using commercial anti-tPSA mAb (CanAg PSA 36 mAb) ............................................................................................................................... 92 2.16 Biotinylation of purified murine anti-fPSA scAb for sandwich assay development .................................................................................................................... 93 2.17 ELISA-based assay for detection of biotinylated murine anti-fPSA scAb B5 ....... 96 2.18 Sandwich assay development for improved assay sensitivity................................. 96 2.19 Biacore sandwich assay using a neutrAvidin-immobilised CM5 dextran surface ............................................................................................................................. 97 2.20 ELISA sandwich assay using a neutravidin-coated ELISA plate ........................... 97 2.21 Neutravidin ELISA plate coating concentration optimisation for improved sandwich assay performance ........................................................................................... 98 2.22 TMB incubation time optimisation ......................................................................... 98 2.23 Inter-day and intra-day studies on sandwich assay for precision and accuracy determination................................................................................................................... 98 2.24 Determination of the limit of detection (LOD) for fPSA in samples ...................... 99 2.25 Sandwich assay in spiked female serum ................................................................. 99 2.26 Patient serum sample analysis ................................................................................. 99 2.27 Immunohistochemical analysis ............................................................................. 100 2.27.1 Sample preparation for murine and avian anti-fPSA analysis (by hand) .......... 100 2.27.2 Immunohistochemistry using avian and murine anti-fPSA recombinant antibody fragments ........................................................................................................ 100 2.27.3 Protocol for immunohistochemistry for diagnostic antibody to PSA (Dako) (automated immunohistochemistry) .............................................................................. 101 Chapter 3 ..................................................................................................................... 102 The generation of recombinant antibodies to human prostate-specific antigen using avian and murine immune systems ................................................................... 102 3.1 Introduction ............................................................................................................. 103 3.2 Murine antibody library generation ........................................................................ 104 3.2.1 Mice immunisation with fPSA antigen ................................................................. 104 3.2.2 Primer selection for murine heavy and light chain gene amplification ............... 105 3.3 Isolation of RNA from murine B cells (spleen) and first-strand DNA synthesis ... 106 3.4 PCR buffer optimisation for murine variable heavy and light chain generation .... 108 3.5 Murine SOE-PCR of variable heavy and light chains ............................................ 109 vii 3.6 Selection of fPSA-specific phage-scFv particles by ‗biopanning‘ ......................... 114 3.7 Murine polyclonal phage ELISA ............................................................................ 115 3.8 Murine monoclonal phage ELISA .......................................................................... 116 3.9 Soluble expression of mouse anti-fPSA scAb fragments ....................................... 119 3.10 Differential expression profile of an anti-PSA scFv and reformatted scAb ......... 121 3.11 Selection of chicken anti-fPSA scFv fragments.................................................... 122 3.12 Optimisation of avian VH and VL chain genes amplification for Fab library construction ................................................................................................................... 123 3.13 Chicken anti-fPSA scFv selection on fPSA-coated ELISA wells ........................ 126 3.14 Soluble expression of chicken anti-fPSA scFv and analysis by ELISA ............... 129 3.15 Discussion ............................................................................................................. 132 Chapter 4 ..................................................................................................................... 135 Screening and characterisation of putative anti-fPSA scFv and Fab antibodies ..... 135 4.1 Introduction ............................................................................................................. 136 4.2 Biacore capture assay design .................................................................................. 137 4.2.1 Preconcentration studies of anti-human C kappa mAb ....................................... 137 4.2.2 Immobilisation of anti-human C kappa mAb on to the CM5 dextran sensor chip surface ................................................................................................................... 137 4.2.3 Designed custom kinetic wizard ........................................................................... 142 4.2.4 Alignment of scAb clones from murine anti-fPSA library.................................... 148 4.2.5 Protein expression, extraction, purification and analysis ................................... 150 4.2.6 Time course expression profile analysis for scAb expression .............................. 153 4.3 Avian clone analysis ............................................................................................... 156 4.3.1 Analysis of the avian anti-fPSA Fab and scFv fragments on the Biacore A100 instrument ............................................................................................................ 156 4.4 Determination of antibody pairs for sandwich assay development ........................ 165 4.5 Discussion ............................................................................................................... 167 Chapter 5 ..................................................................................................................... 172 Assay development and immunohistochemistry studies for fPSA detection .............. 172 5.1 Introduction ............................................................................................................. 173 5.2 Development of a subtractive inhibition Biacore assay for fPSA ........................... 175 5.3 Murine anti-fPSA inhibition ELISA assay with inter-day and intra-day studies .... 175 5.4 Biotinylation of the purified murine anti-fPSA scAb and assay development ........ 176 viii 5.5 Discussion ............................................................................................................... 188 Chapter 6 ..................................................................................................................... 192 Overall Conclusions ..................................................................................................... 192 6.1 Overall conclusions ................................................................................................. 193 Chapter 7 ..................................................................................................................... 196 Bibliography ................................................................................................................. 196 ix Abbreviations Ab – antibody Abs – absorbance AP – alkaline phosphatase bp – base pair BSA – bovine serum albumin BPH – benign prostatic hyperplasia cDNA – complementary deoxyribonucleic acid CDR – complementary determining region CH1 – constant heavy chain 1 CH2 – constant heavy chain 2 CH3 – constant heavy chain 3 CL – constant light chain CM5 – carboxymethylated dextran cfu – colony forming units DMSO – Dimethyl sulfoxide DNA – deoxyribonucleic acid dNTP – Deoxyribonucleic triphosphate E. coli – Escherichia coli EDC – N-ethyl-N'-(dimethylamiopropyl) carbodiimide EDTA – ethylenediaminetetra-acetic acid ELISA – enzyme-linked immunosorbent assay Fv – variable fragment Fab – antibody binding fragment FDA – Food and Drug Administration FCA – Freund‘s Complete Adjuvant FR – framework region fPSA – free prostate-specific antigen HA – haemagglutinin HBS – HEPES buffered saline HV – hypervariable hK11 – human kallikrein 11 x HRP – horseradish peroxidase I – iodine Ig – immunoglobulin IMAC – immobilised metal affinity chromatography IMS – industrial methylated spirits IPTG – isopropyl-β-D-galactopyranoside LAST – Laboratory animal science and training mAb – monoclonal antibody MOPS – (3 (N-Morpholino) propanesulfonic acid mRNA – messenger RNA MW – molecular weight NHS – N-hydroxysuccinimide NTA – nitrilotriacetic acid OD – optical density PAGE – polyacrylamide gel electrophoresis PBS – phosphate buffered saline PBST – phosphate buffered saline tween Pan - biopanning PCa – prostate cancer PCR – Polymerase chain reaction PEG – polyethylene glycol POCT – point-of-care testing pNPP – p-Nitrophenyl phosphate PSA – prostate-specific antigen RNA – ribonucleic acid RI – refractive index RPM – revolutions per minute scAb – single chain antibody scFv – single chain variable fragment SDS – sodium dodecyl sulphate SEER – Surveillance Epidemiology and End Results SERS – surface enhanced raman scattering SOC – super optimal catabolities xi SOE – splice by overlap extension SPFS – surface plasmon field-enhanced fluorescence spectroscopy SPR – surface plasmon resonance tPSA – total prostate-specific antigen TMB – Tetramethylbenzidine dihydrochloride TYE – Tryptone yeast extract medium T – Tween VH – variable heavy chain VL – variable light chain xii Units µg – microgram (k)Da – (kilo) Dalton µL – microlitre o C – degrees Celcius cm – centimetre g – gram h –hour kg – kilogram L – litre m – metre M – molar mg – milligram min – minute mL – millilitre mm – millimetre nM – nanomolar pg – picogram rpm – revolutions per minute RU – response unit sec – second or s v/v – volume per unit volume x ‗g‘ – centrifugal acceleration w/v – weight per unit volume xiii Publications and Poster Presentations (i) A review ‗Biosensor developments: application to prostate-specific antigen detection‘. D. A. Healy, C. J. Hayes, P. Leonard, L. McKenna and R. O‘Kennedy (2007). Trends in Biotech., 3:125-131. (ii) Leonard, P., Säfsten, P., Hearty, S., Butler, J., Jacobson, K. Hayes, C., and O‘Kennedy, R. (2006). High throughput screening of recombinant antibodies. Invited oral presentation at the Biacore UK Protein Interaction User Days, September 26th -27th, Hinxton Hall, Cambridge, UK. (iii) Finlay, W.J.J., Hayes, C., Leonard, P. and O‘Kennedy, R. (2006). Cloning and analysis of specific Fab and scFv fragments for the recognition of diagnostic biomarkers. Cambridge Healthcare Insititute‘s Third Annual Monoclonal Antibodies Conference, Protein Engineering Summit, Boston, M.A., USA, April 24-28th. (iv) Hayes, C., Leonard, P., Le Berre, M., Hayhurst, A., McKenna, L. and O'Kennedy, R. (2007). Engineering Antibody Fragments for Forensic Applications. Accepted poster presentation at the 6th International Congress on Recombinant Antibodies, June 26th28th, Hotel Palace Berlin, Germany. (v) Hayes, C., Leonard, P., Le Berre, M., Hayhurst, A., McKenna, L. and O'Kennedy, R. (2007). Engineering Antibody Fragments for Detection of Prostate Specific Antigen for Forensic Applications. Accepted poster presentation at the Irish Society for Immunology. September 13th-14th. School of Nursing, Dublin City University, Glasnevin, Dublin, Ireland. (vi) Hayes, C., Leonard, P., Le Berre, M., Hayhurst, A., McKenna, L. and O'Kennedy, R. (2007). Engineering Prostate Specific Antigen Antibody Fragments for Forensic Applications. Accepted poster presentation at the Europtrode IX Conference. March 30th-April 2nd. The Crowne Plaza, Santry, Dublin, Ireland. (vii) R. O‘Kennedy, C. Hayes, P. Leonard. Antibodies and Sensors: Technologies and Future Vision for Prostate Cancer Detection. Oral presentation (2007). Prostate Cancer Research Consortium Research Meeting, April 27th. St. James Hospital, Dublin, Ireland. (viii) R. O‘Kennedy, S. Hearty, P. Leonard, B. Byrne, A.D. Sheehan, C. Viguier, N. Gilmartin, E. Tully, S. Stapleton, S. Townsend, C. Hayes, B. McDonnell, G. Donohoe, M. Ryan, E. Darcy, and J. Fitzgerald Antibody engineering. (2006). US-Ireland R&D xiv Partnership Sensors Workshop, February 20th-21st. National Centre for Sensor Research (NCSR), Dublin City University, Glasnevin, Dublin, Ireland. xv 1.0 Abstract Prostate cancer (PCa) is the most prevalent adenocarcinoma and the second highest cause of cancer-related deaths in men. Early diagnosis is required to identify the development of PCa to reduce the risk of the disease metastasising to different regions of the body. Multiple biomarkers in serum have been identified for the diagnosis of PCa. However, prostate-specific antigen (PSA) and its isoforms remain the gold standard test for PCa detection. Many antibodies have been generated for the detection of PSA with good sensitivities and limits of detection. To ensure reliable assay performance a number of biomarkers for PCa are required to increase specificity and sensitivity thus reducing the number of false positive results upon initial examination. The overall aims of the work presented in this thesis were to generate, select and characterise antibodies to PSA, an important biomarker in PCa. The generation and selection of murine and avian recombinant antibody fragments to free PSA (fPSA) was performed. Animals were immunised, RNA extracted and cDNA synthesised. The antibody variable genes were amplified by PCR, gel-purified and cloned into a phagemid vector. Electrocompetent cells were transformed and library sizes of 2.02 x 106 cfu/mL and 3.10 x 108 cfu/mL were obtained for the murine and avian clones, respectively. Specific clones were isolated by ‗biopanning‘ which involved a selection process on fPSA-coated immunotubes, enrichment and affinity maturation. Five and three rounds of selection were performed on the murine and avian anti-fPSA single chain variable fragments (scFv) recombinant antibody libraries, respectively. Preliminary analysis by ELISA indicated a large panel of positive binding clones for both screened libraries with approximately 60% of the analysed clones binding to fPSA. The murine recombinant antibody fragments were reformatted and expressed as single chain antibodies (scAb). This construct greatly enhanced soluble protein expression and stability while also facilitating the rapid kinetic analysis of candidate antibodies by Biacore. A differential expression profile of an anti-fPSA scFv and reformatted scAb was performed by ELISA. Individual antibodies isolated from the ‗biopanning‘ process were then characterised. The murine anti-fPSA antibodies were further analysed using the Biacore 3000 in a xvi capture assay format. This facilitates correct affinity ranking of the selected antibodies where the antibody fragment was captured using the appropriate secondary tag antibody (anti-human constant kappa Ab) and a fixed concentration of fPSA (1 g/mL) passed over the captured antibody fragment. Ninety four antibody fragments were evaluated and ranked on their percentage complex stability values after a 5 minute dissociation period. Eleven of the ninety four were subjected to further analysis and their affinities ranged from 3.99 x 10-9 M to 3.62 x 10-10 M. They were then expressed in host Escherichia coli (E. coli) cells on a large scale and purified by immobilised metal affinity chromatography (IMAC). The cell lysates were characterised by ELISA, SDSPAGE and Western blotting. The antibodies were sequenced and analysed for variability and similarities between the coding regions of the variable domains. Inhibition ELISA-based assays with inter and intra-day analysis were performed on the purified antibodies and a reproducible and accurate assay format was demonstrated with a limit of detection of 2.6 ng/mL. The avian clones were analysed, ranked and affinities evaluated by Dr. Paul Leonard using the Biacore A100 employing the same assay format used for analysis of the murine antibody fragments. Imperative to assay design is the selection of optimal antibody candidates for improved performance. For assay development antibody pairs which bind different regions of the fPSA molecule were determined using the capture assay format on the Biacore 3000. One murine scAb, two avian Fabs and one avian scFv were shown to bind different epitopes of the fPSA molecule with no non-specific binding evident. Following the successful purification of the murine and avian antibody fragments immunohistochemistry studies were performed on PCa tissue samples. These antibodies were shown to bind to different zones of the prostate gland (epithelial and stromal regions) confirming that the antibodies were binding to different epitopes of the fPSA molecule. The purified murine anti-fPSA scAb was biotinylated and this was employed in a sandwich ELISA assay whereby the murine anti-fPSA biotinylated scAb was captured on a neutravidin-coated ELISA plate and fPSA sandwiched using the avian antibody fragments as the detection antibodies. The assay sensitivity was improved using this format with a limit of detection of 1.8 ng/mL in PBST solution. Female serum was spiked with fPSA and inter-day studies performed by sandwich ELISA. Patient serum samples with diagnosed PCa were evaluated by sandwich ELISA to determine levels of xvii fPSA in the sample and data were compared to documented tPSA levels which were comparable to the levels obtained from a commercially available sandwich assay. xviii Chapter 1 Introduction 1 1.1 Section overview The aim of this thesis was to develop a sensitive immunoassay for the detection of prostate cancer (PCa). This chapter describes the importance of early detection of PCa and the impact of the disease on individuals. Potentially useful biomarkers for early detection of PCa and current immunoassay tests available for their detection are critically reviewed. A comprehensive description of antibody production and the methods used for selecting antigen-specific antibodies is given. Sensitive detection methods such as surface-plasmon resonance (SPR) and real-time polymerase chain reaction (PCR) are also reviewed. 1.2 History of PCa PCa was first discovered in 1853 (Lytton, 2001) and was initially regarded as a very rare cancer. However, in the last 30 years an increase in our knowledge of cancer in general and PCa in particular has occurred. Today 85% of new PCa cases are diagnosed at local and regional stages and the 5 year relative PCa survival rate has increased by 20% since 1985 (Denmeade et al., 2004). The most widely used indicator to detect PCa in the general population is a measurement of serum levels of prostate-specific antigen (PSA), a serine protease produced by the prostate epithelium (Nadler et al., 1995). PSA, the most widely used marker in clinical use for detection of PCa (Klotz and Teahan, 2006) became an FDA approved biomarker in 1987. Subsequent data demonstrating that elevated levels of PSA could be used to detect PCa earlier than rectal examination led to widespread PSA testing for early disease detection (Özena and Sözen, 2006). Although raised levels of serum PSA are suggestive of PCa, diagnostic confirmation requires a transrectal prostatic needle biopsy, a costly and invasive procedure requiring prophylactic antibiotics (Roddam et al., 2005). Overall, survival rates have improved when PCa is detected at an early stage but it can metastasize and spread to other regions of the body. PCa is an indolent disease and many more men die with the disease than die from the disease but its diagnosis severely impacts the patient‘s life and contributes to anxiety and stress. 2 1.2.1 Background to PCa Adenocarcinoma of the prostate is now the most frequently diagnosed male malignancy with 1 in every 11 men developing the disease (Hayward, 2000). Each year in the UK more than 25,000 men are diagnosed with PCa and approximately 10,000 die from the disease (Zhu et al., 2006). Excluding skin cancers, PCa is the most common cancer in men in the United States and the leading cause of cancer deaths in American men who do not smoke (Özena and Sözen, 2006). In Europe, an estimated 2.6 million new cases of cancer are diagnosed each year. Prostate cancer constitutes about 11% of all cancers in Europe and accounts for 9% of all cancer deaths among the male population within the EU (Aus et al., 2005). The incidence and mortality numbers world-wide are approximately 679,000 and 221,000, respectively (Aus et al., 2005; Zhu et al., 2006). PCa is classified as an adenocarcinoma, or glandular cancer of the prostate gland. The prostate gland a walnut sized organ located in the front of the rectum and below the bladder (http://www.psa-rising.com/prostatecancer/prostate.htm). Its principle function is the production of seminal fluid that carries sperm. The majority of the seminal fluid is produced by the seminal vesicles which are located directly behind the prostate gland (http://www.mamashealth.com/organs/prostate.asp). The prostate encompasses part of the urethra, the tube that expels urine from the bladder and semen through the penis. Cancer occurs when the prostate cells mutate and start to multiply out of control. These cells may metastasize from the prostate to other parts of the body, especially the bones and lymph nodes causing pain, difficulty in urinating and other symptoms. Early PCa usually produces no symptoms. Sometimes, however, PCa does cause symptoms, often similar to those of non-malignant diseases such as benign prostatic hyperplasia (BPH). These include frequent urination, increased urination at night, difficulty starting and maintaining a steady stream of urine, blood in urine, and painful urination. PCa may also cause problems with sexual function, such as difficulty achieving erection or painful ejaculation (Miller et al., 2003). 3 1.2.2 Environmental factors The specific causes of PCa remain unknown, yet several key factors have been associated with an increasing risk of PCa. Age, diet, lifestyle, medications, race and genetic makeup have long been recognised as contributors to PCa. The primary risk factor is age. Prostate cancer is uncommon in men under 45, but becomes more common with advancing age. The average age at the time of diagnosis is 70 (Hsing and Chokkalingam, 2006). One of the most striking characteristics of PCa is the degree of geographic variation in its patterns of occurrence and progression (Klassen and Platz, 2006). Higher rates are seen in the United States and Western Europe, with peak rates among black men in the United States, while the lowest rates are observed in Asia (Hsing et al., 2000). An interesting observation is the increase risk of PCa among Asians when they emigrate to North America. Thus, the connection between environmental and lifestyle-related factors is further strengthened (Whittemore et al., 1995). The enhanced incidence of PCa is often linked with the high fat intake of Western nations over those of Asia. In the U.S. the diet is rich in animal fats and meats and poor in fruit and vegetables. Previous studies involving 51,529 men have shown that increased total fat intake, animal fat intake, and consumption of red meat are linked with an increased risk of PCa (Giovannucci et al., 1993). Dietary amounts of certain foods such as vitamins and minerals can also contribute to PCa risk. Men with higher serum levels of the short-chain -3 fatty acid linolenic acid have higher rates of PCa (Gann and Giovannucci, 2005). Other dietary factors that may increase PCa risk include low intake of vitamin E, lycopene, omega-3 fatty acids, the mineral selenium and low blood levels of vitamin D (Schulman et al., 2001). Clinical trials have provided evidence that men given tomato sauce-based pasta dishes for three weeks before radical prostatectomy had increased lycopene levels in the blood and the prostate resulting in decreased oxidative genomic damage in leukocytes and prostate cells (Chen et al., 2001). Similar results were observed where high plasma levels of the antioxidant carotenoid lycopene, resulting from high tomato intake, were associated with a reduced risk of PCa (Gann et al., 1999). 4 There are also some links between PCa and medications and medical conditions. Use of anti-inflammatory medicines such as aspirin and chlolesterol-lowering drugs known as the statins may also decrease PCa risk (Jacobs et al., 2005; Shannon et al., 2005). However, recent conflicting studies have shown there may be an increased risk of PCa development with statin use in obese men (Charnow, 2008). The use of statins in patients with high cholesterol levels can mask subclinical PCa by lowering serum PSA levels. Infection or inflammation of the prostate (prostatitis) may increase the chance for developing PCa. In particular, infections with sexually transmitted infections syphilis, chlamydia, and gonorrhea seem to increase PCa risk (Dennis et al., 2002). Increased incidence of PCa found in certain racial groups, in identical twins with PCa and in men with certain genes indicates genetic background contributes to the risk of developing PCa. In the US, PCa affects black men more than white or Hispanic men and is also more deadly in black men (Hoffman et al., 2001). Studies of twins in Scandinavia suggest that forty percent of cancer risk can be explained by inherited factors (Lichtenstein et al., 2000). While no single gene is responsible for PCa, two genes (BRCA1 and BRCA2) are important risk factors for ovarian cancer and breast cancer in women and have also been implicated in PCa (Struewing et al., 1997). 1.2.3 PSA PSA (Figure 1.1) is produced mainly by the prostate gland and its function is thought to be liquefaction of seminal fluid through enzymatic action. The active site of PSA is closely related to that of chymotrypsin (Vihinen, 1994). PSA is not a cancer-specific protein. Its presence is not unique to cancerous cells, as it is present in normal prostate tissue (Jung et al., 2000a). Its use as a marker for PCa is due to the elevated levels detectable in the serum of PCa patients. 5 Figure 1.1: The structure of horse prostate kallikrein (HPK) isolated from stallion plasma as an example of a homologue of Human PSA (PDB accession codes 1PFA). HPK was the first structure of a serine protease purified from seminal plasma and shares extensive sequence homology with human PSA, including the predicted chymotrypsin-like specificity (Carvalho et al., 2002). Image accessed using Protein Data Bank website (http://www.rcsb.org/pdb/home/home.do). The catalytic triad residues (His57, Asp102 and Ser195) within the active site are shown. Under normal conditions, i.e. in non-cancerous tissue, PSA synthesised by the epithelial cells of the prostate gland is secreted into the seminal fluid. PSA can only enter the bloodstream by leaking into the extracellular fluid and diffusing into veins and capillaries. However, in cancerous prostatic tissue, tumour growth disrupts the basal cell layer and cell polarity is altered (Figure 1.2). The direction of PSA secretion is disturbed and PSA is released freely into the circulatory system. 6 Figure 1.2: The prostate is a walnut-sized gland located in front of the rectum and below the bladder. PCa occurs when the cells mutate and begin to multiply out of control and the structure orientation of the gland is breached. This diagram was obtained from http://www.fitness-health.co.uk/prostate-cancer.htm. This explains the elevated PSA levels in the serum of PCa patients (Stenman et al., 1999). Even though PCa cells produce less PSA than non-cancerous cells, more leaks into the serum and, therefore, elevated serum PSA is detected (Jung et al., 2000b). At present, PSA remains one of the best studied PCa-associated markers. The discovery of different forms of PSA and the differences in their relative ratios in PCa patients has refocused research on PSA. However, the practical application of PSA in cancer detection has proved problematic. One major factor contributing to this stems from the fact that PSA is not a truly cancer-specific marker (Levesque et al., 1995). Various other benign disorders have been shown to increase serum PSA levels, and the potential for false positive results has diminished the reliability of PSA assays. One major disorder capable of increasing the level of PSA in serum is benign prostatic hyperplasia (BPH Anderson, 2002). BPH is a non-malignant growth of the prostate 7 gland, characterised by similar distortion of the basal cell layer of the prostate gland and free release of PSA into blood circulation, as previously described for PCa. Current commercially available assays can often fail to distinguish PCa from such benign disorders and, as a result, much research is underway to enhance specificity of PSA assays so as to eliminate the high percentage of false positives. The discovery of various forms of PSA in serum, including the presence of complexes, has provided a new angle on the protein‘s role and significance as a disease marker. In forensics PSA is an acceptable marker in suspected rape criminal cases even in individuals with vasectomises. The marker can be detected in vaginal swabs or extracted from clothing fabric of cadavers. The presence of PSA in the sample can implicate an individual in a specific crime and this may be confirmed by DNA analysis. PSA is a 34 kilo Daltons (kDa) glycoprotein that consists of 237 amino acids, and is found in the serum in several isoforms (Table 1.1). It is a member of the kallikrein (KLK) family which can be divided into 2 groups, plasma and tissue kallikreins. The role of plasma kallikrein in the body includes activation of blood clotting, kinin generation and inflammation. The tissue kallikreins, otherwise known as glandular kallikreins, were discovered initially in urine in 1928 by Frey and Kraut (Garatinni and Shore, 1966). The gene family, located on chromosome 19q13.2-q13.4 (Mahabeer and Bhoola, 2000), are a subgroup of the serine protease family of proteolytic enzymes. Fifteen human tissue kallikrein members have been identified and these proteins are intensely studied because of their association with cancers. 8 Table 1.1: Forms of PSA found in serum PSA form Description Relevance to PSA Reference detection Free PSA Complete PSA protein Levels of PSA increase Kuriyama et al. that has no in the serum of prostate (1980); modifications cancer patients Stowell et al. (1991); Khosravi et al. (1995); Gregorakis et al. (2005). Complexed PSA PSA that has formed Levels of complexed Matsumoto et al. complexes with various PSA decrease in the (1999); Matsumoto serum proteins e.g. 1- serum of prostate cancer et al. (2000); chymotrypsin and 2- patients, and the ratio of Stephan et al. macroglobulin and is free to complexed forms (2007). inactive of PSA can discriminate prostate cancer from benign disorders ProPSA Precursor forms of PSA In serum studies of men Peter et al. (2001); that contains 7, 5, 4 and with PCa, proPSA was Catalona et al. 2 extra amino acids at shown to improve (2003); the start of the protein specificity for PCa Kumar et al. detection. (1997); Mikolajczyk et al. (2001). Benign PSA BPSA contains 2 BPSA concentrations Linton et al. (BPSA) internal peptide bond are elevated in benign (2003); cleavages at lysines 145 prostatic disease Stangelberger et al. and 182 (2007). Intact non-native A non-native form of Serum inPSA levels Nurmikko et al. PSA PSA that is not cleaved decrease in PCa (2000); at lysines 145 Nurmikko et al. (2001); Djavan et al (2004). 9 Kallikrein proteins are useful serum biomarkers for diagnosis and prognosis of cancer (Yousef et al., 2003) and a number of these biomarkers have been linked with ovarian, breast, prostate, testicular, cervical, pancreatic, colon cancers (Obiezu and Diamandis, 2005) and lymphoblastic leukemia. Many of the KLK genes are frequently expressed in certain tissue samples and are secreted into the serum or seminal plasma. The kallikreins share similar homology (40–80%) with the greatest degree of similarity noted between KLK2 and PSA (Pakioures et al., 2007). 1.2.4 PSA and its isoforms Pro PSA is released into the lumen of the prostate and, since it contains a 7 amino acid pro-leader sequence, is also termed (-7)pPSA. (-7)pPSA is converted to mature PSA by human kallikrein 2 (hk2) and human kallikrein 4 (hk4). Partial degradation of (-7)pPSA, leading to truncation of the pro-leader sequence, gives rise to other forms of pPSA including (-2), (-4) and (-5)pPSA. The predominant form of tPSA is an1chymotrypsin PSA complex (PSA-ACT), which appears to form 65 – 95% of the total PSA content of human serum (Zhang et al., 1999; Özena and Sözen, 2006). Other PSAprotein complexes have been found in trace amounts, such as PSA 1-protease inhibitor (PSA-API) and PSA 2-macroglobulin (PSA-AMG) which is inactive. Uncomplexed or ‗free‘ PSA (fPSA) accounts for most of the remaining PSA in serum which is 5–35% of the total PSA content. Free PSA is composed of 3 distinct isoforms, pro PSA (pPSA) isoforms, benign PSA (BPSA) and intact PSA (inPSA) – all of these forms are significant in the diagnosis of patients with PCa (Mikolajczyk et al., 2004) (Figure 1.3). 10 Figure 1.3: ProPSA is activated to PSA by hK2 through cleavage of the 7 amino acid proleader sequence. PSA is mainly secreted directly in the lumen of the prostate gland where it liquefies the seminal plasma, provides nutrients and protects the sperm from the acidic environment of the vaginal secretions. However, gland abnormalities such as BPH and PCa cause an elevation in the levels of detectable PSA in the circulatory system. PSA isoforms such as fPSA and cPSA can be detected to determine for early diagnosis of the disease and the focus has moved towards these isoforms for early detection of PCa. This diagram was obtained from Lilja et al. (2008) PSA testing has revolutionised survival rates among men over the past 20 years (Djavan et al., 2004). According to Surveillance, Epidemiology and End Results (SEER) data, the relative 5 year survival rates in PCa had reached almost 100% by 2000, compared to 67% and 75% during 1973–1976 and 1983–1985, respectively (Weir et al., 2003). A serum total PSA (tPSA) measurement of 4 ng/mL is generally regarded as the threshold above which a prostate biopsy is required. The specificity of this PSA cut-off level is 11 poor, however, especially below 10 ng/mL, and is associated with significant false positive and negative results. It has been estimated that between 60–75% of men in the 4–10 ng/mL range (also termed the diagnostic grey zone) undergo unnecessary biopsy (Catalona et al., 1991, 1994; Brawer et al., 2000; Finne et al., 2000; Andriole et al., 2005) while a PSA threshold of 4 ng/mL misses between 20–40% of PCa positive individuals (Mistry and Cable, 2003; Thompson et al., 2004). A PSA threshold of 2.5 or 3 ng/mL is being adopted by a number of urology clinics and screening studies (Labrie et al., 1999; Krumholtz et al., 2002) as an indication for biopsy in order to maintain high levels of PCa detection, but incurs the cost of a potentially higher proportion of unnecessary biopsies. Despite its importance in significantly decreasing the number of individuals diagnosed with clinically inoperative, non-organ confined disease, alternative biomarkers are required to increase the specificity and reliability of testing, especially at low tPSA serum concentrations (2–10 ng/mL). Increased tPSA levels have been reported in both benign prostatic hyperplasia and prostatitis (Hessels et al., 2004), and as such an increased serum tPSA level is, therefore, only a sensitive marker for prostate gland irregularities (Stephen et al., 2003). Current research is focusing on the use of different PSA isoforms to improve the discrimination between a non-malignant and malignant tumour. Although the majority of tPSA found in serum is PSA-ACT (65-95%), the use of this protein complex as a diagnostic tool may be valuable in early detection of PCa, especially in the 2.5–4.0 ng/mL tPSA range (Naya et al., 2005). However, it still remains unclear whether this marker alone will be sufficient in the diagnosis of PCa. PSA-ACT levels are higher in men with PCa than in healthy normal men. Since its approval by the FDA as a test for monitoring PCa, almost every study performed on PSA-ACT as a specific test marker has shown better diagnosis capabilities than tPSA at tPSA serum levels above 4 ng/mL (Brawer et al., 2000; Jung et al., 2000; Mitchell et al., 2001; Sozen et al., 2005). Investigations into the measurement of percentage (535%) fPSA as an improved and useful diagnostic tool in differentiating PCa from BPH have also been performed, since it is known that a higher percentage of fPSA in serum correlates with a lower risk of PCa. Improved discrimination between PCa and BPH in the 2–10 ng/mL tPSA range can be achieved by measuring f/tPSA ratio (Catalona et al., 1998; Woodrum et al., 1998). It has been reported that by measuring the f/tPSA ratio or 12 PSA-ACT levels, up to 50% and 35% of unnecessary biopsies could be avoided in the tPSA ranges of 4–10 ng/mL and 2–4 ng/mL, respectively, while maintaining PCa detection rates at about 80% (Roddam et al., 2005). Evidence exists that the measurement of proenzyme forms of PSA may improve the detection of PCa (Mikolaczyk et al., 2004). As mentioned earlier, Pro PSA (pPSA), which is converted to active PSA through a cleavage process involving removal of a 7 amino acid sequence from the pro-leader sequence between arginine and isoleucine (Peyromaure et al., 2005), may have an important role to play in PCa detection. Truncated forms of pPSA including (-2) pPSA and (-4) pPSA are of particular interest as they are more resistant to conversion to mature PSA. Scientific data using mass spectrometry investigated levels of intact (-7) pPSA and truncated pPSA in cancer serum and found the combined levels to be more than half of fPSA in the sample (Peter et al., 2001). (-7) pPSA was the major form detected with (-5) pPSA, (-4) pPSA and (2) pPSA present to a lesser extent. In serum studies of men with PCa, pPSA has been shown to improve specificity for PCa detection (Sokoll et al., 2003; Khan et al., 2003). Immunostaining studies in cancerous and benign tissues showed that (-2) pPSA stained stronger in the cancerous tissue extracts than in the benign tissue extracts, indicating its link with cancer. (-2) pPSA represents 25–95% of fPSA in the serum of PCa patients and only 6–19% of fPSA in patients without PCa. The percentage of fPSA that is comprised of the pPSA forms increase in aggressive type cancers and this %pPSA could considerably enhance the specificity for PCa detection in men with tPSA levels of 2 – 10 ng/mL, especially in low tPSA levels (< 4 ng/mL Mikolaczyk et al., 2004; Peyromaure et al., 2005). Catalona and co-workers (Catalona et al., 2003) reported that in the analysis of 1091 serum samples from patients enrolled in PCa screening studies, measurement of %pPSA (ratio pPSA/fPSA) would have spared 19% of unnecessary biopsies for patients with tPSA levels of 2-4 ng/mL, compared to 10% and 11% biopsies spared by measurement of fPSA and PSA-ACT, respectively. At higher tPSA levels (4-10 ng/mL), measurement of % pPSA would have spared 31% of unnecessary biopsies compared to 20% for fPSA and 19% for PSA-ACT. Other PSA isoforms may also have a role to play in the generation of more specific tests that can discriminate between PCa and benign conditions, thereby leading to fewer 13 unnecessary biopsies. A distinct enzymatically-inactive form of PSA containing two internal bond cleavages at Lys145 and Lys182 has been identified in BPH tissue (Zhang et al., 1995; Chen et al., 1997) and is termed BPSA. BPSA has been shown to be significantly higher in the serum of non-cancerous men with elevated PSA (Linton et al., 2003) and relatively lower in cancerous prostate tissue (Mikolajczyk and Rittenhouse, 2003). BPSA analysis in serum in conjunction with pPSA immunoassays may provide additional discrimination between PCa and BPH (Mikolajczyk and Rittenhouse, 2003). In addition to the different proPSA isoforms and BPSA, the remaining fPSA serum appears to be composed of intact PSA, termed inPSA (Noldus et al., 1997; Hilz et al., 1999). It appears that an increase in inPSA is most likely to be associated with benign disease as inPSA levels seem to decrease in cancer (Mikolaczyk et al., 2004). This form of fPSA has shown some promise as a marker for distinguishing PCa from benign disease (Nurmikko et al., 2001). 1.2.5 Other potential markers of PCa PSA and its derivatives are recognised as the best current indicators for the presence of PCa. However, alternative markers that could be used to detect prostate cancer with increased specificity and sensitivity are being investigated, including other members of the human tissue kallikrein family of proteins. The expression of human glandular kallikrein 2 (hk2), which is 78–80% identical to PSA, increases during the change from benign to primary prostate cancer (Darson et al., 1999). It has been reported that hk2 can improve the discrimination of PCa from benign disease when measured in combination with tPSA and fPSA (Kwiatkowski et al., 1998; Partin et al., 1999). Hk2 may also be an important marker for PCa aggressiveness (Haese et al., 2001) and for predicting and staging organ-confined tumours (Hessels et al., 2004). Other kallikrein family members have also been identified as promising new candidate markers for PCa diagnosis (Yousef and Diamandis, 2001), including human kallikreins 4 (hk4) (Obiezu et al., 2002), 11 (hk11) (Diamandis et al., 2002), 14 (hk14) (Yousef and Diamandis, 2001), and 15 (hk15) (Diamandis and Yousef, 2002). For example, a study by Nakamura and co-workers showed that a combination of hk11-to-tPSA ratio with %fPSA values gave a PCa detection specificity of 51.5%, compared with detection specificities of 10–31% and 10–45% using tPSA and %fPSA measurements alone, 14 respectively (Nakamura et al., 2003). The future use of panels of these kallikreins in combination with PSA and other markers may thus provide more specific assays for PCa that reduce the prevalence of unnecessary biopsies. The potential of several other possible prostate cancer markers to improve disease detection have been investigated, and markers reported to be associated with aggressive cancers include E-cadherin (ECAD Rubin et al., 2001), enhancer of Zeste homologue 2 (EZH2 Varambally et al., 2002), hepsin (Magee et al., 2001), -methylacyl-coenzyme A racemase (AMACR Rubin et al., 2002) among others. For example, Rhodes and coworkers have demonstrated that the ratio of E-cadherin (ECAD) to enhancer of Zeste homologue 2 (EZH2) was associated with prostate cancer recurrence after radical prostatectomy in a statistically significantly manner and may be useful in defining a cohort of high-risk patients (Rhodes et al., 2003). High levels of circulating insulin-like growth factor (IGF)-1 and low IGF binding protein-3 (IGFBP-3) have been identified as being associated with increased risk of prostate cancer, although the reliability of these serum markers for use in the detection of prostate cancer has been questioned (Ismail et al., 2003). Serum IGF-11 levels are lower in PCa patients than men with BPH, and it has been suggested that IGF-11 may serve as a useful marker for detecting PCa in men with high PSA values (Trojan et al., 2006). Reports on the usefulness of urinary biomarkers for the detection of PCa have also been published. For example, it has been reported that the ratio of urinary PSA to serum PSA (U/S PSA), when combined with the free to total serum PSA ratio, may be of benefit in detection of PCa (Irani et al., 2005). In addition, the urinary biomarker thymosin 15 has been linked to PCa malignancy and its future recurrence. It has been reported that the specificity of PCa detection using a tPSA cut-off of 2.5 ng/mL can be improved from 55.4% to 70.8% by incorporating thymosin 15 measurements (Hutchinson et al., 2005a). Attempts have been made to increase the value of thymosin 15 as a potential biomarker in PCa detection by developing a quantitative assay to detect this protein in bodily fluids (Hutchinson et al., 2005b). 15 1.3 Introduction to antibodies and immunity 1.3.1 Antibodies and their structure Antibodies are glycoproteins produced when a foreign entity elicits an immune response in the body. The body‘s defence system generates antibodies upon exposure to potentially hazardous pathogens which eliminate and destroy them. The basic structure of an antibody is shown in Figure 1.4. Figure 1.4: (A) Ribbon structure of the immunoglobulin G (IgG) molecule. The molecule consists of two identical heavy (H) chains (approximately 50 kDa) and two identical copies of 16 a light chain (25 kDa). Each light chain is attached to a corresponding heavy chain by a disulphide bond. Both heavy chains are covalently linked by interchain disulphide bridges located at the hinge axis. The heavy chain is divided into 4 domains (1 x VH and 3 x CH domains) and the light chain is divided into 2 domains (1 x VL and 1 x CL). The VH and VL domains confer antigenic binding specificity. (B) The complementary determining regions (CDR) regions which form the antigen binding site, antigen binding fragment (Fab), variable fragment (Fv) and constant fragments (Fc) are highlighted. Polyclonal antibodies are a mixture of immunoglobulins (Ig) that recognise different epitopes of the antigen, while monoclonal antibodies bind a single epitope of the antigen. Monoclonal antibodies (Figure 1.4) were first reported by Georges Kohler and Cesar Milstein in 1975. Since then many advancements in this scientific field have generated recombinant antibody fragments namely, the single chain variable fragments (scFv), the fragment antigen binding (Fab) and the single chain antibody fragment (scAb). These fragments retain antigenic specificity due to the complementary determining region (CDR) of the antibody without comprising binding affinity. 1.3.2 Antibody classes There are 5 major classes of antibodies, IgM, IgG, IgA, IgD and IgE. These immunoglobulins differ in both their structure and immune function. IgMs are pentameric in structure and are clinically significant because they are predominant in early immune responses to most antigens. IgAs are polymeric and are the predominant immunoglobulin in saliva, tears, nasal mucosa, prostatic fluid and many other bodily fluids. IgD antibodies are monomeric and trace amounts are present in serum and are found on the surface of human B lymphocytes. IgE antibodies are present in serum in a monomeric form and represent only a small fraction of total antibodies in blood. They are involved in the production and release of vasoactive mediators e.g. histamine and other chemicals that cause an inflammatory reaction. In healthy adults, the fourpolypeptide chain IgG monomer constitutes approximately 75% of the total serum immunoglobulins (Mayer, 2006). The basic structure of an antibody consists of four polypeptide chains with two identical heavy (H) chains and two identical light (L) chains connected by disulphide bonds. The structure varies depending on the isotype. The amino sequence in the variable region of the antibody varies greatly among 17 different antibodies. It is composed of 110-130 amino acids which give the antibody its specificity for binding to the antigen. The variable region is subdivided into hypervariable (HV) and framework (FR) regions. Within the variable heavy (VH) and variable light (VL) chains three HV regions exist (HV 1, 2 and 3). These HV regions are collectively known as complementary determining regions (CDRs). The CDRs are interspersed by Framework (FR) regions. The FRs provide a backbone structure for the antibody, and can also have an influence on antigenic specificity (Maher, 2006). The conformation of the VH and VL chain CDRs result in six HV loop structures which structurally form antigen-binding surfaces or pockets (Figure 1.4). 1.3.3 Innate and acquired Immunity Immunity is the protection of the body from infectious diseases which may cause harm and sickness. The innate immune system is a non-specific genetically derived system passed on from mother to offspring. The natural body defence system encompasses surfaces barriers e.g. skin, which cannot be penetrated by most organisms unless an opening is present. It also consists of mucosal immunity e.g. saliva, tears, nasal secretions and lysozyme, an enzyme in perspiration that cause cell lysis in Gram positive bacteria. Acquired immunity involves a cascade of events involving B and T lymphocytes (Roitt et al., 2006). B cells lymphocytes (generically referred as B cells) are derived from the stem cells of the bone marrow and produce antibodies. T lymphocytes (also known as T cells) are produced in the bone marrow but processed in the thymus and constitute the basis of cell-mediated immunity. In cell-mediated immunity, macrophages engulf the antigens, process them internally and display part of the antigens on their surface along with their own proteins. This stimulates T cells to recognise the presented antigens. Helper T cells (CD4+) are involved in directing the immune response. They secrete lymphokines that encourage cytotoxic T cells and B cells to grow and divide, attract neutrophils and boost the ability of macrophages to engulf and destroy microbes. Suppressor T cells inhibit the production of cytotoxic T cells and memory T cells recognise and respond to a further infection of the same pathogen. Immature B-lymphocytes are stimulated to maturity when an antigen binds to their surface receptors and T helper cells are in close proximity. 18 Light chain V1V2V3 Vn Heavy chain J1 J2 Jn Ck/ V1 V2 Vn D1 D2 D 5 J1 J2 Jn C 3 n ' ' GermLine DNA D-J recombination Somatic 5 3 ' ' 5 3 ' ' recombination VDJ 5 3 ' ' 5 3 Transcriptio ' ' nPrimary 5 3 ' ' recombination transcript RNA Splicing 5 3 mRNA ' ' Translation 5' 3 ' Polypeptide chain Figure 1.5: Somatic recombination of the separate gene segments for the construction of the variable genes. Light chain genes are constructed whereby a variable (V) and joining (J) gene segment are combined to form a complete light-chain V-region gene. The constant (C) is encoded on a separated gene locus and is attached to the variable region exon through splicing of the light-chain RNA to remove the J and C introns. Heavy chain genes are constructed using three gene segments, diversity (D), J and C. Firstly the D and J gene segments are joined followed by the combination of the V gene segment to form a complete heavy-chain exon. The heavy chain C-region genes are encoded by numerous exons which are spliced to the heavy chain C-region during processing of the heavy-chain transcript. The light and heavy chain genes are translated into large polypeptide chains that associate to form the antibody molecule. The diagram was adapted from Janeway et al., (1999). The capability of the body to generate an enormous diversity of antibodies accounts for the remarkable adaptive nature of the immune system. It is because of the unique genetic mechanisms governing antibody synthesis that the body is capable of producing these highly specific molecules to foreign entities. This diversity is mainly confined to 19 the VH and VL chains. VL chain genes are composed of two loci, the variable (V) and joining (J) loci, while the VH genes are composed of three loci, the V and J loci with an additional diversity (D) locus. During B-cell maturation, these different regions combine randomly to produce a huge range of diverse antibodies. Recombination of the various VDJ sequences in the VH chain and VJ sequences in the VL chain results in the formation of different conformations in the binding sites of the antibody (Storb et al., 2001; Janeway et al., 1999; Figure 1.5). 1.4 Antibody Production 1.4.1 Hybridoma technology Over a century ago Paul Ehrlich (19th Century) coined the phrase ‗magic bullets‘ to explain how antibodies might target and interact with their respective antigens. Seventy five years later, Georges Kohler and Cesar Milstein invented the technology for cloning individual antibodies (Bartal and Hirshaut, 1987). Monoclonal antibodies specifically recognize one epitope of the cognate antigen. They can be generated using hybridoma technology which involves fusing lymphocytes from the spleen of an immunised mouse with an immortal cancer cell line (myeloma). The myeloma cell lines are selected so that they do not possess the capability to produce immunoglobulins unless fused to lymphocytes. This fusion is performed using polyethylene glycol (PEG) which is a polywax solution that enhance adjacent cell fusions and the exchange of nuclei (Little et al., 2000). A mixed population of fused cells, unfused myelomas and lymphocytes results and these are incubated for 7 days before screening. The fused hybridoma cells are selected using HAT (Hypoxanthine, Aminopterin and Thymidine)-containing medium. Myeloma cells lack the HGPRT (hypoxanthine-guanine phosphoribosyltransfer) are enzyme and when the de novo synthesis of purine and pyrimidines is blocked by HAT addition the cells die, while lymphocytes which do not grow in this culture media eventually die. Therefore, in the presence of HATsupplemented media only the hybridoma cells will proliferate. Secreted antibodies in conditioned media from each hybridoma are tested against the immunised antigen by ELISA. Positive hybridomas are scaled up and subsequently cloned (by limiting 20 dilution) to select for a single monoclonal antibody-producing hybridoma cell. The hybridoma cells can be stored indefinitely in liquid nitrogen. 1.4.2 Recombinant antibodies The development of recombinant techniques has facilitated the generation of specifically engineered antibody fragments that have been successfully employed in immunoassays (Dillon et al., 2003). Recombinant antibody fragments are becoming essential tools in research due to their intrinsic properties such as greater penetrability, ability to maintain antigen recognition, small size and ease of production when compared to the complete antibody. In medicine, biotechnology and therapeutics use of recombinant antibody fragments expressed in E. coli cells is becoming increasingly popular. Recombinant antibody fragments can be generated by directly cloning the heavy and light chain variable domains from a monoclonal antibody-producing hybridoma cell or amplifying the variable genes from cDNA derived from mRNA from lymphocytes of an immunised source. The single-chain variable fragment (scFv) consists of heavy and light chain variable domains of the full immunoglobulin antibody joined using a flexible glycine-serine linker (Figure 1.6). The small fragment (30 kDa) retains the specificity and affinity of the parental antibody (150 kDa) despite the exclusion of the constant domains. The antigen-binding fragment (Fab) has generally been seen as a more functional and stable version of recombinant antibodies than the scFv. The Fab consists of the variable heavy and light chain genes joined with the constant heavy and light chain genes developing a more stable antibody construct. Functional stability studies by Hernandez et al., (2006) demonstrated the overall improved stability of a Fab over scFv in guanidinium hydrochloride. In 1 M guanidinium hydrochloride 80% activity for Fab was observed compared to 50% activity for an scFv. The affinity of the Fab and scFv was not drastically affected by the antibody format but the functional stability was significantly improved using the Fab format suggesting that the constant regions of the Fab greatly enhance stability. Disadvantages of using murine scFvs include poor expression levels with low product solubility, cell culture arrest and lysis, misfolding of expressed antibody and protein aggregation (Hayhurst, 2000). Hayhurst and co-workers 21 partially addressed these problems by generating a single chain antibody fragment (scAb) by fusing a human constant kappa light chain constant domain to the scFv and, in addition, significantly improved expression levels were achieved by incorporating a downstream periplasmic Skp chaperone gene. The elevated expression levels of scAb were observed to be 10-fold better than the scFv. Figure 1.6: Genetically-derived recombinant antibodies which retain antigen specificity are shown. The smallest of these recombinant antibody fragments is a scFv fragment consisting of the VH and VL chain of the full IgG molecule bound by a glycine-serine linker. The Fab fragment is similar in structure to the scFv fragment but is a more stable construct. A scAb fragment consists of linking a human constant kappa domain to the scFv fragment. The CDR regions of the antibody fragment are responsible for binding to the epitope on the antigen. 1.4.3 Phage display The selection of antigen-specific recombinant antibody fragments involves a process known phage display technology. This technology was first reported in 1985 by Smith and since then phage display has been extensively and exhaustively implemented in many fields of research, in particular for antibody generation. Five years later McCafferty et al., (1990) demonstrated that an antibody scFv gene could be directly incorporated into the phage genome. The selection of antigen-specific antibodies from recombinant antibody libraries is the most successful use of phage display (Bradbury and Marks, 2004). Phage display involves using a filamentous bacteriophage which is a circular singly stranded DNA genome encapsulated within surface coat proteins. 22 Foreign DNA can be inserted into regions of the genome and expressed on the surface as fusion coat proteins. Thus, there is a direct link between the genotype and phenotype. Figure 1.7: The recombinant scFv is expressed on the surface of a bacteriophage and the gene encoding the fusion protein within the virion. Each pIII protein can display 2 – 3 copies of the recombinant antibody fragment. Each phage infects E. coli cells and multiple copies are propagated and selected using ‘biopanning’. Antigen-specific recombinant antibody fragments are selected from large recombinant antibody libraries. Phage display libraries can be successfully generated using an immunised source or developed synthetically. Naive libraries are generated from unimmunised human V genes, shuffled human V genes or synthetic human V genes while immune libraries are derived from V genes from an immunised source. Normally, cDNA is generated from the mRNA extracted from the spleen or bone marrow of an immunised animal. The heavy and light chain genes are amplified by polymerase chain reaction (PCR) and the antibody fragment inserted into one of the encoding genes for surface coat proteins. The phagemid DNA is transformed into competent E. coli cells and after expression the coat protein fusions are integrated into new phage particles assembled in the bacterium. This enables phage particles to be replicated as ssDNA in the presence of helper phage (NEB). Helper phages provide the necessary viral tools for the initiation and packaging of phagemid DNA into phage particles. Helper phages contain a defective origin of replication thus limiting their own packaging ability to produce more helper phage molecules. This characteristic is important as it reduces the ability of helper phage to replicate resulting in dominant phagemid DNA production. The advantages of recombinant technology for antibody production include the fact that a highly comprehensive library of antibody fragments can be generated from which an antibody 23 with high specificity for the antigen can be selected (Hoogenboom et al., 1998). Phage particles are harvested from the infected cells and positive clones with desired affinities selected by a process known as ‗biopanning‘ (Figure 1.8). The selection process can be achieved by immobilising the antigen on a solid surface, incubating the antibodycontaining phage particles, removing non-specific phage by washing, eluting bound phage with a high salt or low pH solution and re-infection into bacteria for replication, enrichment and further rounds of panning with more stringent selection conditions. The support for antigen immobilisation must possess a low phage binding capacity. The target antigen-coating concentration must be sufficient for selection of specific phage for further rounds of enrichment (Smith, 1985). Figure 1.8: Individual phage particles displaying the scFv – pIII fusion proteins are selected by ‘biopanning’. This process involves incubating the phage-scFv particle in an immunotube coated with the antigen and washing away non-specific phage particles. The antigen-specific phage particles are eluted and re-infected into E. coli cells for propagation and analysis in subsequent rounds of selection. The stringency is increased by decreasing the antigen concentration and increasing the washing steps to select high affinity recombinant antibody fragments. 24 The phage pools from each round of selection are analysed by ELISA to confirm antigen-specific antibody fragment enrichment. Individual clones are analysed, screened, sequenced and affinities determined. Methods for the selection of antigenspecific recombinant antibodies have evolved, of which phage display has emerged as one of the most valuable. The key aspect of phage display is that the genotype of a particular antibody is linked through packaging inside a virion particle to the phenotype of a display antibody fused to a coat protein of the phage particle. Although phage display libraries have been generated using the pVIII major coat phage protein, the pIII minor coat protein of filamentous phage is more widely acceptable (Barbas et al., 2001, Figure 1.7). The clear advantage of pIII over the other coat proteins is its ability to package large insertions, its capability to display monovalent copies of the phage-antibody particle and the availability of suitable vectors. The filamentous bacteriophages are a group of viruses capable of infecting Gram negative bacteria. The most widely used are the Ff class (f1, fd and M13) which have shown to be 98% homologous. The Ff phages infect E. coli cells with the F conjugative pilus and do not kill the host cells during infection. The phage is 6.5 nm in diameter and 930 nm in length with a covalently closed single stranded genome of 6400 nucleotides coding for 11 different proteins (pI-pXI) present within the coat proteins. Five of the 11 proteins (pIII, pVI, pVII, pVIII and pIX) make up the flexible protein cylinder, 2 (pII and pX) are involved in single stranded DNA (ssDNA) replication and 3 are required for phage particle assembly. The length of the cylinder is composed of 2700 molecules of the major coat protein (pVIII) while the minor coat proteins (pVII and pIX) are found at one end of the phage particle with 5 molecules of both gene products. There are approximately 5 copies of the other minor coat proteins (pIII and pVI) at the other end of the phage particle. The package signal is located at the pVII and pIX end of the phage particle and this is the first region to be assembled (Figure 1.7). The infection process is a multistep process involving the F pilus on the surface of the E. coli cell. When the phage comes in contact with the pilus, it retracts drawing the phage particle into the cell. Pilus formation is at its most abundance at mid-exponential phase of growth and decreases with increasing growth profile. The outer membrane bacterial proteins (TolQ, R and A) are needed for translocation of the filamentous phage 25 DNA into the cytoplasm and the translocation of the phage coat proteins into the cytoplasmic membrane. The initiation of infection occurs upon binding of the tip of the F pilus to the pIII phage protein. The pIII phage protein consists of 3 domains (N1, N2 and CT) separated by glycine-rich regions. Each domain is essential for bacterial infection. The N1 and N2 domains are in close proximity and N2 is responsible for binding to the F pilus. The N1 domain interacts with the bacterial membrane protein (TolA-D3) and the phage is retracted towards the bacterium surface where the major and minor phage capsid proteins are disassembled and the phage DNA is translocated into the cytoplasm. The viral (+) phage ssDNA strand enters the cytoplasm and the complementary (-) DNA strand is synthesised by bacterial enzymes. This replicative form (RF) is the template for transcription and phage protein translation. The pII protein cleaves the intergenic region of the positive strand of the RF which acts as a primer for synthesis of a new viral strand via a ‗rolling-circle‘ replication method continuing until 200 copies are within the cell. This continues until pV approaches a crucial level where it binds to newly synthesised viral ssDNA. The newly synthesised ssDNA-pV (800 nm in length and 8 nm in diameter) is assembled in the cytoplasm of the bacterium and the phage particle is subsequently secreted from the cell. This is then cleaved and used as a template for further phage propagation. Approximately 1000 phage per cell are produced in the first hour with 100 – 200 phage particles in subsequent cycles. Consideration of a vector for the display of proteins or peptides as coat protein fusions to pVIII or pIII is important. In phage vectors the gene is inserted directly into the genome and expressed with multiple copies of displayed protein leading to polyvalent display. This causes the selection of lower affinity variants during the biopanning process. To avoid an avidity affect a monovalent phage display process was developed using a phage-plasmid vector (phagemid). This is a plasmid with a plasmid origin of replication and a phage-derived origin of replication. The vector is capable of producing large quantities of pIII-fusion proteins but cannot make phage. This is aided by the addition of helper phage (i.e. M13K07 and VCSM13) that supply all the phage enzymes and proteins needed for phage replication (Barbas et al., 2001). As mentioned previously the phagemid genomes can be packaged in the phage coat preferentially rather than in helper phage as these contain a defective origin of replication, which thus ensure an abundance of pIII fusion protein. In phage assembly only 10% of the 26 packaged phage display the pIII fusion protein with 1–2 fusion proteins displayed on the phage. The low percentage is due to the preferential assembling and packaging of wild type pIII into phage. In contrast to phage vectors, phagemid vectors offer both a plasmid origin of replication and a phage-derived origin of replication with a selectable antibiotic resistance gene for facile selection and propagation of pIII fusion proteins. The emergence of tetracycline resistance in cells not infected with phage was observed. Thus, cultures for phage display should be grown fresh from a reliable frozen stock. The phagemid vector also contains a lac promoter which controls expression levels with ompA and pelB leader sequences directing the protein expression to the bacterial periplasm. Soluble expression of recombinant antibody fragments can be achieved by changing the bacterial strain. An amber stop codon (TAG) is present between the protein gene and the phage gene. During the selection process the phagemid is expressed in a suppressor E. coli strain (XL1-Blue, TG1) which allows for transcription of the pIII fusion protein. By incorporating the phage into a non-suppressor E. coli strain (TOP10F) the amber stop codon is read with soluble expression of the recombinant antibody fragment in the periplasmic space without the pIII gene product. Recombinant antibodies can be expressed with fusion tags (hemagglutinin and His) for downstream detection and purification. Recombinant antibodies can be genetically manipulated by techniques such as affinity maturation (Schier et al., 1996), chain shuffling, error-prone PCR and site-directed mutagenesis (Gram et al., 1992) to further increase affinity and sensitivity. In light chain shuffling the VH gene of the parental antibody is joined with the random VL gene of the generated library stock and the newly generated library screened by phage display. This has been shown to be successful with low affinity antibody fragments (Low et al., 1996) with 20-fold improvements (Marks et al., 1992). Error prone PCR involves introducing random nucleotides in the gene sequence and biopanning against the immunised antigen. It is important not to alter too many nucleotides as it may influence the binding capabilities of the translated antibody fragment. Site-directed mutagenesis involves changing specific amino acid sequences within the known antibody sequence to allow selection of an antibody fragment with improved binding. 27 Tyrosine is an important amino acid in protein recognition and is a common amino acid in the CDR binding region of the antibody variable heavy chain. It has been shown to increase antibody binding to protein molecules and is therefore seen as a favourable amino acid residue. The binding pocket of the antigen should be known prior to sitedirected mutagenesis as key amino acids may be required for antibody/antigen interaction. These amino acids should not be deleted or substituted as they may hinder the binding ability of the antibody fragment. A problem arising with mutagenesis strategies are the library size requirements which may restrict the selection of improved higher affinity antibodies (Irving et al., 1996). Large library sizes are required to ensure the retrieval of antibody fragments with improved binding affinity over the parental clone. By incubating less antigen on the immunotubes the parental antibody fragment should not be selected or propagated in sequential rounds of biopanning (Smith, 1985). This approach should ensure the selection of recombinant antibody fragment with improved binding characteristics towards the antigen. 1.5 Immunoassays for PCa detection The application of antibodies for the detection of a variety of analytes in medicine, agriculture and food industries is an area of major significance. It is possible to produce antibodies against a very broad range of antigens, including conformationally intact or denatured proteins, short peptides, carbohydrates, drugs, hormones and small chemicals (Delves et al., 1997). Indeed, immunological detection with antibodies is perhaps the only technology that has been successfully employed for such a range of analytes. While the versatility of such assays is widely recognised, advancement in their quality and usefulness arises from the more recent availability of antibodies with higher specificity. This achievement can largely be attributed to several key discoveries involving novel methods of antibody production (Kohler and Milstein, 1975; Krebber et al., 1997). Today, three main types of antibody are used for immunodiagnostic applications i.e. polyclonal, monoclonal and recombinant antibodies. 28 1.5.1 Current immunoassays for PSA detection Advances in immunoassay technology have facilitated the development of sensitive immunoassays for the measurement of PSA in blood samples to detect the presence of PCa. PSA is currently the best available marker for PCa detection and is an FDA approved marker as previously described in Section 1.2.3. The major application of PSA measurement is for screening men of 45 years and older for the presence of PCa. A tPSA measurement above a cut-off value of 4.0 ng/mL is generally regarded as positive and indicates the need for biopsy. PSA testing is also used to monitor the response of PCa patients to ablative therapy such as radical prostatectomy. In this situation tPSA should be undetectable, and subsequent detectable levels of tPSA is a sign of disease recurrence. For this reason, more sensitive PSA assays have been developed to facilitate earlier detection of recurrence. Although there are different ways to assemble immunoassays, the most common format is a solid phase, two-site sandwich immunoenzymatic assay for use on high throughput immunoassay analysers. The capture antibody is immobilised onto coated paramagnetic beads, and an enzyme-labelled secondary antibody directed against a distinct antigenic site allows colorimetric determination of tPSA concentration. The use of paramagnetic beads as the solid phase allows efficient washing steps, since the beads can be captured from a solution upon application of a magnetic field, but at they do not themselves become magnetised they are resuspended upon removal of the magnetic source. 1.5.2 History of PSA immunoassay development for clinical use Before the distinction of the various forms of tPSA (i.e. free and bound forms) was established as an important factor in PCa detection, the available antibodies did not distinguish between such forms, and assays detected tPSA levels only. The first immunoassay for the measurement of tPSA was reported in 1980 (Kuriyama et al., 1980), and the first commercial immunoassays for tPSA used clinically were available in the late 1980s, namely the Hybritech Tandem-R and the Yang Pros-Check radioimmunometric assays. These assays were manually performed and had analytical sensitivities (i.e. the lowest concentration of measured PSA that could be distinguished 29 from zero samples) of the order of 0.5 ng/mL. These ‗first generation‘ assays were sensitive enough to distinguish elevated PSA from normal levels, but not adequate for the early detection of disease recurrence following, for example, radical prostatectomy. During the early to mid 1990s a large body of research both by clinical researchers and diagnostic immunoassay manufacturers led to the development of numerous immunoassays with improved limits of detection. Table 1.2: Commercially available tests commonly used for PSA determinations in serum Manufacturer PSA isoform Claimed Claimed detected sensitivity range (ng/mL) (ng/mL) Free PSA < 0.008 0–30 Total PSA < 0.008 0–100 Free PSA < 0.02 0–10 Total PSA < 0.04 0–50 IMx Total PSA < 0.04 0–50 Immunlite Total PSA < 0.04 0–150 Products Free PSA < 0.02 0–25 Corporation 3rd Generation PSA < 0.003 0–20 Total PSA < 0.002 0–100 Free PSA < 0.01 0–50 Access- Total PSA < 0.008 0–150 Hybritech Free PSA < 0.005 0–20 ADVIA PSA-ACT < 0.03 0–100 Abbott Assay Architect Diagnostics AxSYM Diagnostic Roche Beckman Coulter Bayer Elecsys Diagnostics These ‗second generation‘ assays, also termed hypersensitive or ultrasensitive, provided detection limits in the region of 0.05 ng/mL, i.e. roughly a 10–fold improvement in analytical sensitivity compared to first-generation assays. At the same time, automated immunoassay analysers were developed, providing a platform for the processing of hundreds of samples per day with reduced inherent error compared to earlier manual 30 tests. The first ‗third generation‘ PSA immunoassay was available to the U.S. market in 1997, with the subsequent introduction of numerous other PSA tests in the following years. Third generation assays offer analytical sensitivities in the region of 0.005 ng/mL, and are generally performed on high throughput analysers allowing the processing of hundreds or thousands of samples per day if required. Today, there are numerous PSA assays available in the marketplace (Table 1.2), although it should be noted that only a limited number have attained FDA approval for clinical use, for example the Tandem E and Tandem R assays (Formerly Hybritech, now Beckman Coulter), IMx assay (Abbott), and Immunlite assay (Diagnostic Products Corporation). Measurement of tPSA using a cut-off level of 4 ng/mL, is associated with a significant rate of false-positive and false-negative results and is unable to distinguish PCa from BPH. Therefore, at the same time as assay sensitivity was being improved, more specific assays were also being developed. The increased assay specificity was based on the detection of different molecular forms of PSA, including PSA complexed to α1antichymotrypsin (PSA-ACT) and free (uncomplexed) PSA (fPSA), discovered in the early 1990s (Lilja et al., 1991). In a landmark paper, Catalona et al. demonstrated that measurement of % fPSA within the so-called diagnostic grey zone of 4–10 ng/mL significantly improved the specificity of cancer detection compared to tPSA measurement, such that 20% of unnecessary biopsies could be avoided (Catalona et al., 1995). By 1998, assays specific for the complexed form of PSA were reported with sensitivities as low as 0.05 ng/mL (Allard et al., 1998; Wu et al., 1998). A year later an ELISA capable of detecting the levels of uncomplexed, fPSA in serum was first reported by Matsumoto and co-workers (Matsumoto et al., 1999). The detection limit for fPSA established for this assay was 0.008 ng/mL. Today many commercial assays are available that discriminate between different PSA isoforms, allowing better discrimination between benign and malignant conditions than by measurement of tPSA alone. However, studies have shown significant differences in the sensitivities of these assays, indicating a lack of reliability (Oberpenning et al., 2002). It is for this reason that current research is still intensely focused on the development of more reliable and accurate assays for the presence of PCa. 31 One aspect of such research is the development of immunoassays that can demonstrate higher specificities for the various forms of PSA. Assays able to specifically detect these isoforms could provide further accuracy in discriminating PCa from benign disorders (Steuber et al., 2002). In 2000, Nurmikko et al. produced a PSA-specific monoclonal antibody that did not detect the internally cleaved form of PSA (Nurmikko et al., 2000). A year later, the same group reported the development of an immunofluorometric sandwich assay for the measurement of intact, fPSA, but not internally cleaved forms (Nurmikko et al., 2001). The detection limit of this assay was determined to be 0.035 ng/mL. To date as mentioned in Section 1.2.4, limited research seems to indicate that measurement of the ratio of internally cleaved to normal forms of PSA may be useful to discriminate benign from malignant prostatic disease (Steuber et al., 2002). Hence, an immunoassay capable of quantifying these forms could potentially increase the accuracy of detection of PCa. 1.6 PSA assays for forensic applications In addition to its use in the diagnosis of PCa, detection of PSA has also become the method of choice for determining the presence of semen in the absence of sperm and when sperm count is low, in forensic sexual assault cases. Measurement of PSA in such samples is associated with many variables not applicable to normal serum samples. Problems encountered with forensic samples include contamination with other body fluids or dirt, scarcity of sample, the need for extraction from different types of fabric leading often to low extraction efficiencies and sample decomposition in burnt cadavers or following laundry of fabrics. Other problems associated with the detection of PSA include specificity, false positive/negative results, sensitivity to pH/temperature, interference from matrix components and PSA stability. In sexual assaults, PSA may be exposed to harsh environmental conditions for prolonged periods of time that will alter the protein. This can happen before analysis is carried out and can lead to conformational changes, reduction of enzymatic activity, and degradation of the molecule. Furthermore, this can result in non-detection of the molecule by the antibody used in the test, giving false negative results. The vaginal environment and secretions are also acidic, which may also contribute to denaturation of PSA. Methods for the detection of PSA in semen include Ouchterlony double diffusion, crossover 32 electrophoresis, rocket immunoelectrophoresis, radial immunodiffusion and ELISA (Graves et al., 1985). PSA immunoassays involving an ELISA format commonly used in tests for clinical measurement of serum PSA have been studied for their potential use in the forensic identification of semen (Johnson and Kotowski, 1993; Jimenez-Verdego et al., 1994; Simich et al., 1999). These high sensitivity assays are relatively time consuming and expensive for the analysis of small numbers of samples, and unsuitable for forensic applications because of the aforementioned difficulties associated with forensic samples. It has been reported that the specificity and sensitivity of ELISA-based PSA tests becomes compromised for forensic semen samples that have been subjected to laundry treatment or exposed to contaminants including dishwashing detergents, shampoo, toothpaste and cleansers (Johnson and Kotowski, 1993). Also, PSA is expressed in female tissue at very low levels and, therefore, antibodies which are too sensitive would not be desired candidates for assay purposes. The possibility of antibody cross-reactivity with female PSA was investigated by Laux and Custis (2004) as potential cross-reactivity would lead to false results. This study demonstrated that the level of PSA in female tissue varies with the tissue type. Current PSA tests used in detection of semen are generally immunochromatographic membrane tests (Hochmeister et al., 1999; Yokota et al., 2001; Maher et al., 2002; Sato et al., 2002; Khaldi et al., 2004). The sensitivity issues of a commercial immunochromatographic test were addressed in the study by Laux and Custis (2004) with respect to the presence of PSA in female body fluids. Levels of PSA reported prior to extraction were 3.7 ng/mL in female urine and 0.53 ng/mL in female serum. Post-extraction, levels were 0.558 ng/mL and 0.08 ng/mL, for urine and serum, respectively. These results indicated that female PSA levels are negligible in comparison to male PSA (820 000 ng/mL in semen and 123 000 ng/mL in semen post-extraction). Validated immunochromatographic membrane tests include the OneStep ABAcard p30 test (Abacus Diagnostics, California, USA), Seratec PSA Semiquant Kit (Seratec, Gottigen, Germany), BioSign PSA membrane test (Biosign), SMITEST PSA card (manufacturer) and Onestep PSA test Strip (FF Diagnostics, Cologne, Germany). These assays are simple in nature, suitable for use on small numbers of samples, give rapid 33 results, and have been reported to be as sensitive and specific as ELISA tests (Hochmeister et al., 1999). The membrane tests work on the basis that PSA binds to a mobile monoclonal anti-PSA antibody in the membrane, forming a mobile antigenantibody complex that migrates through the test area due to the capillary effect of the membrane. A second labelled anti-PSA antibody that is immobilised in the test area binds the migrating antibody-PSA complex and leads to the formation of a coloured band in the presence of sufficient PSA (generally 1–4 ng/mL). Although membrane tests are far less sensitive than the clinically used second or third generation PSA assays, the nature of forensic PSA detection is such that an assay that is too sensitive may yield false positive results, because PSA has been detected in many female tissues and fluids, including breast tissue and milk (Papotti et al., 1989; Yu and Diamandis, 1995), amniotic fluid (Yu and Diamandis, 1995) and urine (Breul et al., 1994). Although more suitable than current ELISA tests for use with forensic samples, membrane tests still suffer from a number of problems related to specificity, false positives, interference from matrix components and PSA instability (Hochmeister et al., 1999; Yokota et al., 2001; Khaldi et al., 2004). We envisage that future PSA tests for use in forensic work will contain antibodies capable of detecting multiple epitopes on the PSA antigen: for example, a combination of antibodies directed against pH-stable, temperature stable and detergent stable PSA elements may improve the detection of PSA in difficult forensic samples. These tests are likely to be delivered in the form of handheld portable devices, and similar to future PSA tests for clinical use, may incorporate new assay formats and detection technologies such as those discussed below in Section 1.7. 1.7 Novel immunoassay formats reported in the literature Over the past few years many novel-format PSA measurement assays have been reported in the literature that aim to facilitate more sensitive and specific measurement of PSA in serum and semen samples (Table 1.3). Research is now focused on more mechanised, rapid and reliable assays. Biosensor technology has emerged as a useful tool in immunodetection strategies for a variety of analytes. Biosensors use a combination of biological receptor compounds (antibody, enzyme, nucleic acid, etc.) 34 and a physical or physico-chemical transducer directing, in most cases, ―real-time‖ observation of a specific biological event e.g. antibody–antigen interaction. In recent years, new immunoassay readout strategies have been devised that have potential to provide greater sensitivity than more conventional approaches. Strategies including surface plasmon resonance (SPR), immuno-PCR, microcantilevers, quantum dots, surface-enhanced Raman scattering (SERS), and atomic force microscopy (AFM) have shown promise in PSA assay formats. Although this section does not document every assay reported, selected assays of particular interest are included. Table 1.3: Novel PSA assay formats recently reported in the literature Detection Assay format method PSA Reported isoform sensitivity Reference detected Surface Self-assembled plasmon monolayer on resonance commercial SPR PSA-ACT 18.1 ng/mL Cao et al. (2006); Total PSA 1 ng/mL biochip with signal Huang et al. (2005); enhancement using a 0.15 ng/mL sandwich assay Besselink et al. (2004). format Surface Sandwich plasmon immunoassay format fluorescence on commercial CM5 spectroscopy (Biacore) chip Surface- Sandwich assay using enhanced gold nanoparticles Raman coated with a Raman scattering scatterer Time- Fluorescently-labelled resolved nanoparticles Free PSA 80 fM Yu et al. (2004). Free PSA 1 pg/mL Grubisha et al. (2003). Total PSA 7.0 pg/mL Ye et al. (2004); fluorescence 0.4 ng/mL Ye et al. (2005); Free PSA 35 0.83 pg/mL Huhtinen et al. (2004); 0.04 pg/mL Soukka et al. (2001). Electrical Resonance frequency sensing shift in a Total PSA 10 pg/mL Lee et al. (2005). nanomechanical microcantilever Free PSA 0.2 ng/mL Hwang et al. (2004); Conductance change Free PSA 100 fg/mL in silicon nanowire sensor chip array Wu et al. (2001); PSA-ACT 100 fg/mL Zheng et al. (2005). Real-time Sandwich assay with PCR DNA label on Total PSA 4.8 x 105 Lind et al. molecules (2005). 100 fg/mL Drukier et al. detection antibody Multi-photon Sandwich assay with detection I-streptavidin on Total PSA (2005). detection antibody Disposable Strip format one-step Free and assay lateral flow assay total PSA 1 ng/mL FernandezSanchez et al. (2005). 1.8 Biosensor devices This approach has many advantages over traditional methods for the detection of minute amounts of target analyte. It allows label-free, rapid monitoring of biomolecular reactions in ‗real-time‘, and requires only a small amount of sample. The SPR device consists of a sensor chip containing flow cells, a light source, a prism and a detector (Figure 1.10). SPR sensor chips are usually composed of a carboxymethyl dextran matrix linked to a thin layer (~50 nm) of gold substrate on the sensor surface (Wilson, 2002). SPR measures changes in refractive index and thus in the resonance angle at which polarised light is reflected from a surface, which is in turn related to a change in layer thickness or mass (Wilson, 2002). Thus, if polarised light strikes a gold layer at 36 the interface between media (e.g. glass and buffer) of differing refractive indices at a fixed wavelength above a critical angle, the photons are absorbed into surface plasmons (electron density waves) resulting in resonance and no reflection of light. If the refractive index changes at one side of the surface due to immobilisation of, for example, a ligand to its receptor, the resonance angle is changed and the intensity of the reflected light is increased (Figure 1.10). A B 37 Figure 1.10: Schematic of the typical set-up for a surface plasmon resonance (SPR) biosensor. SPR detects changes in the refractive index in the immediate vicinity of the surface layer of a sensor chip. SPR is observed as a shadow in the reflected light from the surface at an angle dependent on the mass of material at the surface. (A) When antigen is passed over the flow cell it binds to the receptor resulting in a change in the mass of the surface layer, leading to a shift in the resonance angle (from I to II in the diagram). This change in signal is proportional to the amount of ligand bound to the receptor, which enables detection of a ligand and determination of the ligand–receptor interaction affinities and kinetics. (B) The change in resonance angle can be monitored non-invasively in ‘real-time’ as a plot of resonance signal (proportional to mass change) versus time. Huang and co-workers reported an immunosensor based on the use of a commercial SPR biosensor (Biacore 2000) for the detection of PSA concentrations as low as 1 ng/mL (Huang et al., 2005). Biacore SPR chips are generally composed of a carboxymethyl dextran matrix linked to a gold substrate on the sensor surface. However, self-assembled monolayers (SAMs) have been used as the immobilisation substrate and have been shown to give better sensitivities than dextran layers (Frederix et al., 2003). The PSA sandwich assay reported by Huang et al. (2005) used a camel anti-PSA antibody fragment attached to a mixed SAM of thiols for PSA capture, and a biotinylated mouse monoclonal anti-PSA antibody and streptavidin-modified gold nanoparticles for amplification of the detection signal. Cao and co-workers subsequently reported a SPR-based immunoassay specific for PSA-ACT, in which the limit of detection was enhanced simply by using a sandwich strategy in which an intact PSA monoclonal antibody was used as an amplifying agent (Cao et al., 2006). Yu and co-workers reported the use of a CM5 Biacore chip as a dextran surface on which a sandwich assay for free PSA was conducted (Yu et al., 2004). The assay combined SPR and fluorescent labelling, i.e. surface plasmon field-enhanced fluorescence spectroscopy (SPFS). This technology used surface plasmons to excite the fluorescent label and produce the signal. The limit of detection observed was 80 fM for a 40 minute contact time, which is many orders of magnitude lower than detection limits of third generation PSA assays commercially available today. Related to SPR, surface-enhanced Raman scattering (SERS) has also been used in the detection of minute amounts of target molecules. In Raman spectroscopy the light scattered inelastically from a molecule provides information about the molecule's vibrational quantum states. The Raman effect 38 can be greatly strengthened if the molecules are attached to nm–sized metal structures, similar to the gold sheets on SPR biosensor chips. SERS has also been used in a sandwich assay format immunoassay for the detection of PSA (Grubisha et al., 2003). The assay used tracer nanoparticles created by the physisorption of antibodies and Raman reporter molecules (RRMs) on gold colloids. It has been speculated that SERS could be used for the sensitive detection of multiple analytes by immobilising a mixture of different capture antibodies and using nanoparticles coated with different sets of tracer antibodies and RRMs (Ni et al., 1999). Such an approach is possible because Raman bands are sufficiently narrow to reduce the likelihood of spectral overlap when multiple labels are used. There has been considerable interest in harnessing the very high sensitivity of ‗realtime‘ PCR for use in diagnostic applications. As early as 1992, Sano and co-workers described a new technique using PCR for the analysis of protein analytes, which they called immuno-PCR (Sano et al., 1992). This technique was based on a sandwich assay format, in which the detection antibody was coupled to a DNA molecule, which was then amplified after washing steps. The amount of product formed during the exponential phase of DNA amplification, assessed by gel electrophoresis or PCRELISA, reflected the amount of target protein bound by the capture antibody. However, the use of real-time PCR for DNA quantification, first demonstrated by Sims and coworkers, led to a significant improvement in sensitivity and accuracy over a wide concentration range (Sims et al., 2000). Recently, the development of real-time immuno-PCR systems for the specific detection of PSA was reported (Lind et al., 2005). The most sensitive assay could detect 4.8 x 105 PSA molecules in a 5 µl sample, equivalent to a sensitivity of approximately 0.2 pg/mL. ‗Real-time‘ immuno-PCR assays could be applied to simultaneous multi-analyte detection, since primers binding specific DNA sequences could be used for each of a number of different detection antibodies. Much research into immunoassay development is currently focused on the miniaturisation of assays for future use in disposable format tests. Some disposable format PSA tests have recently been reported. A disposable immunosensor based on a lateral flow format has been successfully designed for the detection of free and total 39 forms of PSA (Fernandez-Sanchez et al., 2005). The lateral flow system was based on a non-competitive sandwich assay format. The membrane was coated with anti-free-PSA or anti-total-PSA antibodies with a monoclonal anti-PSA urease conjugate as the probing molecule. The system consisted of a polymer-coated electrode system to which each immunostrip was placed in contact. Upon completion of the immunoassay on the strip, hydrolysis of urea by the urease label generated ammonia and an increase in pH, which resulted in degradation of the polymer and subsequent determination of the amount of analyte in the sample, PSA could therefore be quantified. A lower detection limit of 10 ng/mL of both free and complexed PSA species was observed with this method. Considerable interest exists in biosensors that use microelectromechanical system (MEMS) and nanoelectromechanical system (NEMS) detection technologies (Cherian et al., 2003). Most nanomechanical sensors use silicon-based cantilevers that bend due to increased surface stress when the target molecule binds to specific receptors on the cantilever surface. Lee et al., (2005) reported the development of an immunoassay for PSA using a piezoresistive nanomechanical microcantelever. Electrical detection, via surface stress changes, of antigen-antibody binding was achieved through microfabricated self-sensing micro-cantilevers. When binding occurred on a functionalised gold surface, surface stress was induced resulting in cantilever bending and resistance change of the piezoresistive layer. The authors reported that the novel electrical measurement system facilitated label-free detection of PSA with a reported achievable sensitivity of 10 pg/mL. The last 30 years have seen a significant improvement in PCa diagnosis, general welfare and life expectancy of the PCa patient. This has coincided with the discovery of PSA, the most widely used cancer-associated marker for the disease. Despite its achievements, some researchers have questioned its value in PCa diagnosis, since increased serum levels are also associated with non-malignant conditions such as BPH. Immunoassay development is currently focusing on the detection of the various isoforms of PSA to increase assay specificity and reduce the rate of false positives and unnecessary biopsies. While numerous reports of assays for the detection of various forms have been reported, it is still unclear whether these assays will improve the 40 detection of PCa in the presence of non-malignant disease, and therefore further research is required. It is likely that the specific detection of PCa will require a panel of antibodies against multiple markers for PCa including other members of the kallikrein family of proteases, and even non-protein markers such as genes upregulated in PCa progression. The advent of proteomics and microarray technology will likely involve the amalgamation of current antibody-based assays with antibodies against novel biomarkers monitoring the change in levels of numerous factors thus giving a more accurate assessment of the disease state and facilitating earlier diagnosis. 1.9 Thesis Outline The aim of the work presented in this thesis was focused on the generation, selection, characterisation and application of novel recombinant antibody fragments for the detection of fPSA. The work involved generating 3 recombinant antibody libraries from immunised sources and selecting antigen-specific antibody fragments using phagedisplay technology (Chapter 3). Recombinant murine antibody fragments selected from ‗biopanning‘ were reformatted and expressed as scAb for enhanced protein expression and stability due to problems incurred with poor expression levels of murine scFvs. Individual clones (murine and avian clones) were ranked using a capture assay approach on the Biacore system and kinetic analysis performed on the most suitable candidate clones displaying the greatest percentage stability (Chapter 4). All antibodies were purified and characterised by ELISA, Biacore analysis, SDS-PAGE and Western blotting. Epitope mapping using a sandwich assay on the Biacore system revealed potential clones for immunoassay development. ELISA and Biacore inhibition-based assays were evaluated. However, low sensitivity proved to be an issue. A sandwich assay approach was investigated using a biotinylated murine anti-fPSA scAb and an avian anti-fPSA scFv as a detection antibody. This approach improved assay sensitivity; female serum samples spiked with fPSA were assessed to evaluate assay sensitivity in the serum matrix. Serum samples from patients presenting with PCa were analysed to determine levels of fPSA in the serum sample. PCa tissue samples were analysed by immunohistochemistry using purified murine and avian anti-fPSA recombinant antibodies and positive staining was observed (Chapter 5). 41 Chapter 2 Materials & Methods 42 2.1 Materials & Methods 2.1.1 Materials All reagents were purchased from Sigma-Aldrich, Airton Road, Tallaght, Dublin 24, Ireland except where otherwise stated below. Reagent Supplier Bacteriological Agar Cruinn Diagnostics Ltd., Hume Centre, Yeast Parkwest Business Park, Nanogor Road, Tryptone Dublin 12, Ireland. DNA ligase ISIS Ltd., Unit 1 & 2, Ballywaltrim Helper phage Business Centre, Boghall Road, Bray, Co. Restriction enzymes Wicklow, Ireland. EZ-Link Biotinylation Kit Fischer Scientific Ireland, Suite 3, Plaza 212, Blanchardstown Corporate Park 2, Ballycoolin, Dublin 15, Ireland. fPSA Lee Biosolutions Inc., 2924 Mary Avenue, St. Louis, Missouri, (63144-2730) USA. Methanol Lennox, John F. Kennedy Drive, Naas Road, Dublin 12, Ireland. PCR primers Eurofins MWG Operon, 318 Worple Road, Raynes Park, London, SW20 8QU, UK. Perfect Prep gel cleanup kit Unitech, Unitech House, Magna Drive, Magna Business Park, City West, Dublin 24, Ireland. Prestain full range rainbow marker GE Healthcare Life Sciences, Amersham Place, Little Chalfont, Buckinghamshire, HP7 9NA, UK. Superscript system Bio-sciences, 3 Charlemont Terrace, Crofton Road, Dun Laoghaire, Dublin, Ireland. Trypsin AGB Scientific Ltd., Dublin Industrial 43 Estate, Dublin 11, Ireland. Wizard Plus Mini-prep kit Medical Supply Co Ltd., Damastown, Promega cDNA synthesis kit Mulhuddart, Dublin 15, Ireland. Promega gel clean up kit 2.1.2 Culture media formulation Media Ingredients 2x Tryptone Yeast extract medium (2xTY) Tryptone 16 g/L Yeast Extract 10 g/L Super Broth Medium (SB) Super Optimal Catabolite Medium (SOC) NaCl 5 g/L Tryptone 30 g/L Yeast 20 g/L MOPS 10 g/L Tryptone 20 g/L Yeast Extract 5 g/L NaCl 0.5 g/L KCl 2.5 mM MgCl2 20 mM Glucose 20 mM 2.1.3 Buffer compositions 2.1.3.1 Phosphate buffered saline (PBS) (150 mM, pH 7.4) 0.15 M NaCl 2.5 mM KCl 10 mM Na2HPO4 18 mM KH2PO4 All the listed reagents were dissolved in 800 mL of ultra pure H2O, adjusted to pH 7.4 and made up to 1 litre with ultra pure H2O. 44 2.1.3.2 PBS-Tween (PBST) Tween 20 detergent (Sigma) was added to PBS to give a final concentration of 0.05 % (v/v). 2.1.3.3 HEPES buffered saline (HBS) (240 mM, pH 7.4) 2.38 g HEPES 8.77 g NaCl 1.27 g EDTA 0.5 mL Tween HEPES, NaCl and EDTA were dissolved in 800 mL of ultra-pure water. The pH was adjusted to pH 7.4 by the addition of NaOH. The solution was filtered through a 0.2 µm filter. The Tween 20 was added to the solution and the solution was degassed. 2.1.3.4 Tris buffered saline (TBS) (150 mM, pH 7.6) 0.05 M Trizma base 0.15 M NaCl The Trizma base and NaCl were dissolved in 800 mL ultra-pure water. The pH was adjusted to pH 7.6 using NaOH and made up to 1 litre with ultra pure H2O. 2.1.3.5 Tris-acetic acid-EDTA buffer (TAE) (400 mM, pH 8.3) A 10X stock solution of TAE buffer, supplied as a proprietary reagent by Sigma, was diluted to 1 litre with distilled water. All agarose gels for DNA visualisation were prepared and resolved in 1X TAE buffer. 2.1.3.6 Tris-EDTA buffer (TE) (10 mM, pH 8.0) 0.01 M Trizma hydrochloric acid 0.001 M EDTA 45 2.1.4 Equipment list Equipment Supplier Balances (Chyo JK-180, Mettler PJ300) Medical Supply Company Ltd, Damastown, Mulhuddart, Dublin 15, Ireland. Biacore 3000TM Pharmacia Biosensor AB, Uppsala, CM5 chips Sweden. Biometra TGRADIENT PCR machine AGB Scientific Ltd., Dublin Industrial Orbital shaker Estate, Glasnevin, Dublin 9, Ireland. Millipore Ultrapure H2O Milli-Q academic Sanyo Orbital Incubator Bio Rad Gene Pulser Xcell Alpha Technologies, The Leinster SDS Bio-Rad Min-Protean® 3 Cell Technology Centre, Blessington Industrial Trans-Blot®SD Semi-Dry Transfer cell Estate, Blessington, Co. Wicklow, Ireland. Bio-Rad Powerpac Basic Bio-Rad DNA gel apparatus (Wide-MiniSub® CellGT) Blood tube rotator SB1 Stuart Scientific, Beacon Road, Stone, Staffordshire, ST15 0SA, UK. Gelaire BSB4 laminar unit Gelman Ltd, 71 Broomhill road, Tallaght Industrial Estate, Tallaght, Dublin 24, Ireland. HermLe Z233MK-2 refrigerated HermLe Labortechnik GmbH, 25 centrifuge Siemensstrasse, Wehingen, 78564, Germany. Homogeniser (Ultra-Turrax) Janke & Kunkel IKA-Werk Ultra-Turrax, Staufen, 79129, Germany. Camag Linomat 5 (Lateral Flow Mason Technologies, Greenville Hall, 228 Instrument) South Circular Road, Dublin 8, Ireland. New Brunswick Scientific-Excella E24 Incubator Shaker Series (plate shaker) ND-1000 Spectrophotometer (Nanodrop) 46 Tomy Autoclave SX-700E High Pressure Steam Sterilizer) pH meter (Orion 3 star) Medical Supply Company Ltd, Damastown, Mulhuddart, Dublin 15, Ireland. Plate incubator shaking IKA® MTS 2/4 Lennox, John F. Kennedy Drive, Naas digital Road, Dublin 12, Ireland. Platform shaker STR6 Tecan Saffire2 Plate reader Tecan Austria GmbH, Untersbergstrasse, 5082 Grödig, Austria. Water bath (Y6 model) Grant instruments (Cambridge) Ltd, 29 Station Road, Shepreth, Royston, Herts., SG8 6PZ, UK. 2.1.5 Bacterial strains used All bacterial strains cultured and expressed in different nutrient broth solutions are listed below. Bacterial Strain Product details Suppliers E.coli XL-1 Blue cells Stratagene RecA endA1 Techno-path Ltd., gyrA96 thi-1 hsdR17supE44 Holland Road, National relA1 lac (F proAB lacIq Z Technology Park, r M15 Tn10 (Tet )) Plassey, Limerick, Ireland. E.coli TOP10F' cells Invitrogen F'[lacIq Tn10(tetR)] Techno-path Ltd., mcrA Δ(mrr-hsdRMS-mcrBC) Holland Road, National φ80lacZΔM15 ΔlacX74 deoR Technology Park, nupG recA1 araD139 Δ(ara- Plassey, Limerick, leu)7697 galU galK rpsL(StrR) Ireland. endA1 λE.coli Tuner cells Novagen – Merck F– ompT – hsdSB (rB mB–) 47 gal dcm lacY1 Techno-path Ltd., Holland Road, National Technology Park, Plassey, Limerick, Ireland. 2.2 Production of murine recombinant antibody fragments 2.2.1 Preparation of bacterial stocks All bacterial stocks were streaked on 2xTY agar plates overnight at 37oC for individual colony isolation. Individual colonies were picked and cultured in 2xTY media overnight at 37oC with shaking at 200 rpm. The overnight cultures were subcultured into fresh 2xTY medium (Section 2.1.2) and grown until reaching the desired cell density (OD600 nm ~ 0.8). The cultures were centrifuged at 4,000 rpm (3,220 x g) for 10 minutes and resuspended in sterile glycerol (20% v/v final concentration)-supplemented media. The cell suspension was transferred aseptically to sterile 1.5 mL centrifugation tubes and flash frozen using liquid nitrogen. Bacterial strains were stored at -80oC. 2.2.2 Immunisation of Balb/c mice with fPSA and antibody titre determination The Laboratory Animal Science and Training (LAST) Ireland Animal handling training course was attended and the associated examination passed. The proposed work was passed by the ethics committees of the School of Biotechnology and the University and the licence subsequently granted by the Department of Health and Children. Extreme care was taken to minimise stress levels to the animals. Balb/c mice aged between 5–8 weeks were used for production of recombinant antibodies to fPSA. These were initially immunised in the peritoneal cavity with a final concentration of 50 g/mL fPSA in 200 µl PBS, mixed in a 1:1 ratio with Freund‘s complete adjuvant. The mixture was vigorously shaken to ensure a complete homogenous solution. Freund‘s Complete Adjuvant (FCA) is an emulsified mineral oil antigen solution used as an immunopotentiator. Subsequent ‗booster‘ injections (4, 8 and 12 weeks) were performed with 25 g/mL fPSA mixed in a 1:1 ratio with Freund‘s Incomplete Adjuvant (FICA). A tail bleed was taken from the mice 7 days after the second booster administration to confirm the animals were responding to the immunisations and 48 antigen-specific antibodies were being produced. Once the titre reached an appropriate level (1:100,000 dilution) as determined by ELISA, the mice were boosted 7 days prior to sacrifice (Krebber et al., 1997). 2.2.3 Extraction and isolation of total RNA from the spleen of an immunised Balb/c mouse All work was performed in the Bioresource facility in DCU, Dublin, Ireland. Prior to use the laminar unit (Gelaire BSB 4) was cleaned with Industrial Methylated Spirits (IMS) and ‗RNase-free‘ wash solution thus ensuring an uncontaminated environment. The mouse was sacrificed by cervical dislocation and transfer to the laminar unit. Its spleen was removed and homogenised in TRI reagent (2 mL) using a pre-baked (180oC, overnight) sterile homogeniser. The mixture was incubated for 5 min at room temperature to permit the total dissociation of nucleoprotein complexes. Chloroform (400 L) was added and the solution shaken vigorously for 15 seconds. The resulting mixture was stored for 15 min at room temperature and after incubation centrifuged at 12,000 rpm (Eppendorf centrifuge 5810R) for 15 min at 4oC. Three layers were observed, a lower red phenol/chloroform phase, a protein interphase and a colourless liquid upper phase containing the RNA. The aqueous upper phase was precipitated using isopropanol (1 mL) in a fresh ‗RNase-free‘ tube. This mixture was stored for 10 min at room temperature and centrifuged at 12,000 rpm for 8 min at 4oC. RNA forms a white pellet at the bottom of the tube. The supernatant was removed, the pellet washed with 75% (v/v) ethanol (2 mL) and centrifuged at 7,500 rpm for 5 min at 4oC (Eppendorf centrifuge, 5810R). The pellet was allowed to dry and resuspended in molecular grade water (140 L). 2.2.4 Reverse transcription of total RNA to cDNA A PCR was performed for 1 hour at 42oC to transcribe total RNA to cDNA. The synthesis of cDNA was performed using a Promega cDNA synthesis kit containing oligo (dT) primers. The PCR reaction contained the following components 49 Components Stock Concentration Final Conc. Volume (20L) MgCl2 25 mM 5 mM 4 L 10X Buffer 10 X 1X 2 L dNTP mix 10 mM 1 mM 2 L RNase 40 U/L 1 U/L 0.5L Oligo (dT) Primer 0.5 g/L 0.2 g/rxn 0.4 L AMV RT* 25 U/L 10 U/rxn 0.4 L RNA 1.5 g/L 11.25 g/rxn 7.5 L 3.2 L Molecular Grade H20 * Avian myeloblastosis virus reverse transcriptase All the components, excluding the enzymes, were placed on ice and slowly thawed. The enzymes were removed from the -20oC freezer immediately before use to minimise loss of activity. The components were added to sterile PCR tubes and the PCR programme initiated. Temperature (oC) Time (min) 1 42 15 2 95 5 3 4 pause Steps 2.2.5 PCR Primers The primers listed below were obtained from Sigma-Genosys Ltd and are compatible with the primers set described by Krebber for the pAK vector system (Krebber et al., 1997). The FLAG tag (gactacaaaGAY) and Bam H1 restriction site (ggatcc) were incorporated into the designed primers for amplifying the antibody variable domains. Variable light chain reverse primers LB1 5gccatggcggactacaaaGAYATCCAGCTGACTCAGCC3 LB2 5gccatggcggactacaaaGAYATTGTTCTCWCCCAGTC3 LB3 5gccatggcggactacaaaGAYATTGTGMTMACTCAGTC3 LB4 5gccatggcggactacaaaGAYATTGTGYTRACACAGTC3 50 LB5 5gccatggcggactacaaaGAYATTGTRATGACMCAGTC3 LB6 5gccatggcggactacaaaGAYATTMAGATRAMCCAGTC3 LB7 5gccatggcggactacaaaGAYATTCAGATGAYDCAGTC3 LB8 5gccatggcggactacaaaGAYATYCAGATGACACAGAC3 LB9 5gccatggcggactacaaaGAYATTGTTCTCAWCCAGTC3 LB10 5gccatggcggactacaaaGAYATTGWGCTSACCCAATC3 LB11 5gccatggcggactacaaaGAYATTSTRATGACCCARTC3 LB12 5gccatggcggactacaaaGAYRTTKTGATGACCCARAC3 LB13 5gccatggcggactacaaaGAYATTGTGATGACBCAGKC3 LB14 5gccatggcggactacaaaGAYATTGTGATAACYCAGGA3 LB15 5gccatggcggactacaaaGAYATTGTGATGACCCAGWT3 LB16 5gccatggcggactacaaaGAYATTGTGATGACACAACC3 LB17 5gccatggcggactacaaaGAYATTTTGCTGACTCAGTC3 LB 5gccatggcggactacaaaGATGCTGTTGTGACTCAGGAATC3 Variable light chain forward primers LF1 5ggagccgccgccgcc(agaaccaccaccacc)2ACGTTTGATTTCCAGCTTGG3 LF2 5ggagccgccgccgcc(agaaccaccaccacc)2ACGTTTTATTTCCAGCTTGG3 LF4 5ggagccgccgccgcc(agaaccaccaccacc)2ACGTTTTATTTCCAACTTTG3 LF5 5ggagccgccgccgcc(agaaccaccaccacc)2ACGTTTCAGCTCCAGCTTGG3 LF 5ggagccgccgccgcc(agaaccaccaccacc)2ACCTAGGACAGTCAGTTTGG3 Variable heavy chain reverse primers HB1 5ggcggcggcggctccggtggtggtggatccGAKGTRMAGCTTCAGGAGTTC3 HB2 5ggcggcggcggctccggtggtggtggatccGAGGTBCAGCTBCAGCAGTC3 HB3 5ggcggcggcggctccggtggtggtggatccCAGGTGCAGCTGAAGSASTC3 HB4 5ggcggcggcggctccggtggtggtggatccGAGGTCCARCTGCAACARTC3 HB5 5ggcggcggcggctccggtggtggtggatccCAGGTYCAGCTBCAGCARTC3 HB6 5ggcggcggcggctccggtggtggtggatccCAGGTYCARCTGCAGCAGTC3 HB7 5ggcggcggcggctccggtggtggtggatccCAGGTCCACGTGAAGCAGTC3 HB8 5ggcggcggcggctccggtggtggtggatccGAGGTGAASSTGGTGGAATC3 HB9 5ggcggcggcggctccggtggtggtggatccGAVGTGAWGYTGGTGGAGTC3 51 HB10 5ggcggcggcggctccggtggtggtggatccGAGGTGCAGSKGGTGGAGTC3 HB11 5ggcggcggcggctccggtggtggtggatccGAKGTGCAMCTGGTGGAGTC3 HB12 5ggcggcggcggctccggtggtggtggatccGAGGTGAAGCTGATGGARTC3 HB13 5ggcggcggcggctccggtggtggtggatccGAGGTGCARCTTGTTGAGTC3 HB14 5ggcggcggcggctccggtggtggtggatccGARGTRAAGCTTCTCGAGTC3 HB15 5ggcggcggcggctccggtggtggtggatccGAAGTGAARSTTGAGGAGTC3 HB16 5ggcggcggcggctccggtggtggtggatccCAGGTTACTCTRAAAGWGTSTG3 HB17 5ggcggcggcggctccggtggtggtggatccCAGGTCCAACTVCAGCARCC3 HB18 5ggcggcggcggctccggtggtggtggatccGATGTGAACTTGGAAGTGTC3 HB19 5ggcggcggcggctccggtggtggtggatccGAGGTGAAGGTCATCGAGTC3 Variable heavy chain forward primers HF1 5ggaattcggcccccgaggcCGAGGAAACGGTGACCGTGGT3 HF2 5ggaattcggcccccgaggcCGAGGAGACTGTGAGAGTGGT3 HF3 5ggaattcggcccccgaggcCGCAGAGACAGTGACCAGAGT3 HF4 5ggaattcggcccccgaggcCGAGGAGACGGTGACTGAGGT3 2.2.6 Components and PCR conditions for antibody variable domain genes amplification using Krebber-specific primers The reaction components and volumes for mouse variable chain amplification were as follows: Component 20 L volume cDNA 0.8 L dNTP mix 0.4 L 5 X Buffers (A, B, D, G) 4.0 L VL/VH Forward Primer 0.4 L VH/VL Reverse Primer 0.4 L H2O 13.2 L Sigma Red Taq polymerase 0.8 L 52 PCR buffer compositions: buffer A (7.5 mM MgCl2, pH 8.5), buffer B (10 mM MgCl2, pH 8.5), buffer D (17.5 mM MgCl2, pH 8.5), buffer G (12.5 mM MgCl2, pH 9.0) (Figure 3.3). The PCR conditions for the amplification of the mouse antibody variable chain genes by PCR were as follows: Steps Temperature (oC) Time (seconds) 1 94 60 2 63 55 3 72 60 Repeat Steps 1 3 seven times 4 94 60 5 69 30 6 72 60 Repeat Steps 4 6 twenty three times 7 72 600 8 4 Pause After successful amplification of both the variable heavy (VH) and variable light (VL) chain gene fragments, the PCR products were resolved on a 1% (w/v) agarose gel and purified using the Promega gel clean up kit. 2.2.7 Purification of VH and VL variable gene fragments using the Promega gel clean up Kit The VH and VL chain genes were purified from a 1% (w/v) agarose gel by excising the correct size fragments, 386–440 bp for VH chain, and 375–402 bp for VL chain. The gel slices were weighed and binding buffer (add three times the volume of the excised gel fragment) added. The mixture was incubated in a water bath for 10 min at 50oC. The guanidine isothiocyanate-containing binding buffer allows for sufficient binding of the DNA to the silica membrane columns. An equal volume of isopropanol to the weight of the excised fragment was added and the resulting mixture added to a collection column. 53 Isopropanol encourages precipitation of the excised DNA fragment. The column was centrifuged at 14,000 rpm for 1 min to remove remaining residual buffer and the flowthrough discarded. The column was washed with 750 L wash buffer, centrifuged at 14,000 rpm for 1 min and washed with 250 L wash buffer. The column was centrifuged at 14,000 rpm for 2 min and the DNA eluted from the column using 30 L molecular grade H2O into a clean 1.5 mL sterile centrifugation tube. The absorbance of the purified DNA was measured at 260 nm using the Nanodrop ND-1000 spectrophotometer and the concentration calculated using the Beer-Lambert law. 2.2.8 Splice by Overlap extension PCR (SOE-PCR) The VH and VL gel purified fragments were joined with a glycine-serine linker (G4S)4 using SOE-PCR to produce an 850 bp DNA fragment. The SOE primers for the 850 bp amplification were:- Single chain forward 5ggaattcggcccccgag3 Single chain reverse 5ttactcgcggccagccggccatggcggactaccccg3 Initially the PCR was performed using different VH and VL template ratios with Phusion polymerase (Finnzymes). The standard 1 x DNA template concentration for each chain was 10 ng. The VH and VL ratios for the SOE-PCR amplification were as follows; (i) 10 ng VH: 10 ng VL, (ii) 20 ng VH: 20 ng VL, (iii) 5 ng VH: 5 ng VL, (iv) 5 ng VH: 10 ng VL, (v) 5 ng VH: 2.5 ng VL, (vi) 5 ng VH: 1.25 ng VL, (vii) 10 ng VH: 5 ng VL, (viii) 2.5 ng VH: 5 ng VL, (ix) 1.25 ng VH: 5 ng VL. The concentrations of all other components remained equal to give a final concentration of 20 µL per reaction. 54 20 L volume Component Buffer G* 4 L 10 mM dNTP mix 0.4 L scForward Primer 0.4 L scReverse Primer 0.4 L Phusion polymerase 0.2 L VH chain (10 ng DNA) 2 L VL chain (10 ng DNA) 2 L H20 10.6 L * For composition of buffer G, see page 51. The reaction was performed using the following PCR conditions: Steps Temperature (oC) 1 98 60 2 98 20 3 55 30 4 72 30 Time (seconds) Repeat Steps 2 4 six times 5 98 20 6 63 30 7 72 30 Repeat Steps 5 7 twenty times 8 72 300 9 4 Pause A non-specific 1,000 bp band was preferentially amplified over the desired 800 bp scFv product and, therefore, optimisation was required (Figure 3.4). DMSO and BSA were added to the PCR mix to decrease the levels of non-specific amplification and improve concentration of the desired PCR product. A touchdown PCR was incorporated to facilitate the amplification of the product. 55 L volume Component 5X Buffer G* 10 L scForward Primer 1 L scReverse primer 1 L dNTP mix 1 L VH chain 2.66 L VL chain 2 L Red Taq Polymerase 2 L DMSO - BSA - H20 - *For composition of Buffer G, see page 51 The PCR amplification was performed using the listed components with varying concentrations of DMSO, BSA and H2O. The PCR amplified reactions were as follows (i) supplemented with 5% (v/v) DMSO (ii) supplemented with 50 µg/mL BSA (iii) supplemented with 5% (v/v) DMSO and 50 µg/mL BSA (iv) no additives (v) positive control (in-house scFv) and (vi) negative control (no VH and VL DNA template). The reaction was performed using the following PCR conditions Steps Temperature (oC) 1 94 30 2 63 30 3 72 60 Time (seconds) Repeat Steps 1 3 five times 4 94 60 5 69 30* 6 72 60 Repeat Steps 4 6 thirty times 7 94 60 56 8 50 50 9 72 60 Repeat Steps 7 9 six times 10 72 600 11 4 Pause *Note: An annealing temperature decrease of 0.5oC after each successive cycle. The non-specific band was preferentially amplified over the desired product and therefore the VH and VL DNA template concentrations were increased with a touchdown PCR amplification. A range of DMSO concentrations (2%, 4%, 6%, 8% and 10% v/v) were used in the PCR with Red Taq polymerase and Phusion polymerase, respectively. Component L volume 5X Buffer G* 10 L scForward Primer 1 L scReverse primer 1 L dNTP mix 1 L VH chain (20 ng DNA) 4 L VL chain (20 ng DNA) 4 L Red Taq/Phusion polymerase 2 L /0.5 L DMSO - H2O - *For composition of Buffer G, see page 51 The PCR amplification was performed using the above components with an increased DNA concentration of VH and VL (20 ng) with varying concentrations of DMSO and H2O investigating 2 different polymerases in the PCR. The PCR amplified reactions were as follows (i) supplemented with 2% (v/v) DMSO (ii) supplemented with 4% (v/v) DMSO (iii) supplemented with 6% (v/v) DMSO (iv) supplemented with 8% (v/v) DMSO (v) supplemented in 10% (v/v) DMSO (vi) positive control (in-house scFv) and (vii) negative control (no VH and VL template) for each tested polymerase. 57 The PCR conditions using the Red Taq polymerase were as follows; Steps Temperature (oC) 1 94 60 2 69 30* 3 72 60 Time (seconds) Repeat Steps 1 3 thirty times 4 72 600 5 4 pause *Note: An annealing temperature decrease of 0.5oC after each successive cycle. The PCR conditions using the Phusion polymerase were as follows: Steps Temperature (oC) 1 98 30 2 98 10 3 69 20* 4 72 30 Time (seconds) Repeat Steps 2 4 thirty times 5 72 600 6 4 pause *Note: An annealing temperature decrease of 0.5oC after each successive cycle. Other polymerases with proof reading capabilities were investigated. A number of polymerases were added to the PCR reaction to determine their influence on amplification. The various polymerases were (i) Red Taq, (ii) Phusion, (iii) Vent (NEB) and (iv) Pfu (Promega) polymerase. A touchdown PCR was performed using the above polymerases using the following reaction component compositions: 58 L volume Component 5X Buffer G* 10 L scForward Primer 1 L scReverse primer 1 L dNTP mix 1 L VH chain (20 ng DNA) 4 L VL chain (20 ng DNA) 4 L Polymerase 0.25 L DMSO (5 % (v/v)) 2.5 L H20 - *For composition of Buffer G, see page 51 The PCR amplification was performed using the above components with various polymerases (Red Taq, Phusion, Vent and Pfu) and H2O. The PCR amplifications were (i) supplemented with 5% (v/v) DMSO using Red Taq polymerase (ii) supplemented with 5% (v/v) DMSO using Phusion polymerase (iii) supplemented with 5% (v/v) DMSO using Vent polymerase in buffer G (iv) supplemented with 5% (v/v) DMSO using Pfu polymerase in buffer G (v) supplemented in 5% (v/v) DMSO in Vent polymerase in Vent buffer (vi) supplemented with 5% (v/v) DMSO in Pfu polymerase in Pfu buffer (vii) positive control (in-house scFv) and (viii) negative control (no VH and VL template). The PCR conditions using the Red Taq polymerase were as follows; Steps Temperature (oC) 1 94 60 2 69 30* 3 72 60 Time (seconds) Repeat Steps 1 3 thirty times 4 72 600 5 4 pause 59 *Note: An annealing temperature decrease of 0.5oC after each successive cycle. The PCR conditions using the Phusion, Vent and Pfu polymerase were as follows; Steps Temperature (oC) 1 98 30 2 98 10 3 69 20* 4 72 30 Time (seconds) Repeat Steps 2 4 thirty times 5 72 600 6 4 pause *Note: An annealing temperature decrease of 0.5oC after each successive cycle. The proof reading polymerases did not enhance the amplification of the desired product. Therefore, the PCR amplification was performed with a number of DMSO concentrations (3%, 5%, 8% and 12% (v/v)) using Red Taq polymerase and the desired 800 bp band was successfully amplified and large scale amplification performed. Components 50 L volume 5X buffer G* 10 L Sc Forward primer 1 L Sc Reverse primer 1 L dNTP mix 1 L VH 4 L VL 4 L H2O 24.5 L 5% (v/v) DMSO 2.5 L Red Taq polymerase 2 L *For composition of Buffer G, see page 51 The PCR conditions using the Red Taq polymerase were as follows; 60 Steps Temperature (oC) 1 94 60 2 69 30* 3 72 60 Time (seconds) Repeat Steps 1 3 thirty times 4 72 600 5 4 pause The amplified SOE-PCR product (scFv) was separated on a 1% (w/v) agarose gel, DNA excised and gel purified. The DNA was quantified using the Nanodrop ND-1000 spectrophotometer. The gel-purified DNA was incubated with the restriction enzyme SfiI (NEB) in parallel with the pAK 100 phagemid vector. 2.2.9 pAK 100 vector purification using Wizard miniprep DNA purification system A single colony of E. coli XL1-Blue cells harbouring the pAK 100 (all vectors were donated by Andreas Pluckthun, University of Zurich, Switzerland) was picked from a streaked agar plate and grown overnight at 37oC at 200 rpm in 2xTY media supplemented with 25 µg/mL chloramphenicol. The overnight culture was centrifuged at 4,000 rpm for 10 min at room temperature. The supernatant was discarded and the pellet thoroughly resuspended in 250 L cell resuspension solution. The suspension was transferred to a 1.5 mL sterile centrifuge tube and 250 L cell lysis solution added, inverted 4 times and incubated at room temperature for 5 min. The mixture was homogenised by inversion and incubated at room temperature for 5 min. Neutralisation solution containing guanidine hydrochloride, potassium acetate and glacial acetic acid (350 L) was added and mixed by inverting the tube, as indicated before. This solution causes the genomic DNA and proteins present in the solution to precipitate. The bacterial lysate was centrifuged at 14,000 rpm at room temperature for 10 min and the supernatant-containing plasmid DNA carefully transferred to a spin column and centrifuged at 14,000 rpm at room temperature for 1 min. The plasmid DNA binds to the membrane of the column which was washed using 750 L column wash solution (previously diluted in 100% (v/v) ethanol). The column was re-washed using 250 L column wash solution and centrifuged at 14,000 rpm at room temperature for 2 min. 61 The DNA membrane spin column was transferred to a new sterile 1.5 mL centrifuge tube and the plasmid DNA eluted using 100 L molecular grade H2O. The eluted plasmid DNA was stored at -20oC. 2.2.10 SOE-PCR restriction using SfiI enzyme and ligation into pAK 100 vector for phage display analysis and ethanol precipitation The gel-purified scFv fragment and pAK 100 vector for phage selection were cut using SfiI restriction enzyme. The SfiI restriction enzyme allows for the unidirectional cloning of scFv fragment into the pAK 100 phage display vector. It recognises 8 bases which are interrupted by 5 non-recognised nucleotides (5ggccnnnnnggcc3), thus virtually eliminating internal digestion in antibody sequences. The fragments were incubated at 50oC for 5 hours in a water bath. The pAK 100 vector and SOE-PCR fragment were digested using SfiI as follows: 40 L volume Components SOE-PCR product 1.23 L NEB 10 X buffer 2 4 L 10 X BSA 4 L H2O 24.77 L SfiI 6 L 40 L volume Components pAK 100 vector 6 L NEB 10 X buffer 2 4 L 10 X BSA 4 L H2O 25 L SfiI 1 L Following large scale digestion, the restricted pAK 100 vector and scFv fragment were gel-purified from a 1% (w/v) agarose gel using the Promega gel clean up kit. The DNA concentrations were determined on the Nanodrop ND-1000 spectrophotometer and the 62 purified scFv fragment ligated into the purified pAK 100 vector using a ratio of 3:1 (insert to vector) using T4 DNA ligase overnight at 16oC. The ligation mixture contained the following components; Components 60 L volume pAK 100 vector 24 L scFv fragment 14.62 L 5 X ligase buffer 12 L H2O 7.28 L NEB T4 DNA ligase 2 L After ligation the mixture was ethanol precipitated to remove any salt which would interfere with the electroporation and cause arcing. Components 218 L volume Ligation mixture 60 L 3 M sodium acetate 6 L 100% (v/v) ethanol 150 L Novagen pellet paint 2 L The ligation mixture was stored overnight at -20oC. It was centrifuged at 14,000 rpm for 20 minutes at 4oC and the pellet washed using 70% (v/v) ice cold ethanol. The mixture was centrifuged at 14,000 rpm for 10 min at 4oC and the pellet resuspended in 10 L of molecular grade H2O. After precipitation the DNA was resuspended in a small volume of water sterile molecular grade H2O to ensure a high DNA concentration for the transformation. 2.2.11 Preparation of electrocompetent E. coli cells The surface, pipettes, shaking incubator, culture flasks, centrifugation bottles and rotor were thoroughly bleached prior to electrocompetent cell preparation thus ensuring a clean working environment free of phage contamination. A 10 mL volume of SB medium supplemented with 30 µg/mL tetracycline was inoculated with a single E. coli colony picked aseptically from a freshly streaked agar plate and grown overnight at 63 37oC at 200 rpm. Three conical flasks each containing 500 mL of SB with 20% (w/v) glucose and 1 M MgCl2 were individually inoculated with 2.5 mL aliquots of the overnight culture. The flasks were shaken at 200 rpm at 37oC until the optical density (600 nm) reached 0.7. The conical flasks were then incubated on ice and chilled with the centrifugation bottles at 4oC for 15 minutes. The culture was carefully transferred into the pre-chilled centrifugation bottles and centrifuged at 4,000 rpm for 20 min at 4oC. The supernatant was removed and the pellets resuspended in 25 mL of ice cold 10% (v/v) glycerol. Two pellets were combined with 10% (v/v) glycerol and centrifuged at 4,000 rpm for 20 min at 4oC. The pellets were resuspended in 10% (v/v) glycerol and centrifuged at 4,000 rpm for 20 min at 4oC. The supernatant was removed, the pellet resuspended in 25 mL 10% (v/v) glycerol, transferred to prechilled 80 mL polypropylene tubes and centrifuged at 3,500 rpm for 15 min at 4oC. The supernatant was carefully removed without disturbing the pellet. It was resuspended in 5 mL 10% (v/v) glycerol, transferred to 1.5 mL centrifugation tubes, flash frozen in liquid nitrogen and stored at -80oC. 2.2.12 Electro-transformation of XL-1 blue E. coli cells with an scFv-containing plasmid An aliquot of electrocompetent XL-1 blue E. coli cells were transformed with the ethanol precipitated ligation mixture (Section 2.2.10). This was executed using a Gene Pulser Xcell electroporation system with the controls set at 25 F, 1.25 kV and the Pulse Controller at 200 . The E. coli cells were thawed on ice and the ligated product carefully added to the cells, mixed, incubated on ice for 1 min, the mixture was transferred to an ice-cold 0.2 cm electroporation cuvette. The mixture was gently tapped to the bottom of the electroporation cuvette. The cuvette was placed in the ShockPod and the chamber lid closed. A pulse was passed through the suspension in the cuvette chamber. The cuvette was removed from the chamber and quickly flushed with prewarmed SOC medium (37oC). The electroporated cells were quickly resuspended using a sterile 1 mL filter tip. The period between applying the pulse and transferring the cells to the growth media is crucial for recovering E. coli transformants. A delay in transfer, even by 1 min, causes a 3-fold drop in transformation. The 1 mL suspension was transferred to a 20 mL sterile centrifugation tube and the cuvette flushed with a further 2 64 mL of SOC media. The transformed cells were shaken for 1 hour at 250 rpm (Sanyo orbital incubator) at 37oC. Serial dilutions of the transformed library were performed by diluting the library in SOC media and spreading 100 µL of each dilution on 2xTY plates supplemented with 25 g/mL chloramphenicol and 1% (w/v) glucose. The 3 mL library suspension was centrifuged at 4000 rpm for 10 min, resuspended in SOC media and uniformly spread on 2xTY plates supplemented with 25 g/mL chloramphenicol and 1% (w/v) glucose. A vial containing non-transformed XL-1 blue E. coli cells (negative control) was plated out on agar plates with 25 g/mL chloramphenicol and 1% (w/v) glucose. The plates were incubated overnight at 37oC. Individual colonies were counted on the dilution plates and the library size determined. The transformed colony stock plates were scraped using a spreader and the suspension centrifuged at 4,000 rpm for 10 min. The cells were resuspended in 20% (v/v) glycerol-supplemented 2xTY media, transferred to sterile 1.5 mL centrifugation tubes and flash frozen in liquid nitrogen. The cell library stocks were stored at -80oC. 2.2.13 Rescue of scFv-displaying phages and precipitation of phage-displayed fragments To rescue the scFv-displaying phages, a conical flask containing 200 mL 2xTY media with 2% (w/v) glucose and 25 g/mL chloramphenicol was inoculated with 200 L from the glycerol library stocks. The flask was shaken at 200 rpm and 37oC until the mid-exponential phase of growth was reached (OD600 ~ 0.400) and the contents then centrifuged at 4,000 rpm for 10 min at room temperature. The pellet was resuspended in fresh 200 mL 2xTY media with chloramphenicol (25 g/mL) and 2 x 1011 pfu/mL helper phage. The mixture was incubated without shaking for 30 min at 37oC enabling the initiation of the infection process (F pilus conjugation) of the E. coli cells. The culture was shaken at 200 rpm at 37oC for 2 hours, 50 g/mL kanamycin (helper phage resistance gene) added and the culture incubated overnight at 200 rpm at 30oC. 2.2.14 Phage precipitation using PEG/NaCl The 200 mL culture was transferred to a sterile 250 mL Sorvall and centrifuged at 4,000 rpm for 15 min at 4oC to remove the bacterial pellet. The phage were precipitated by 65 transferring the supernatant to a clean, sterile 250 mL Sorvall centrifuge tube and adding 8 g of PEG 8,000 (4% w/v) and 6 g of sodium chloride (3% w/v). The PEG/NaCl was dissolved by shaking the suspension at 180 rpm for 10 min at 37oC. The 250 mL centrifuge tube was placed on ice for 1 hour at 4oC to precipitate and concentrate the phage-scFv particles. After incubation the centrifugation tube was spun at 8,000 rpm (7,649 x g) for 25 min at 4oC. The phage scFv pellet was resuspended in 2 mL TE buffer in 2% (w/v) BSA solution. The resuspended phage scFv pellet was transferred to sterile 1.5 mL centrifuge tubes and centrifuged at 14,000 rpm (19,500 x g) for 5 min at 4oC. The supernatant containing phage scFv was placed on ice and stored at 4oC until required for selection against the required antigen. 2.2.15 Selection of antigen-binding phage scFv fragments by biopanning using immobilised antigens Immunotubes (Nunc, Maxisorp) were coated with 500 L of 50 g/mL fPSA overnight at 4oC. After blocking with 3% (w/v) BSA in PBS (150 mM, pH 7.4) for 2 hours at room temperature, 500 L of rescued phage were added and the tubes incubated on a roller mixer SRT1 (Stuart) for 2 hours at room temperature. The immunotubes were washed with PBST (x 3) and PBS (x 3). The washing steps remove any non-specific phage particles that do not bind the immobilised antigen which if not removed could be propagated in subsequent rounds of selection. PBST removes phage adsorbed due to non-specific hydrophobic interactions, while PBS removes phage adsorbed by nonspecific ionic interactions. Bound phages were eluted using 500 L of 100 mM glycineHCl, pH 2.2, for 10 min, removed and transferred to a sterile 1.5 mL centrifugation tube containing 30 L of 2 M Tris-HCl, pH 8.0, for neutralisation purposes. Half of the eluted phage (265 L) was added to mid-exponential phase (OD600 ~ 0.4) XL-1 Blue cells and allowed to infect for 30 min at 37oC without shaking for pilus retraction. After 30 min the culture was shaken at 200 rpm for 1 hour at 37oC and centrifuged at 4,000 rpm (3,824 x g) for 10 minutes at room temperature. The pellet was resuspended in 600 L fresh 2xTY media and spread on 2xTY agar plates supplemented with 25 µg/mL chloramphenicol and 1% (w/v) glucose and incubated overnight at 37oC at 200 rpm. Input and output titres for each round of selection were performed by infecting E. coli cells and performing serial dilutions of the phage. The infected cells were incubated for 66 15 min at 37oC and 100 µL of each dilution spread on 2xTY agar plates containing 25 g/mL chloramphenicol and 1% (w/v) glucose. The serial dilution plates were incubated overnight at 37oC. The input and output titres were calculated by counting the number of cells present on the dilution plates. The stock plates containing the eluted phage were scraped using 2xTY media for the next round of selection. The infected cells were grown in 100 mL of 2xTY media with 2% (v/v) glucose and 25 g/mL chloramphenicol until the midexponential phase of growth was attained (OD600 ~ 0.4). The enriched cells were centrifuged and resuspended in glucose-free 2xTY media supplemented with helper phage. The helper phage were allowed infect the E. coli cells for 30 min at 37oC and the culture vigorously shaken for 2 hours at 37oC. After 2 hours, kanamycin was added and the culture was shaken at 200 rpm overnight at 30oC. In the second round of selection the antigen coating concentration on the surface of the immunotubes for antigen binding phage remained the same (500 µL of 50 µg/mL fPSA) but more stringent washes (PBST (x 5) and PBS (x 5)) were utilised to reduce non-specific binding of phage to the immobilised antigen. The phage were eluted in the same manner as described in the previous round of selection. Input and output phage plates were incubated at 37oC and XL-1 Blue cells infected with half of the eluted phage, incubated for 30 min at 37oC without shaking initially, then with shaking for 1 hour at 37oC and the cells were subsequently uniformly spread on 2xTY agar plates. The third round of selection involved using 100 mL 2xTY media containing 2% (v/v) glucose and 25 g/mL chloramphenicol with less antigen (500 L of 10 g/mL fPSA) coated on the surface of the immunotubes and an increased number of washes (PBST (x 10) and PBS (x 10) to enhance the selection process and ensuring enrichment of positive clones. The fourth round of selection involved rescuing the phage-scFv fragments in 100 mL of 2xTY media with 2% (v/v) glucose and 25 g/mL chloramphenicol and biopanning against 500 L of 2 g/mL fPSA immobilised on the surface of the immunotubes with a further increase in the number of washing steps (PBST (x 10–15) and PBS (x 10–15)). After washing, an antigen challenge was performed which involved incubating fPSA (500 µL of 10 µg/mL) in the immunotubes for 10 min to remove any weakly binding phage-scFv fragments. Phage-scFv particles bound to the fPSA-coated immunotube were eluted and the phage infected into mid-exponential phase E. coli cells. During the fifth round of 67 biopanning, the immunotube was coated with 500 µL of 5 g/mL fPSA and washed (PBST (x 13) and PBS (x 13)). 2.2.16 Polyclonal and monoclonal phage ELISA and scFv check. An ELISA plate was incubated with 100 L of 2 g/mL fPSA overnight at 4oC. The fPSA was removed from the wells and a blocking solution of 3% (w/v) BSA in PBS (150 mM, pH 7.4) added to each well, plates were incubated for 1 hour at 37oC. The cells containing the rescued phage from the original library and each round of selection were grown in 10 mL 2xTY media until a culture OD600 ~ 0.4 was reached, and then centrifuged. The cells were resuspended in 2xTY media supplemented with helper phage, incubated for 30 min at 37oC without shaking, shaken at 200 rpm for 2 hour at 37oC, kanamycin added and cultures incubated overnight at 30oC at 200 rpm to ensure protein expression. The cultures were centrifuged at 4,000 rpm (3,824 x g) for 10 min at room temperature and the phage supernatant diluted 10-fold in 1% (w/v) BSA PBST. One hundred L of diluted phage supernatant were added in duplicate to the plate and the plate was incubated for 1 hour at 37oC. After incubation the wells were washed with PBST (x 3) and PBS (x 3). After washing, 100 L of 1/3,000 dilution of a HRPconjugated anti-M13 antibody (Pharmacia) in 1% (w/v) BSA PBST were added and the plate incubated for 1 hour at 37oC. Binding of HRP-conjugated anti-M13 antibody was detected using TMB as substrate for HRP. This solution was left to develop for 20 min and the reaction quenched using 50 L of 10% (v/v) HCl. The absorbance was then read at 450 nm. Colonies (20) were randomly picked from the fifth round of biopanning and a colony pick PCR performed to ensure the presence of the scFv insert. The PCR amplification was performed as follows: 68 Volume (154 L) Components Buffer G 88 L sc forward primer 8.8 L sc reverse primer 8.8 L DMSO 22 L dNTPs 8.8 L Red Taq polymerase 17.6 L Individual colonies were picked using sterile tips and mixed with the PCR components (above) and a PCR profile executed as follows: Steps Temperature (oC) 1 94 60 2 69 30 3 72 60 Time (seconds) Repeat Steps 13 thirty times 4 72 600 5 4 Pause The amplified scFv fragments were resolved on a 1% (w/v) agarose gel. The polyclonal phage ELISA showed the high absorbance signal from round 5 of biopanning. Therefore, a monoclonal phage ELISA was performed on individual colonies selected from rounds 4 and 5 of biopanning. Individual colonies were grown in 200 L 2xTY media supplemented with 2% (v/v) glucose and 25 g/mL chloramphenicol in sterile 96 well plates until the mid-exponential growth phase. The plates were centrifuged at 4,000 rpm for 10 min at room temperature and the pellets resuspended in fresh 2xTY media (180 L) containing 25 g/mL chloramphenicol and helper phage. After 30 minutes incubation at 37oC the cultures were shaken for 2 hours at 200 rpm at 37oC. After the 2 hour incubation period 2xTY media (20 L) supplemented with 500 g/mL kanamycin was added to each well and incubated overnight at 30oC. 69 An ELISA plate, coated overnight with 100 L of 2 g/mL fPSA, was blocked with 200 L of 3% (w/v) BSA in PBS (150 mM, pH 7.4) for 1 hour at 37oC. The overnight phage-scFv expressed cultures were centrifuged at 4,000 rpm for 10 min at room temperature. One hundred µL of the supernatant was incubated with 100 µL of 1% (w/v) BSA in PBST for 1 hour at 37oC. The pre-blocked phage (100 L) were added to single wells for 1 hour at 37oC. Following washing with PBST (x 3) and PBS (x 3), 100 L of HRP-conjugated anti-M13 antibody (1:3,000 dilution) were added to each well and the plate incubated for 1 hour at 37oC. The wells were washed using PBST (x 3) and PBS (x 3). The substrate, TMB, was added, incubated for 20 min and quenched using 10% (v/v) HCl. The absorbance was read at 450 nm. 2.2.17 Inhibition phage ELISA Three individual clones were grown in 20 mL 2xTY media containing 2% (v/v) glucose and 25 g/mL chloramphenicol until the mid-exponential phase of growth was reached. The cultures were centrifuged at 4,000 rpm for 10 min at room temperature and the pellet resuspended in fresh 2xTY media (20 mL) supplemented with 40 L commercial helper phage (NEB) and 25 g/mL chloramphenicol. The mixture was placed at 37oC for 30 minutes without shaking and then shaken at 200 rpm for 2 hours at 37oC. The antibiotic kanamycin (20 µl of 50 µg/mL) was added, and the cultures shaken overnight at 30oC. An ELISA plate coated overnight with 100 L of 2 g/mL fPSA was blocked using 3% (w/v) BSA in PBS for 1 hour at 37oC. The cultures containing individual phage scFv clones were centrifuged at 4,000 rpm for 10 min at room temperature. The supernatant was incubated with various fPSA concentrations in 1% (w/v) BSA in PBS (final concentration 5 g/mL, 1 g/mL and 200 ng/mL fPSA) for 30 min at room temperature in 1.5 mL centrifugation tubes. Varying fPSA concentrations were incubated with the phage-scFv supernatant to determine if the selected clones were binding fPSA in solution. The supernatant for each clone containing no fPSA was diluted to ensure the same concentration for each selected clone. The phage supernatants (100 L) were added in triplicate to the ELISA plate and incubated for 1 hour at 37oC. Any phage binding to the fPSA in solution was removed by the washing steps (PBST (x3) and PBS (x3)). An anti-M13 bacteriophage HRP-labelled antibody (1/3,000 dilution) was added to detect any phage-scFv fragments binding positively to 70 the adsorbed fPSA on the surface of the ELISA plate. The substrate, TMB, was added and the reaction quenched after 20 min by adding 50 L of 10% (v/v) HCl. The absorbance was read at 450 nm. 2.2.18 ScAb expression vectors (pMoPAC 16 & 53), plasmid preparation and gel purification The pMoPAC series of vector DNA were kindly donated by Dr. Andrew Hayhurst (Department of Virology and Immunology, Southwest Foundation for Biomedical Research, 7620 NW Loop 410, San Antonio, TX 78227, USA). The DNA was transformed into E. coli XL-1 Blue cells using the Gene Pulser using the same procedure as previously described (Section 2.2.12). The transformed cells were rescued, spread uniformly on 2xTY agar plates, containing 25 g/mL carbenicillin and 2% (w/v) glucose, and incubated overnight at 37oC. A single colony containing the plasmid DNA was picked, used to inoculate 2xTY media containing 25 g/mL carbenicillin, cultures were grown overnight at 37oC. The plasmid preparation was performed using a Wizard miniprep kit as described in Section 2.2.9. Vector DNA was stored at -20oC until required. 2.2.19 Vector and SOE-PCR product digestion The plasmid DNA and the scFv fragment were cut by incubation with the restriction enzyme SfiI for 5 hours at 50oC in a water bath. For soluble expression the scFv was subcloned into the pMoPAC vector from the pAK 100 phagemid vector. Plasmid purification was carried out on an overnight culture of the pan 5 rescue glycerol cell stock. The E. coli cells containing the scFv fragments from the fifth round of selection were chosen due to the large number of positive fragments represented in the polyclonal phage pool as determined by ELISA (Figure 3.9) The plasmid DNA was subsequently amplified by PCR using the SOE primers to generate the scFv fragment. The murine anti-fPSA scFv was amplified from the plasmid DNA using the following components and PCR conditions: 71 Volume (500 L) Components Steps Plasmid DNA 20 L Buffer G 50 L sc forward primer 10 L sc reverse primer 10 L dNTPs 10 L H2O 365 L 3% (v/v) DMSO 15 L Red Taq polymerase 20 L Temperature (oC) Time (seconds) 1 94 60 2 69 30* 3 72 60 Repeat Steps 13 thirty times 4 72 600 5 4 Pause * 0.5oC decrease in annealing temperature after each consecutive completed cycle. The amplified scFv was gel-purified from a 1% (w/v) agarose gel and DNA quantified using the Nanodrop ND-1000 spectrophotometer. The recombinant pMoPAC vector DNA and the gel-purified scFv were digested using the restriction enzyme, SfiI, for 5 hours at 50oC in a water bath. After digestion the fragments were resolved on a 1% (w/v) agarose gel and gel purified. The DNA was quantified using the Nanodrop ND1000 spectrophotometer and the scFv subcloned into the pMoPAC vectors. 72 Volume (100 L) Components pMoPAC 16 & 53 Vector 50 L NEB 10 X buffer 2 10 L 10 X BSA 10 L H2O 20 L SfiI restriction enzyme 10 L Volume (50 L) Components Mouse scFv insert 16.88 L NEB 10 X buffer 2 5 L 10 X BSA 5 L H2O 7.12 L SfiI restriction enzyme 16 L 2.2.20 Sub-cloning the restricted scFv fragment into the restricted vector fragments for scAb expression The gel-purified SfiI restricted scFv insert fragment was subcloned into scAb soluble expression vectors overnight. Components Volume (30 L) pMoPAC 16 & 53 vector 4.66 L & 4.57 L scFv fragment 12 L NEB 5 X ligase buffer 6 L H2O 2.34 L & 2.43 L T4 ligase enzyme 5 L The ligated product was ethanol-precipitated and transformed into Tuner cells (Novagen Section 2.1.5) which were previously transformed with the pRARE plasmid (supplied by Dr. Paul Clarke, DCU, Ireland). The transformed cells were rescued using SOC media (3 mL) with shaking for 1 hour at 200 rpm (37oC). Serial dilutions were uniformly spread on 2xTY agar plates supplemented with 25 g/mL carbenicillin 73 (pMoPAC vector resistance) and stock plates incubated overnight at 37oC. The stock plates were scraped to prepare glycerol stocks which were stored in the -80oC freezer until required. 2.2.21 Soluble expression of scAb fragments in pMoPAC 16 and pMoPAC 53 vectors Individual colonies were picked and grown in 2xTY media supplemented with 25 g/mL carbenicillin overnight at 180 rpm at 37oC, in sterile 96 well plates. These colonies were used as back-up stocks for each positive clone selected. Twenty L from the overnight cultures were inoculated into fresh 2xTY media (180 L) containing 0.5 % (v/v) glycerol and 0.05 % (v/v) glucose (final concentration) (1 x 505 – allows autoinduction of cells without changing growth media) (Studier et al., 2005), 1mM MgSO4 and 25 g/mL carbenicillin. The sterile 96 well plates were shaken at 37oC at 180 rpm until a cell density of ~0.600 (cells appear turbid) was achieved. A final concentration of 1 mM IPTG was added to each individual well and plates were shaken overnight at 180 rpm at 30oC. An ELISA plate coated overnight with 100 L of 1 g/mL fPSA was blocked with 3% (w/v) BSA in PBS for 1 hour at 37oC. The expressed antibody culture lysate was extracted by placing the plates at -80oC for 10 min until frozen and then thawing the plates at 37oC. This freeze-thaw method of extraction bursts the E. coli cells releasing periplasmic proteins. This procedure was repeated 3 times in total and the plates centrifuged at 4000 rpm for 10 minutes at room temperature. One hundred L of scAb was added to the ELISA plate and incubated for 1 hour at 37 oC. The plates were washed using PBST (x 3) and PBS (x 3) to remove non-specific proteins. The scAb was detected using 100 L of anti-human kappa C light chain antibody labelled with alkaline phosphatase (1/2000 dilution). The substrate, 4pNPP, was added, the plate incubated for 30 minutes at 37oC and the absorbance read at 405 nm. 74 2.3 Generation of chicken-derived anti-fPSA recombinant antibody library 2.3.1 Removal of spleens from chickens immunised with fPSA, RNA extraction and cDNA synthesis Chickens (Leghorn females) were immunised between the ages of 5–8 weeks. When the titre was deemed sufficient (after 3rd booster administration with over 1:100,000 response determined by ELISA) for recombinant antibody library generation the spleens (2) were removed, RNA extracted, as described in Section 2.2.3, and cDNA synthesised by reverse transcription, as outlined in Section 2.2.4. The cDNA acted as the template for the generation of both chicken anti-fPSA scFv and Fab antibodies by PCR. Preparation of the chicken anti-fPSA scFv library involved amplifying the VH and VL chains separately and linking them with a glycine-serine linker, as described in Section 2.2.8. The generation of the chicken anti-fPSA Fab library was more complicated and involved a number of essential steps: (i) amplifying the VH and VL chain genes separately, (ii) performing the first PCR-overlap of the VH with the constant heavy chain (CH) and VL with the constant light chain (CL), (iii) final PCR-overlap of the VH/ CH with VL/ CL to generate the Fab. 2.3.2 PCR Primers VH Primers CHybVH (forward) 5-GCT GCC CAA GCC ATG GCC GCC GTG ACG TTG GAC GAG TCC 3 CHybIg-B (reverse) 5-CGA TGG GCC CTT GGT GGA GGC GGA GGA GAC GAT GAC TTC GGT CCC 3 Vλ Primers CSCVK (forward) 5-GTG GCC CAG GCG GCC CTG ACT CAG CCG TCC TCG GTG TC 3 CHybL-B (reverse) 75 5-AGA TGG TGC AGC CAC AGT TCG TAG GAC GGT CAG GGT TGT CCC GGC 3 CH Primers HIgGCH1-F (forward) 5-GCC TCC ACC AAG GGC CCA TCG GTC 3 dpseq (reverse) 5-AGA AGC GTA GTC CGG AAC GTC 3 CL Primers HKC-F (forward) 5-CGA ACT GTG GCT GCA CCA TCT GTC 3 Lead-B (reverse) 5-GGC CAT GGC TGG TTG GGC AGC 3 VH /CH Overlap Primers LeadVH (forward) 5-CGA ACT GTG GCT GCA CCA TCT GTC 3 Dpseq (reverse) 5-AGA AGC GTA GTC CGG AAC GTC 3 Vl /Cl Overlap Primers CSC-F (forward) 5-GAG GAG GAG GAG GAG GAG GTG GCC CAG GCG GCC CTG ACT CAG 3 Lead-B (reverse) 5-GGC CAT GGC TGG TTG GGC AGC 3 Primers for assembly of chimeric heavy chain with chimeric light chain sequence CSC-F (forward) 5-GAG GAG GAG GAG GAG GAG GTG GCC CAG GCG GCC CTG ACT CAG 3 dp-Ex (reverse) 5-GAG GAG GAG GAG GAG GAG AGA AGC GTA GTC CGG AAC GTC 3 76 The primers listed above were obtained from Eurofins MWG (Anzinggerstr. 7a, 85560, Ebersberg, Germany) and are compatible with the primers set described by Barbas for the pComb vector system (Barbas et al., 2000). 2.3.3 Amplification of VH and VL chain genes for Fab library generation Each PCR amplified chain (VH or VL) was resolved on an agarose gel, gel-purified and the purified DNA quantified using the Nanodrop ND-1000 spectrophotometer. The variable region genes were amplified using different concentrations of MgCl2 (1.5, 2.0, 2.5 and 3.0 mM), respectively. MgCl2 promotes activity of the DNA polymerase and is a crucial component for successful amplification. The VH and Vλ chain genes were amplified by PCR using the following components and conditions: Component L volume cDNA 1 L dNTP 1 L 5 X Buffer (Promega) 5 L Forward Primer 1 L Reverse Primer 1 L H2O - Taq polymerase 0.25 L MgCl2 - Steps Temperature (oC) 1 94 300 2 94 15 3 56 15 4 72 90 Time (seconds) Repeat Steps 2 4 thirty times 5 72 600 77 The constant heavy and light chains were amplified from the pComb3XTT vector by a co-worker (Dr. William Finlay) and used in the first SOE-PCR by joining the VH chain with the CH and VL chain with the CL chain genes. Component 20 L volume Variable region 1 L Constant region 1 µL dNTP 1 L 5 X Buffer (Promega) 5 L Forward Primer 1 L Reverse Primer 1 L H2O 6.75 µL Taq polymerase 0.25 L MgCl2 3 µL Steps Temperature (oC) 1 94 300 2 94 15 3 56 15 4 72 120 Time (seconds) Repeat Steps 2 4 fifteen times 5 72 600 The heavy and light chain constructs were gel-purified, ethanol-precipitated and quantified using the Nanodrop ND-1000 spectrophotometer. The final SOE-PCR involved joining the 2 gel-purified domains to generate the Fab construct. The final overlap was performed using a Platinum Taq high fidelity polymerase (Bio Sciences Ltd., 3 Charlemont Terrace, Crofton Road, Dun Laoghaire, Dublin, Ireland) to eliminate the introduction of mutations in the construct which may inhibit binding to the antigen. 78 Component 50 L volume Heavy chain region 1 L Light chain region 1 µL dNTP 1 L 10 X Buffer 5 L Sense Primer 0.5 L Reverse Primer 0.5 L H2O 38.8 µL Platinum Taq polymerase 0.2 L MgSO4 2 µL Steps Temperature (oC) 1 94 300 2 94 15 3 56 15 4 72 180 Time (seconds) Repeat Steps 2 4 Twenty five times 5 72 600 After large scale amplification, the construct was resolved on a 2% (w/v) agarose gel, purified and quantified using the Nanodrop ND-1000 spectrophotometer. The next stage involved SfiI digestion of both the gel-purified Fab construct and pComb3XSS phagemid vector at 37oC for 5 hours. Component 200 L volume Fab construct 60 µL 10 x buffer 2 20 µL 10 x BSA 2 µL SfiI 8 µL H20 110 µL 79 Component 200 L volume pComb3XSS 60 µL 10 x buffer 2 20 µL 10 x BSA 2 µL SfiI 8 µL H20 110 µL After gel purification, the digested overlap PCR product was ligated into the pComb3XSS vector (supplied by Barbas). The ligation was performed overnight at room temperature and the plasmid DNA ethanol precipitated at -20oC. The ethanol precipitate was centrifuged at 4oC and the pellet resuspended in molecular grade H2O. Component 400 L volume pComb3XSS vector 120 µL Fab construct 90 µL 5 x ligase buffer 80 µL Ligase enzyme 10 µL H2O 100 µL 2.3.4 Transformation into XL-1 Blue E. coli cells The ligation mixture was transformed into electrocompetent E. coli XL-1 Blue cells and the library size determined by counting the number of colonies from plate dilutions. Both chicken anti-fPSA scFv and Fab antibodies were selected by biopanning the phage against fPSA adsorbed onto the wells of ELISA plates. In the first round of biopanning 8 wells of an ELISA plate were coated with 100 L of fPSA (10 g/mL), blocked with 3% (w/v) BSA in PBS (150 mM, pH 7.4), and the eluted phage, after PEG precipitation, added to each well and allowed to bind for 1 hour at 37oC. Each well that contained phage was washed with PBST (x 3) and PBS (x 3). After washing 100 L of 10 mg/mL trypsin in PBS (150 mM, pH 7.4), was added to each well and the plate incubated for 30 min at 37oC. The pComb 3XSS vector has a trypsin-sensitive site that is cut following the addition of trypsin. Phage were aspirated from the wells by pipetting the content of the wells vigorously i.e. removing the liquid in each and releasing it again into the well. 80 Half of the eluted phage (400 L) were added to mid-exponential phase E. coli XL-1 Blue cells. The culture was placed at 37oC for 15 min and then shaken for 1 hour at 200 rpm and 37oC. Input and output phage titres were determined, as previously described in the mouse anti-fPSA scFv biopanning protocol. In round two of selection, 2 ELISA plate wells were coated with 100 L of fPSA (10 g/mL) and in round three of selection, 1 ELISA plate well was coated with 100 L of fPSA (10 g/mL). The numbers of washing steps for each round of selection were PBST (x 5–10), PBS (x 5– 10 second round) and PBST (x 10–15) and PBS (x 10–15 third round), respectively. Gradually decreasing the concentration of antigen and increasing the washing stringency encourages the selection of positive antibody recombinant fragments from the generated library. Phage were eluted as before using 100 L of 10 mg/mL trypsin per well and used to infect E. coli XL-1 Blue cells. After each round of selection the infected E. coli cells were spread on 2xTY agar plates containing 100 g/mL carbenicillin and 2% (v/v) glucose. Individual colonies (5) from each round of biopanning were selected and a scFv check performed. This involved picking a colony and performing a scFv-specific PCR. The components and PCR conditions were as follows: Volume (20 L) Components Steps Forward primer 0.2 L Reverse primer 0.2 L dNTPs 0.4 L Buffer G 2 L H2O 16.4 L Red Taq polymerase 0.8 L Temperature (oC) Time (seconds) 1 94 60 2 69 30 3 72 60 Repeat Steps 13 thirty times 4 72 600 5 4 Pause 81 A polyclonal phage ELISA was performed, as described previously (Section 2.2.16), and colonies from round 3 of biopanning analysed for soluble expression of chicken anti-fPSA scFv fragments. Individual clones were selected from 2xTY agar plates, supplemented with 100 g/mL carbenicillin, and functionally expressed. The positive clones binding to fPSA on the surface of the ELISA plate were subjected to further examination. A DNA fingerprinting assay was performed on 5 anti-fPSA Fab clones selected from the original library and 5 clones from the final round of selection. The clones were cut using a high frequency cutting enzyme, AluI to check for diversity. The DNA from the clones was resolved on a 3% (w/v) agarose gel. The eluted phage from both the chicken anti-fPSA scFv and Fab antibodies were used to infect E. coli TOP10F cells for soluble expression of the antibodies. Individual clones were picked, grown overnight at 37oC, sub-cultured into fresh 2xTY media supplemented with 1 x 505 (50 % (v/v) glycerol and 5 % (v/v) glucose solution) and 1 mM MgSO4 and grown until turbid (OD600 ~ 0.6). Media supplemented with IPTG (1mM) was added to each clone and the plate shaken overnight at 30oC. Antibodies expressed in the periplasmic space were extracted from the E. coli cells using the freeze-thaw method and individual clones analysed by ELISA. The positive clones were stored in 2xTY media supplemented with 20% (v/v) glycerol at -80oC. High throughput screening of selected positive anti-fPSA clones were evaluated on the Biacore A100 by Dr. Paul Leonard and the data analysed. 2.4 Murine anti-fPSA scAb analysis using the Biacore 3000 instrument Analysis of murine anti-fPSA scAb was performed on a Biacore 3000TM instrument using a CM5 dextran chip. The running buffer was HBS buffer, pH 7.4, containing 10 mM HEPES, 150 mM NaCl, 3.4 M EDTA, and 0.05% (v/v) Tween 20. The running buffer was passed through a 0.22 µm nylon filter and degassed using a Millipore filtration unit directly before use. 2.4.1 Mouse monoclonal anti-human kappa light chain purification The mouse monoclonal anti-human kappa light chain clone KP-53, supplied in ascites fluid from Sigma, was purified by Protein G affinity chromatography. The Protein G82 Sepharose 4B fast flow slurry resin (500 µL) was equilibrated using 20 mL of PBS (300 mM, pH 7.4). An aliquot (200 L) of anti-human kappa light chain monoclonal antibody, diluted in 5 mL of PBS (300 mM, pH 7.4) was added to the resin. The solution was passed through the resin and re-circulated 5 times in total. The resin was washed using 20 mL of PBS (300 mM, pH 7.4), to remove the ascites fluid. The captured monoclonal antibody was eluted in 0.1 M glycine-HCl solution, pH 2.4. Collected fractions were neutralised with 1 M Tris-HCl solution, pH 7.5. The fractions were pooled and passed through a PD-10 desalting column (removal of low-molecular weight compounds) to remove any NaCl residue that may affect downstream processing (for successful immobilisation the ligand must be in a pure form). The PD-10 column was equilibrated with 20 mL H2O and the eluted protein G-purified fraction applied to the resin. The anti-human kappa light chain mAb passes quickly through the resin by size exclusion; 500 L fractions were collected. An Immunodot blot was performed using an AP-labelled anti-mouse antibody to determine the fractions containing the purified antibody. The fractions were pooled and centrifuged in a Millipore Amicon ultrafiltration device (Vivaspin 6 – 10,000 molecular weight cut-off) to ensure a high concentration for preconcentration studies and immobilisation. 2.4.2 Biacore maintenance The Biacore instrument was cleaned using a ‗super desorb‘ program at 25oC. This method involves cleaning the unit thoroughly prior to starting an assay. It is imperative that the instrument is properly sanitized to remove any residual protein, thus, ensuring high-quality data and complete confidence in results generated. A maintenance chip was docked and primed 5 times with desorb solution 1 (0.5 % v/v sodium dodecyl sulphate (SDS)), once with water, 5 times with desorb solution 2 (50 mM glycine, pH 9.5), and, finally, 5 times with water. 2.4.3 Preconditioning of the CM5 dextran sensor chip After docking the CM5 chip, the dextran layered surface was cleaned and hydrated using 2 consecutive injections each of 100 mM HCl, 50 mM NaOH and 0.5% (v/v) 83 SDS. These solutions were passed over each of the 4 flow cells for 10 seconds at a flow rate of 100 µL/min. 2.4.4 Preconcentration studies To achieve maximum immobilisation of the desired protein to the sensor surface, preconcentration studies were performed. This important step results in an electrostatic interaction between the negatively charged carboxyl group-rich sensor surface and positively charged amine groups on the protein surface. At pH values lower than the isoelectric point (pI), the protein carries a ‗net‘ positive charge and this facilitates the binding event. The protein G-purified anti-human kappa light chain antibody diluted in 10 mM sodium acetate solution at pH values of 4.0, 4.2, 4.4, 4.6, 4.8 and 5.0, was injected over a blank surface for 1 min at a flow rate of 5 µL/min. The pH at which the highest ‗apparent‘ binding was observed, compared to the underivatised surface and was used for immobilisation purposes. 2.4.5 Immobilisation conditions The CM5 dextran surface is a three dimensional surface containing carboxyl groups (COOH) attached to the gold surface layer. It was activated by mixing equal volumes of 400 mM EDC (N-ethyl-N-(dimethyl-aminopropyl) carbodiimide hydrochloride) and 100 mM NHS (N-hydroxysuccinimide) and injecting the mixture over the surface for 7 min at a flow rate of 10 µL/min. The EDC solution strips a H ion from the carboxyl group resulting in a negatively charged sensor surface. The NHS solution encourages binding of the amine groups in the protein to the COO- -rich (negatively charged) surface. The anti-human kappa light chain, diluted in the appropriate pH buffer, was injected over the activated surface for 30 min at 10 µL/min. Unreactived NHS groups were capped, and deactivated and non-covalently bound proteins removed using 1 M ethanolamine hydrochloride, pH 8.5, for 10 minutes at 10 µL/min. After capping the surface was regenerated using 10 µL of 10 mM NaOH solution. 84 2.4.6 High throughput analysis of murine anti-fPSA scAbs using Biacore 3000 A fPSA custom kinetics wizard program was created to analyse and appropriately rank the murine anti-fPSA scAbs selected from the library after biopanning. A capture assay format was designed for the analysis and subsequent ranking of the murine anti-fPSA scAbs. Each murine anti-fPSA scAb clone was grown in 20 mL 2xTY media supplemented with 100 g/mL carbenicillin, 1 x 505 (50 % (v/v) glycerol and 5 % (v/v) glucose solution), 1 mM MgSO4 and expressed overnight at 30oC after induction with ITPG. The cultures were centrifuged at 4,000 rpm (3,824 x g) for 10 min at 4oC, pellets were resuspended in 900 L PBST and the protein lysates extracted using the freezethaw method. The bacterial pellets were centrifuged at 14,000 rpm (13,384 x g) for 10 min and the lysate (900 µL) added to 100 L 10X BSA/CM dextran (120 mg/mL) solution to reduce the chance of non-specific binding to the dextran chip surface. The assay involved capturing three scAb clones on flow cells 2, 3 and 4 while leaving flow cell 1 blank for reference subtraction. Each murine anti-fPSA scAb was injected over the anti-human kappa mAb-immobilised surface for 5 min at a flow rate of 20 µL/min. After capturing 3 murine anti-fPSA scAbs, the fPSA (30 nM) was passed over the captured scAbs for 3 minutes at a flow rate of 30 µL/min. The fPSA molecule was allowed dissociate for 5 min over the captured scAbs and early and late stability responses recorded. HBS was passed over the captured scAbs and the binding response subtracted from the fPSA binding response. Thus, double subtractive referencing was employed. The chip surface was regenerated using a 30 second pulse of 20 mM NaOH at a flow rate 30 L/min. The percentage stability of each clone was determined by exporting the raw data for each curve generated using BIAevaluation software and evaluating it in Excel. Early stability report points were calculated for a 10 second period (between 10 and 19 seconds) starting 10 seconds after the end injection of 30 nM fPSA, while the late stability percentage report point was calculated for a 10 second period between 291 and 300 seconds. The complex stability was determined by dividing the stability late by the stability early. If the stability early was less than 5 RU it was discarded. Using a 68% complex stability cut-off of 30 nM fPSA remaining after 5 minute dissociation, selected murine anti-fPSA scAbs were further analysed by kinetic analysis. 85 2.4.7 Kinetic analysis of selected clones using Biacore 3000 instrument The best performing murine anti-fPSA scAbs were captured on the anti-human kappa mAb-immobilised surface and various concentrations of fPSA (1, 0.5, 0.25, 0.125, 0.063 and 0.03 g/mL) injected over the captured antibody using HBS buffer as a negative control for reference subtraction. The data were analysed using BIAevaluation software and the kinetic curves generated. Association and dissociation rate constants were calculated in addition to the affinities of each selected scAb. The calculated affinities were very similar to those determined by kinetic analysis. 2.5 Murine anti-fPSA scAb clone sequencing The murine anti-fPSA scAbs were sent to Eurofins MWG for sequencing. The clones were sent as agar stab cultures in 1.5 mL centrifugation tubes supplemented with carbenicillin (100 µg/mL) and chloramphenicol (25 µg/mL) antibiotics. The DNA sequences for each scAb was received and the amino acid sequences determined. The heavy and light chain regions of the antibody were identified using the Kabat rules for antibody CDR regions. The programmes used for the amino acid sequence determination and alignment of the clones were found in the Expasy website. Expasy translate tool: http://www.expasy.ch/tools/dna.htmL ClustalW: http://www.ebi.ac.uk/Tools/clustalw2/index.htmL 2.6 Selection of most sensitive murine anti-fPSA scAb by ELISA The affinities determined by Biacore were generally quite similar, with a 10-fold difference between the best and worst analysed scAb. Therefore, to determine the most suitable clones for assay development an ELISA was performed. The clones were expressed and the protein extracted by freeze-thawing. A plate coated overnight with 1 µg/mL fPSA was blocked with 3% (w/v) BSA and serial dilutions of each scAb incubated for 1 hour at 37oC. Any binding scAbs were detected using AP-labelled antihuman kappa antibody and 4pNPP as substrate. The absorbances were read at 405 nm. Once the optimal scAb dilution was calculated, an inhibition assay was performed with 500, 333, 222, 148, 98, 66, 44, 29, 19.5, 13 and 8.67 ng/mL fPSA. These fPSA dilutions 86 were incubated with the optimal murine anti-fPSA scAb dilution for each clone for 1 hour and added to the ELISA plate. After 1 hour incubation the wells were washed and any bound scAb detected using AP-labelled anti-human kappa antibody and 4pNPP as substrate. The absorbances of the wells were read at 405 nm and the scAb with the highest degree of inhibition selected for immunoassay design. 2.7 Large-scale expression of a murine anti-fPSA scAb and subsequent purification using immobilised metal affinity chromatography (IMAC) Centrifugation tubes containing a culture of cells which have been induced overnight (500 mL SB media), was spun at 12,000 rpm (11,472 x g) for 10 min at 4oC. The pellets were resuspended in 30 mL of sonication buffer (1X PBS + 0.5 M NaCl + 20 mM imidazole). The resuspended pellets was transferred into thirty 2 mL centrifugation tubes (thirty one mL samples) and each sample sonicated on ice using the appropriate setting (40 amp for 45 seconds with 3 second intervals). The sonicated-scAb containing samples were centrifuged at 14,000 rpm (13,384 x g) for 10 min at 4oC and the lysate was passed through a 0.2 m sterile filter. A 4 mL column of nickel NTA resin slurry (Qiagen) was prepared and equilibrated with 30 mL of running buffer (sonication buffer + 1% v/v Tween). The filtered lysate was passed through the column once and the flowthrough collected. The column was washed with 30 mL of running buffer to remove any non-specifically adsorbed proteins. The recombinant antibody fragment was eluted with 20 mL of 100 mM sodium acetate solution, pH 4.4. The eluted antibody fragment was collected in 1.5 mL centrifugation tubes (final volume 500 L in each tube) containing 50 L 10X PBS (filtered through a 0.2 m filter) and 50 L 100 mM NaOH. An immunodot blot was performed on each eluted fraction where 2.5 L was spotted onto Whatman chromatography paper (0.45 µm Protran BA 85 nitrocellulose). The blot was blocked with 3% (w/v) BSA for 1 hour at room temperature on a rotation table. The membrane was washed with PBST (x 1) and PBS (x 1) and an AP-labelled anti-human C kappa antibody diluted in 1% (w/v) BSA in PBS was added to the blot for 1 hour at room temperature. After washing the blot with PBST (x 3) and PBS (x 3) the liquid substrate (BCIP) was added. The positive fractions were pooled together and added to a spin column with a 5,000 Dalton MWCO. This column was centrifuged at 4,000 rpm (3,824 x g) at 4oC until concentrated to a volume of 500 L. Five mL of filtered PBS 87 was added to the column. This was stored on ice overnight at 4oC. The following day the sample was centrifuged at 4,000 rpm (13,384 x g) at 4oC until concentrated to a 500 L volume. The concentration of protein was calculated on the Nanodrop ND-1000 spectrophotometer and an SDS-PAGE and Western blot performed. The recombinant antibody was stored in 20 L aliquots at -20oC until required. 2.8 Induced scAb cell expression profile A culture (100 mL SB media) supplemented with 100 g/mL carbenicillin, 25 g/mL chloramphenicol, 1 x 505 and 1 mM MgSO4 was inoculated with 500 L of the murine anti-fPSA scAb clone B5 culture and grown until an OD600 of 0.7 was reached. A 1 mL sample was removed from the culture and stored at -20oC. The culture was then induced with IPTG (1 mM final concentration) and shaken overnight at 30oC. One mL samples were removed from the culture every hour and stored at -20oC. The protein was extracted from each sample using the freeze-thaw method (-80oC, 37oC (x3)). Periplasmic extracts were analysed by SDS-PAGE, Western blot and ELISA. 2.9 Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) The following stock solutions were prepared for gel casting: 30% (w/v) acrylamide containing 0.8% (w/v) bis-acrylamide, 1.5 M Tris-HCl, pH 8.8, 0.4% (w/v) SDS, 0.5 M Tris-HCl, pH 6.8, 0.4% (w/v) SDS and 10% (w/v) ammonium persulfate. The glass plates were washed with acetone and sealed with a gasket and grips. Free radicalinduced polymerisation of the resolving gel acrylamide (Table 2.1) was catalysed by addition of ammonium persulphate and the accelerator TEMED and the gel added to the space between the plates and covered with a layer of ethanol. Following polymerisation of the gel, the ethanol was removed and the stacking gel placed directly onto the resolving gel. A plastic comb was placed in this gel creating the wells for sample application. Once the gel had fully polymerised, the plates were then placed in an electrophoresis chamber, the comb was removed and the chamber and wells filled with electrophoresis buffer (25 mM Tris, 250 mM glycine (electrophoresis grade), pH 8.3 and 0.1% (w/v) SDS). 88 Table 2.1: SDS-PAGE gel reagents Solution Resolving Gel Stacking Gel 30 % Acrylamide (w/v) 10 mL 1.66 mL Distilled Water 5.0 mL 5.75 mL Resolving Gel Buffer 5.0 mL - Stacking Gel Buffer - 2.5 mL Ammonium Persulphate 200 mL 70 mL TEMED 20 µL 14 µL The gel was removed from the casting tray apparatus and stained using Coomassie blue solution. The gel was transferred to destain solution and incubated until the protein bands were clearly visible. 2.10 Western blotting The SDS-PAGE gel was resolved and the proteins transferred to nitrocellulose paper. The gel, extra thick blotting paper and nitrocellulose were incubated in transfer buffer for 20 min before the actual transfer. Extra thick blotting paper (Sigma - #P7176) was placed on the bottom of the transfer apparatus, the nitrocellulose was carefully placed on top of the blotting paper, the SDS-PAGE gel was added on top of the nitrocellulose and extra thick blotting paper placed over the SDS-PAGE gel. The proteins were transferred to the nitrocellulose using 15 V for 20 min. The nitrocellulose was carefully removed from the apparatus and placed in 3% (w/v) BSA solution for 1 hour. The membrane was washed and incubated with anti-human C kappa AP-labelled antibody in 1% (w/v) BSA in PBST for 1 hour. The membrane was washed and BCIP liquid substrate carefully added to the blot. The reaction was quenched using 100 mM EDTA to stop the development of background colour. 2.11 Avian clone analysis on Biacore A100 The avian anti-fPSA scFvs and Fabs were analysed by Dr. Paul Leonard in Uppsala, Sweden using the A100 Biacore. An anti-Cλ surface was created on the surface of all four flow cells using amine coupling chemistry. Ninety six anti-fPSA Fab antibodies 89 were screened with each antibody tested against three fPSA concentrations (30, 5 and 0 nM). The analysis consisted of 3 ‗start-up‘ cycles with buffer followed by 72 sample cycles. Each analysis cycle consisted of a 5 minutes injection of antibody across the whole flow cell. Free PSA (fPSA) was then injected for 2 minutes with a 10 minutes dissociation time. The surface was finally regenerated with a 30 second injection duration of 10 mM NaOH. The time required to analyse 96 anti-fPSA Fab was 26 hours in total. All sensorgrams were reference subtracted against an unmodified CM5 surface. The zero concentration was then subtracted from the 30 and 5 nM sensorgrams (double referencing) to remove any systematic instrument disturbances. A 1:1 Langmuir equation with a mass transfer term and two separate Rmax for the two surfaces was globally fitted to the data. Twenty six series were found acceptable (classified OK) by the software and the determined kinetic constants can be trusted. Seventeen were classified as inspect by the software but the reasons were either a large residual segment or low signal to noise ratios. Since a capture system was used and the baseline drift after the capture injection was significant a double referencing procedure was used before the % remaining calculation was performed. The report point at 0 nM fPSA was subtracted from the same report point collected at 30 nM fPSA. The % remaining after dissociation was calculated by dividing the response after 5 min dissociation (stability late response) with a report point placed immediately after the end of the sample injection (stability early response). 2.12 Epitope Mapping of murine and avian anti-fPSA recombinant antibody fragments using the Biacore 3000 instrument Using an anti-human C kappa mAb-immobilised CM5 dextran chip (4-3 subtractive referencing) epitope mapping was investigated using fPSA sandwiched between a captured murine anti-fPSA scAb and avian anti-fPSA scFv/Fabs. The purified murine anti-fPSA scAb clone B5 was passed over the surface of the sensor chip for 6 min at a flow rate of 5 L/min. An aliquot containing 30 nM fPSA (1 g/mL) was passed over the captured scAb for 2 min at a flow rate of 5 µL/min. The extracted avian clone lysates were passed over the captured fPSA molecule for 2 min at a flow rate of 5 µL/min and the data analysed for binding. The surface was regenerated using 10 mM 90 NaOH solution for 1 min using an increased flow rate of 30 µL/min. Two negative controls were employed to ensure no non-specific binding to the anti-human C kappa mAb-immobilised surface or to the captured scAb fragment. FPSA (30 nM) was passed over the immobilised anti-human kappa mAb surface to ensure no non-specific binding to the surface had occurred. The other negative control involved passing the avian clone lysate directly over the captured murine anti-fPSA scAb with no fPSA intermediate molecule. No non-specific binding was observed. 2.13 Biacore subtractive inhibition assay for the detection of fPSA using the purified murine anti-fPSA scAb After preconcentration studies, a CM5 dextran chip was immobilised with fPSA using EDC/NHS chemistry with approximately 6,968 RU binding. Regeneration studies were performed to ensure a reliable, reusable working surface. The murine anti-fPSA scAb was injected over the fPSA surface with 40 regeneration cycles using 10 mM NaOH solution. The binding signal after each injection was measured to verify a suitable and stable working surface for assay development. Serial dilutions of the purified murine anti-fPSA scAb (1:50, 1:100, 1:200, 1:400, 1:800, 1:1,600, 1:3,200, 1:6,400, 1:12,800, 1:25,600, 1:51,200 and 1:102,400) diluted in HBS buffer were injected over the fPSA immobilised surface for 2 min at a flow rate of 10 µL/min. The contact time of the murine anti-fPSA was evaluated by increasing the injection time from 2 min to 4 and 6 min, respectively. The flow rate was evaluated to determine if more antibodies could potentially bind to the surface at a higher flow rate. The inhibition assay was performed using 2 murine anti-fPSA scAb dilutions (1:10,000 and 1:20,000 dilution) with a number of fPSA concentrations (1.56 g/mL, 312, 62.5, 12.5, 6.25, 2.5, 1.0 and 0.5 ng/mL). These samples were incubated for 1 hour at 37oC and injected over the surface. A decrease in binding response was observed with increasing fPSA concentrations. Inter-day studies were performed at both scAb dilutions with coefficients of variation and percentages accuracies (determined for each concentration) back-calculated from the calibration curve. 91 2.14 Murine anti-fPSA scAb inhibition ELISA The optimal working dilution was determined by incubating serial dilutions (1:400, 1:800, 1:1,600, 1:3,200, 1:6,400, 1:12,800, 1:25,600 and 1:51,200) of the murine antifPSA in 1% (w/v) BSA PBST for 1 hour at 37oC on a pre-blocked fPSA-coated ELISA plate. The murine anti-fPSA scAb was detected using anti-human kappa AP-labelled antibody and 4pNPP as substrate. The plate was incubated for 30 min at 37oC and the absorbance read at 405 nm. The inhibition assay was performed using 2 scAb dilutions (1:10,000 and 1:20,000) with a number of fPSA concentrations (100, 66, 44, 29, 19, 13, 8.7, 5.6 and 3.9 ng/mL). These samples were incubated for 1 hour and then added to a fPSA-coated plate which was subsequently incubated for 1 hour at 37oC. Binding was detected using anti-human C kappa AP-labelled antibody and 4pNPP as substrate. The absorbances of each well was determined at 405 nm. Inter-day studies were performed at both scAb dilutions with coefficients of variation and percentages accuracies, determined for each concentration, back-calculated from the calibration curve. 2.15 Biacore sandwich assay using commercial anti-tPSA mAb (CanAg PSA 36 mAb) After pre-concentration studies, the anti-tPSA mAb was successfully immobilised (approximately 17,000 RU) onto the surface of a CM5 dextran chip using EDC/NHS chemistry. Aliquots of fPSA (10, 5, 1 g/mL, 500, 250, 125, 62.5, 31.25, 15.63 and 7.81 ng/mL) were injected over the anti-tPSA mAb-immobilised surface and the data points exported into Excel and a graph plotted. Various dilutions of a commercial anti-fPSA mAb (CanAg PSA 30 mAb) (1:40,000, 1:32,000, 1:16,000, 1:8,000, 1:4,000 and 1:2,000) and murine anti-fPSA scAb (1:40,000, 1:25,000, 1:20,000, 1:15,000, 1:10,000, 1:8,000, 1:4,000, 1:2,000, 1:1,000 and 1:500) in HBS buffer containing 10X CM Dextran/BSA (120 mg/mL) were passed over 60 ng/mL captured fPSA at 20 µL/min for 4 min. Non-specific binding was observed by injecting the diluted antibodies over the anti-tPSA mAb surface without the fPSA interface so the assay was unreliable as the detection antibody was binding non-specifically to the immobilised anti-tPSA mAb. 92 2.16 Biotinylation of purified murine anti-fPSA scAb for sandwich assay development The purified murine anti-fPSA scAb was biotinylated using the Pierce EZ-link biotinylation kit. The concentration of antibody and the levels of successful biotinylated were calculated as follows: A 20-fold molar excess of biotin reagent was incubated with the antibody to ensure a high ratio level of biotin per antibody molecule. The millimoles (mmol) of biotin reagent to add to the antibody to have a 20-fold molar excess were calculated using equation (A): (A) 560 µL scAb x 1.6 mg scAb x 1 mmol scAb 560 µL scAb 45,000 mg scAb x 20 mmol biotin 1 mmol scAb = 0.000711 mmol biotin To calculate microlitres of 10 mM biotin reagent solution to add to the scAb reaction equation B (below) was used: (B) 0.000711 x 557 mg mmol biotin x 400 L 2.2 mg = 72 µL biotin added to 560 µL scAb 557 = molecular weight of Sulfo-NHS-LC-biotin 400 = microlitres of H2O in which 2.2 mg Sulfo-NHS-LC-biotin is dissolved to make a 10 mM solution A vial of sulfo-NHS-LC-biotin was removed from the freezer and equilibrated at room temperature until required. The concentration of purified scAb (PBS) was calculated using the Nanodrop ND-1000 spectrophotometer and the scAb then placed on ice. A 10 mM sulfo-NHS-LC-biotin solution was prepared immediately before use by dissolving 2.2 mg sulfo-NHS-LC-biotin in 400 L ultra pure H2O. Twenty seven L of this mixture was incubated with the protein for 2 hours on ice. The NHS esters react to form stable amide bonds with primary amino groups of proteins. After an incubation period 93 of 2 hours the protein was buffer-exchanged to remove excess non-protein bound biotin. This was performed using a ZebraTM desalt spin column. The column was initially centrifuged at 1,000 x g for 2 minutes to remove the storage buffer. The column was equilibrated with 2.5 mL of PBS 150 mM, pH 7.4 and centrifuged at 1000 x g for 2 min. The flow-through was discarded and this equilibration step was repeated twice. The biotinylated scAb sample was applied to the resin and allowed adsorb to the resin. The column was centrifuged at 1,000 x g for 2 min and the purified protein collected in the flow-through. This was immediately dispensed into 20 L volumes and stored at -20oC. The degree of biotin incorporation was determined using the HABA assay test (http://www.piercenet.com/files/1776gj4.pdf). This involved mixing the biotinylated antibody with a mixture of HABA and avidin. Due to the higher affinity of avidin for biotin, the HABA is displaced and this can be evaluated at 500 nm. One mg of avidin was mixed with 60 L of 10 mM HABA and 1.94 mL of PBS. One hundred and eighty L of this solution was dispensed into the well of a Nunc microtitre plate and the absorbance measured at 500 nm (A500 HABA/avidin). Twenty L of the biotinylated protein were added to the well and the solution was mixed using a plate shaker (IKAMTS-2/4 digital-Lennox). The HABA/avidin/biotin absorbance at 500 nm was determined. The moles of biotin per mole of protein was calculated using the Beer Lambert Law A=bC where A is the absorbance at a particular wavelength-no units for absorbance is the absorptivity or extinction coefficient at the wavelength (). For HABA/Avidin at 500 nm the extinction coefficient is equal to 34000 M-1cm-1. b is the cell path length expressed in centimetres (cm). For the microtitre plate format the path length is 0.5 cm. C is the concentration of the protein sample expressed in molarity (= mol/L = mmol/mL) Other values needed to calculate moles of biotin per moles of scAb were Concentration of scAb in the reaction expressed as mg/mL, 94 Molecular weight of the scAb in grams per mole, Absorbance of HABA/avidin solution at 500 nm (A500 H/A) Absorbance of HABA/Avidin/biotin solution at 500 nm (A500 H/A/B) Calculation of mmol protein per mL mmol protein per mL = 1.18 mg/mL 45,000 = 0.00003 The biotin was incubated with murine anti-fPSA scAb, the complex purified and the concentration of biotin linked to the scAb determined by adding the biotinylated scAb (B) with a HABA (H)/avidin (A) solution. Biotin has a higher affinity for avidin and the HABA becomes displaced. This can be measured spectrophotometrically at 500 nm. Calculation of change in absorbance after adding the biotinylated scAb to the HABA/avidin solution: ∆ A500 = ((A500 (0.969) – (A500 (0.747)) = 0.222 Calculation of the concentration of biotin in mmol per mL of reaction mixture mmol biotin____ = mL reaction mixture ∆ A (0.222) ( 34,000 x 1) = 0.00000653 Calculation of mmol of biotin per mmol of protein = (0.00000653) (10) (dilution factor) 0.00002622 mmol per mL protein in original sample = 2.49 mmol biotin per mmol scAb 95 2.17 ELISA-based assay for detection of biotinylated murine anti-fPSA scAb B5 The biotinylated scAb was analysed by ELISA to determine the optimal working dilution for an inhibition assay. The unbiotinylated scAb was used as a negative control to ensure no non-specific binding between the secondary extrAvidin-HRP-labelled antibody and the purified murine anti-fPSA scAb. A plate was coated with 1 g/mL fPSA, blocked with 3% (w/v) BSA in PBS and serial dilutions (1:100, 1:200, 1:400, 1:800, 1:1600, 1:3200, 1:6400, 1:12800 and 1:51200) of the biotinylated and unbiotinylated scAb in 1% (w/v) BSA in PBST added in duplicate to replicate wells. An extrAvidin-HRP-labelled antibody was added and this was detected using TMB substrate. The reaction was quenched after a 10 minute period and the absorbance read at 450 nm. 2.18 Sandwich assay development for improved assay sensitivity A sandwich ELISA using the purified anti-fPSA biotinylated scAb as the capture antibody and avian anti-fPSA clone lysates (1 scFv and 4 Fab) as detection antibodies was investigated. A plate was coated using 5 g/mL neutravidin in PBS (150 mM, pH 7.4) for 1 hour at 37oC. The wells were blocked using 3% (w/v) BSA in PBS for 1 hour at 37oC, washed with PBST (x 1) and PBS (x 1) and incubated with biotinylated murine anti-fPSA scAb for 1 hour at 37oC. The wells were washed with PBST (x 2) and PBS (x 2) and a number of fPSA concentrations (100, 10, 1 and 0.1 ng/mL) were added to individual wells, in duplicate, for 1 hour at 37oC. Any binding of fPSA was detected using avian anti-fPSA clone lysates to determine whether the selected clones bind different epitopes on the fPSA molecule. The wells were washed again with PBST (x 3) and PBS (x 3) and an anti-HA-HRP-labelled secondary antibody added for 1 hour at 37oC. The wells were washed with PBST (x 3) and PBS (x 3). The substrate, TMB, was added and plates were incubated for 10 min at room temperature. The reaction was quenched using 10% (v/v) HCl and the absorbance read at 450 nm. The avian anti-fPSA scFv was successfully purified by IMAC and utilised in a sandwich assay format. 96 2.19 Biacore sandwich assay using a neutrAvidin-immobilised CM5 dextran surface After pre-concentration studies the neutravidin was successfully immobilised onto the surface of a CM5 dextran chip (24425 RU). The biotinylated murine anti-fPSA scAb was injected over flow cell 2 and the captured levels recorded. Aliquots of fPSA (1 g/mL, 500 ng/mL, 250 ng/mL, 125 ng/mL, 62.5 ng/mL, 31.75 ng/mL, 15.75 ng/mL, 7.5 ng/mL, 3.9 ng/mL, 1.9 ng/mL and 0.8 ng/mL) in HBS buffer were injected over the captured biotinylated scAb at a flow rate of 10 L/min for 2 min using subtractive referencing (2-1). The purified avian anti-fPSA scFv (1:500 dilution) was injected over the captured fPSA using a flow rate of 30 µL/min for 2 min. The surface was regenerated using a 30 second pulse of 2.5 mM NaOH solution. 2.20 ELISA sandwich assay using a neutravidin-coated ELISA plate A plate (Nunc Maxisorp) coated with 5 µg/mL neutrAvidin in PBS (150 mM, pH 7.4) for 1 hour at 37oC was blocked using 3% (w/v) BSA in PBS for 1 hour at 37oC and washed using PBST (x 1) and PBS (x 1). An aliquot containing 1 µg/mL biotinylated scAb (1:1,000 dilution) was incubated for 1 hour at 37oC and the plate washed with PBST (x 2) and PBS (x 2). Serial dilutions of fPSA (25, 18.75, 14, 10.5, 7.9, 5.9, 4.45, 3.33, 2.5, 1.8, 1.4, 1.0, 0.79 and 0.59 ng/mL) in 1% (w/v) BSA in PBST were incubated for 1 hour at 37oC. After washing with PBST (x 3) and PBS (x 3), the four anti-PSA antibodies, (i) avian anti-fPSA scFv (1:100 dilution), (ii) commercial anti-fPSA mAb (PSA 30) (1:1,000 dilution), (iii) commercial anti-tPSA mAb (PSA 36) (1:1,000 dilution) and (iv) commercial anti-cPSA mAb (PSA 53) (1:1,000 dilution) were incubated in individual wells for 1 hour at 37oC in 1 % (w/v) BSA in PBST. The plate was washed with PBST (x 3) and PBS (x 3) and each antibody detected. The avian antifPSA scFv was detected using HRP-labelled anti-HA antibody (1:2,000 dilution) and the commercial antibodies with HRP-labelled anti-mouse IgG (1:2,000 dilution) prepared in 1% (w/v) BSA in PBST, following incubation for 1 hour at 37oC. The plate was washed with PBST (x 3) and PBS (x 3), substrate, TMB was added and the plate incubated for 10 min and the reaction quenched using 10% (v/v) HCl. Absorbance values were read at 450 nm. 97 2.21 Neutravidin ELISA plate coating concentration optimisation for improved sandwich assay performance Previous experience within the Applied Biochemistry Group indicated that a high coating concentration of neutravidin was required for a workable assay format. Therefore, a plate was coated with 10 µg/mL and 5 µg/mL neutrAvidin for 1 hour at 37oC. After the plate was blocked with 3% (w/v) BSA in PBS, a 1 µg/mL solution of biotinylated scAb was added to plate wells and the plate incubated for 1 hour at 37oC. After washing, a range of fPSA concentrations (25, 18.75, 14, 10.5, 7.9, 5.9, 4.45, 3.33, 2.5, 1.8, 1.4, 1.0, 0.79 and 0.59 ng/mL) were incubated (in duplicate) for 1 hour at 37oC. After washing the avian anti-fPSA scFv (1:1,000 dilution) was added for 1 hour at 37oC and this was detected using anti-HA-HRP-labelled antibody and TMB as substrate. The reaction was quenched using 10% (v/v) HCl and absorbance read at 450 nm. The optimal coating concentration determined was 5 µg/mL neutravidin. 2.22 TMB incubation time optimisation The sandwich assay was repeated using the optimised conditions, as described in Section 2.21, and the TMB incubation time was evaluated. The substrate was incubated for 10 and 20 min, respectively, before quenching with 10% (v/v) HCl. The absorbance was read at 450 nm. The sensitivity of the assay was improved using a longer TMB incubation period with no increase in background signal. Therefore, by incubating the substrate for 20 min a more sensitive assay was achieved. 2.23 Inter-day and intra-day studies on sandwich assay for precision and accuracy determination Inter-day and intra-day assay studies were performed on the sandwich ELISA using the optimised neutravidin coating concentration and biotinylated scAb dilution with a 20 min TMB incubation period. Calibration curves were generated for both assay formats using BIAevaluation software and the data points exported. The curves were generated in Excel and the coefficient of variation and percentages accuracies calculated for each concentration of fPSA. 98 2.24 Determination of the limit of detection (LOD) for fPSA in samples A sandwich ELISA was performed using 20 blank replicates (1% (w/v) BSA in PBST) with 20 replicates of 2.5 and 1.8 ng/mL fPSA diluted in 1% (w/v) BSA PBST (150 mM, pH 7.4), respectively. Binding of fPSA was detected using the avian anti-fPSA scFv clone (1:500 dilution), plates were washed with PBST (x 3) and PBS (x 3), then incubated with HRP-labelled anti-HA antibody, washed again using PBST (x 3) and PBS (x 3), and finally detected with TMB substrate, the reaction was quenched after 20 min using 10% (v/v) HCl. The absorbance was read at 450 nm. The LOD of the assay is defined as where one can detect with 95% confidence at least 19 out of 20 positive samples mixed with 20 negative samples. It is the lowest concentration of analyte where a response can be reliably distinguished from the background. 2.25 Sandwich assay in spiked female serum Blood was kindly donated by female members within the DCU faculty and centrifuged at 3,500 rpm for 10 min. The serum was aliquoted and stored at -20oC. An inter-day sandwich assay using the biotinylated scAb as the capture antibody and the avian scFv as the detection antibody was performed using fPSA-spiked female serum. A number of concentrations of fPSA (25, 18.75, 14, 10.5, 7.9, 5.9, 4.45, 3.33, 2.5, 1.8, 1.4, 1.0, 0.79 and 0.59 ng/mL) were added to the plate. The assay was repeated over 3 days and the coefficient of variation and percentages accuracies determined from the calibration curve. 2.26 Patient serum sample analysis Serum samples from patients with PCa were kindly donated by Professor William Watson, Conway Institute, UCD, Belfield, Dublin and stored at -80oC. The individual samples with known tPSA concentrations, but unknown fPSA concentrations, were analysed in duplicate using the sandwich assay format. A calibration curve was generated and the unknown concentrations back-calculated from the curve data. The concentration of fPSA was determined for each sample analysed and compared to the tPSA concentration for the appropriate sample. 99 2.27 Immunohistochemical analysis 2.27.1 Sample preparation for murine and avian anti-fPSA analysis (by hand) Representative full face sections from 2 radical prostatectomy cases of PCa were examined for PSA expression. The sections consisted of histologically benign glands and prostate adenocarcinoma. Suitable formalin-fixed paraffin embedded blocks from the cases were collected, sections (4 µm thick) from the formalin fixed paraffin embedded blocks were cut using a microtome (Leica RM2135 Rotary Microtome) and mounted onto VBS Glass Slides Plus (S21.1910.110, Leica). The sections were dried overnight at 37°C. 2.27.2 Immunohistochemistry using avian and murine anti-fPSA recombinant antibody fragments Parraffin sections were deparaffinized with two 5 min incubations in clean xylene followed by three washes with absolute ethanol and finally by distilled water. Antigen retrieval was carried out by incubating the sections in tri-sodium citrate (10 mM), pH 6.0, for 15 min, at high power, in a microwave oven prior to immunohistochemical staining. After incubation the slides were rinsed for 5 min (x 2) in Tris buffer saline (TBS – 150 mM, pH 7.6) followed by endogenenous peroxidase blocking in blocking buffer (Envision Kit (HRP), K4006, Dako) at room temperature. The sections were then washed for 5 minutes (x 2) in TBS and incubated with primary antibody (1:100 dilution avian anti-fPSA scFv and murine anti-fPSA scAb) for 1 hour. After rinsing in TBS for 5 min (x 3) the sections were incubated with HRP-labelled anti-HA antibody. The sections were washed with TBS for 5 min (x 3) and incubated with DAB chromagen substrate (Envision Kit (HRP), K4006, Dako) for 10 min at 37oC. The reaction was quenched by immersing the sections in distilled water. The sections incubated with the DAB chromogen (Envision Kit (HRP), K4006, Dako) were counterstained with haematoxylin (RBA-4213-00A, Reagecon), dehydrated, and the cover slip applied using an automated coverslip machine (Shandon Consul Coverslipper). The tissue sections were visualised under high magnification (100 x magnification). 100 2.27.3 Protocol for immunohistochemistry for diagnostic antibody to PSA (Dako) (automated immunohistochemistry) Deparrafinization, antigen retrieval and immunohistochemistry were performed on the paraffin-embedded 4 µm tissue sections using an automated IHC platform (Bond MaxTM - Leica Microsystems). A polymer-based detection system with 3,3diaminobenzidine (DAB) as the chromogen was used for the generation of a brown colour. The optimal antigen retrieval, concentration of primary antibody and antigen retrieval for PSA (anti-PSA monoclonal A0562, Dako) of 1:15,000 ER 1 for 20 min (Epitope retrieval method 2 Bond MaxTM - Leica Microsystems) were determined by serial dilution. The maximum signal without background immunostaining using the appropriate positive control was determined. Sections were counterstained with haematoxylin, dehydrated, and cover slip applied. The tissue samples were visualised under high magnification (100X magnification). 101 Chapter 3 The generation of recombinant antibodies to human prostate-specific antigen using avian and murine immune systems 102 3.1 Introduction This chapter gives an insight into the generation of recombinant antibody fragments, phage library construction and includes an in-depth overview on the process of selecting positively binding anti-fPSA-specific antibodies. Phage display is the predominant technique utilised in recombinant antibody selection. A phage is a virus capable of infecting male E. coli cells and replicating without causing cell lysis. Random segments of DNA can be inserted into the phage particle and amplified in bacterial cells. Antibody variable genes are commonly inserted into the coat protein of the phage and expressed on the surface of the phage after superinfection with helper phage (Oh et al., 2007). The phage-scFv particle can be isolated using solid phase selection with the cognate antigen. A major advantage of using phage display is the link between the phenotype (protein) and corresponding genotype DNA (Schmitz et al., 2000) allowing for selection of antigen-specific phage clones. The gene encoding the displayed protein is within the phage-scFv particle and this can easily be re-infected into E. coli cells for propagation and further affinity maturation. Recombinant antibody technology is an alternative approach to hybridoma technology for generating antibodies. Recombinant antibodies can be developed using many animal sources as long as the genome in known (Schmitz et al., 2000). Chickens have become an ideal immunological host for the generation of recombinant antibodies due to the selection of high affinity antibodies against highly conserved mammalian proteins (Leonard et al., 2007). Desired clones may be selected using high throughput automated screening facilities whereby 1,000–10,000 clones or more can be rapidly identified (Buckler et al., 2008). It is important to note that since there is less germ-line diversity in chickens than in mice and humans, fewer primers are required for amplifying heavy and light chains. (Barbas et al., 2001). The major advantage of using immunised animals over the conventional naïve antibody library is that the host animal immune system refines specificity towards the cognate antigen. This increases the possibility of selecting high affinity antibodies from a generated recombinant antibody fragment library due to in vivo affinity maturation (Schmitz et al., 2000). Although naïve libraries are becoming more popular for 103 recombinant antibody selection they lack the diversity of the immune system (somatic hypermutation) for generating a large panel of different antibodies. Mice and chickens were immunised with the fPSA molecule according to protocols described by Krebber et al. (1997) and Barbas et al. (2001), respectively. The animals were sacrificed, the spleens were harvested and the ribonucleic acid (RNA) extracted with Trizol reagent using a well established protocol (Section 2.2.3 Barbas et al., 2001). The amplified antibody variable genes were assembled by splice by overlap extension polymerase chain reaction (SOE-PCR Section 2.2.8) and ligated into the pAK 100 (murine scFv) and pComb3XSS (avian scFv/Fab) vectors for ‗biopanning‘ (Section 2.2.15). This is a selection process whereby antigen-specific clones are recovered and enriched for subsequent rounds of panning. 3.2 Murine antibody library generation The Krebber system for recombinant antibody fragment generation allows for the reliable cloning of functionally active antibody variable domains from a secreting hybridoma cell or spleen primed cells (lymphocytes Krebber et al., 1997). This system was successfully employed to generate a murine recombinant antibody fragment library (Section 2.2.12) from the spleen cells of Balb/c mice immunised with fPSA. 3.2.1 Mice immunisation with fPSA antigen Balb/c mice were subcutaneously immunised with fPSA over a period of 3 months and the serum extracted from the tail of the mouse analysed by ELISA to determine the antibody titre (level of immune response Section 2.2.2). The antibody serum titre was determined to ensure fPSA-specific antibodies were being generated and the titre was sufficiently high for recombinant antibody library production. The specific antibody titre obtained was 1/100,000 when screened against fPSA (Figure 3.1). The mouse was sacrificed to harvest the B cells from the spleen for recombinant antibody library generation and selection of positive antibodies to fPSA. 104 Figure 3.1: Titration of serum antibodies. Serum was harvested from the tail of mice immunised with fPSA and an antibody serum titre determined by ELISA. Bound murine antibodies were detected using alkaline phosphatase (AP)-labelled anti-mouse antibody followed by the addition of 4pNPP substrate. The absorbance was read at 405 nm. A high signal was observed at the 1:4,000 dilution and gradually decreases with decreasing concentration of antibody. At 1:128,000 dilution the signal is twice the background and sufficiently high for recombinant antibody library generation. 3.2.2 Primer selection for murine heavy and light chain gene amplification The primer sets for the amplification and assembling of the mouse variable light and variable heavy chain domains are listed in Section 2.2.5. The respective gene fragments (VH and VL) were amplified by PCR using these primers after successful RNA extraction and cDNA synthesis. The gene fragments were gel purified, and coupled with a 20 amino acid glycine-serine repeat linker. The PCR amplified recombinant antibody fragment was compatible for use with the Krebber pAK series of vectors for selection. The gel-purified scFv fragment and pAK 100 vector were digested for 5 hours at 37 oC using the SfiI restriction enzyme. The restricted fragments were gel-purified, quantified 105 using a Nanodrop ND-1000 spectrophotometer and ligated overnight at room temperature using T4 DNA ligase. This enzyme catalyses the formation of covalent phosphodiester bonds between the 3 hydroxyl end and the 5 phosphate end. The ligated mixture was precipitated using ethanol to concentrate the ligated DNA and to remove any contaminating salts which may interfere with the efficiency of electroporation (arcing is a problem often associated with electroporation where the sample is too conductive due to salt concentration and therefore for library construction it is vitally important to remove any residual salt prior to transformation). The DNA was transformed into electrocompetent E. coli XL-1 Blue cells, rescued and recovered on chloramphenicol (pAK 100 vector resistance gene) supplemented agar plates. The library size (2.02 x 106 cfu/mL) was calculated, as described in Section 2.2.12, and the library subjected to 5 rounds of selection on fPSA-immobilised immunotubes. Immunotubes offer high binding capacity with a large surface area for maximum antigen immobilisation and recovery of potential clones. The phage-scFv particles were incubated in the fPSA-coated immunotube, eluted after washing and propagated in E. coli cells. 3.3 Isolation of RNA from murine B cells (spleen) and first-strand DNA synthesis The spleen was removed from the immunised mouse and carefully homogenised in Trizol reagent. The total RNA was extracted from the tissue sample, quantified using a Nanodrop and first-strand cDNA synthesised by reverse transcription. The RNA and cDNA were resolved on a 1% (w/v) agarose gel (Figure 3.2) to confirm the presence of good quality templates for further gene amplification. 106 Figure 3.2: Preparation of cDNA by reverse transcription of RNA isolation from the B lymphocytes from a mouse spleen. Lane 1, 1 kb+ DNA molecular weight marker; lane 2, the cDNA and lane 3, the extracted RNA. Good quality RNA and DNA were extracted from the spleen of the immunised mouse (Figure 3.2). The cDNA template was used in the amplification of the mouse variable heavy and light chain genes using a range of different buffers. The variable genes were successfully amplified by PCR (Figure 3.3) and large scale amplification was performed. 107 3.4 PCR buffer optimisation for murine variable heavy and light chain generation Figure 3.3: VH and VL chain optimisation using different buffer compositions. The buffer composition used for each variable chain PCR amplification was as follows: buffer A (7.5 mM MgCl2, pH 8.5), buffer B (10 mM MgCl2, pH 8.5), buffer D (17.5 mM MgCl2, pH 8.5), buffer G (12.5 mM MgCl2, pH 9.5). Lane 1 and 10, 1 kb+ DNA molecular weight marker; lanes 2–5: VH chain amplification using buffers A, B, D and G, respectively, and lanes 6–9 represent VL chain amplification using buffer A, B, D and G, respectively. The buffer composition that yielded the greatest amplification was buffer G. The role of MgCl2 in the PCR is to promote DNA/DNA interactions and to form complexes with dNTPs. The gel-purified DNA encoding VH and VL fragments were linked by splice-by overlap extension PCR (SOE-PCR) whereby the purified fragments were joined by an oligonucleotide encoding 20 amino acid glycine-serine linker (G4S)4. A smaller linker would cause multimerisation of the scFv and this longer linker provides sufficient flexibility for the VH and VL domains to associate predominantly as scFv monomers. The SOE-PCR protein product, commonly known as the scFv fragment of the IgG molecule, is much smaller than the IgG antibody but still retains its antigen binding specificity to the target molecule and can be exploited for immunoassay development. 108 3.5 Murine SOE-PCR of variable heavy and light chains The gel-purified mouse variable heavy and light chain genes were used for the generation of the 800 bp scFv by PCR amplification. The initial PCR involved amplifying different VH and VL template ratios to generate the desired 800 bp product. Figure 3.4: SOE-PCR optimisation of murine variable domain gene DNA. Lane 1 contains the 1 Kb+ ladder for DNA size determination. Lanes 2 – 10 represent different concentrations of VH: VL in the SOE-PCR. Lane 2, 10 ng VH : 10 ng VL; lane 3, 20 ng VH : 20 ng VL; lane 4, 5ng VH : 5 ng VL; lane 5, 5 ng VH: 10 ng VL; lane 6,5 ng VH : 2.5 ng VL; lane 7, 5 ng VH : 1.25 ng VL; lane 8, 10 ng VH: 5 ng VL; lane 9, 2.5 ng VH : 5 ng VL and lane 10 contains a PCR template ratio of 1.25 ng VH : 5 ng VL. The PCR was performed using Phusion polymerase using the PCR conditions described in section 2.2.8. A non-specific band of approximately 1,000 bp was amplified in addition to the 800 bp SOE-PCR product (Figure 3.4). A number of additives have been proven to enhance PCR amplification (Frankman et al., 1998) increasing specificity and maximising yield of the desire product. Therefore, after comprehensively searching the literature it was decided to evaluate two of these additives (DMSO and BSA) which could potentially eliminate the non-specific 1,000 bp band. The PCR annealing temperatures of the primers can have a major effect on the amplification of a product. A touchdown PCR 109 was also performed in conjunction with the addition of DMSO (5% v/v) and BSA (50 g/mL) to the PCR amplification reaction. A touchdown PCR (Don et al., 1991) enables binding of the primers at high initial annealing temperatures without nonspecific amplification. The annealing temperature decreases after each successive cycle but the desired template should be abundantly present and is preferentially amplified. Fixed concentrations of 10 ng of the VH and VL chains were utilised during the PCR (Figure 3.5). Figure 3.5: SOE-PCR optimisation using DMSO and BSA additives with a 0.5oC primer annealing temperature decrease after each consecutive cycle. Lane 1 contains the 1 Kb+ DNA ladder for DNA size determination; lane 2 represents the PCR amplification product in the presence of 5 % (v/v) DMSO; lane 3 represents the PCR amplification product in the presence of BSA (50 µg/mL); lane 4 represents the PCR amplification product in the presence of 5 % (v/v) DMSO and BSA (50 µg/mL); lane 5 represent the PCR amplification product without any additives and lane 6 is a positive scFv fragment control (internal antibody fragment). The presence of DMSO in the PCR amplification did have a positive effect on reducing the amplification of the non-specific 1,000 bp band. Therefore, a touchdown PCR was performed using varying concentrations of DMSO (2%, 4%, 6%, 8% and 10% v/v) in the PCR reaction. The touchdown PCR was performed using Phusion and Red Taq polymerase to investigate if the type of polymerase had an effect on amplification of the 110 desired product. Betaine was another additive used for the PCR amplification. However, no improvement in amplification was observed (results not shown). Figure 3.6: An SOE-PCR was performed with varying DMSO concentrations using Phusion polymerase and Red Taq polymerase. Lane 1 and 8 contain the 1 Kb+ DNA ladder for size determination; lanes 2–6 represent the PCR amplification in the presence of 2%, 4%, 6%, 8% and 10% (v/v) DMSO using Red Taq polymerase, respectively. Lane 7 is blank. Lanes 9–13 represent the PCR amplifications in the presence of 2%, 4%, 6%, 8% and 10% (v/v) DMSO using Phusion polymerase, respectively. Due to the clear difference in amplified PCR product quality provided using DMSO and the difference between Taq and Phusion polymerases (Figure 3.6) the SOE-PCR was also evaluated with additional polymerases with proof reading capabilities. A touchdown PCR was performed using a number of different polymerases - Red Taq, Phusion, Vent and Pfu polymerases in the presence of a fixed concentration of DMSO (5%). The objective was to determine if amplification could be enhanced by using a number of high fidelity DNA polymerases (Figure 3.7). 111 Figure 3.7: Optimisation of SOE-PCR using different polymerases for the amplification of the scFv product. Lane 1 and 8 contain the 1 Kb+ universal ladder for DNA size determination; lane 2 represents the PCR amplification using Red Taq polymerase; lane 3 represents the PCR amplification using Phusion polymerase; lane 4 represents the PCR amplification using Vent polymerase in buffer G; lane 5 represents the PCR amplification using Pfu polymerase in buffer G; lane 6 represents a positive scFv PCR amplification using Red Taq polymerase; lane 7 represents a negative control; lane 9 represents the PCR amplification using Vent polymerase in Vent buffer and lane 10 represents the PCR amplification using Pfu polymerase using Pfu buffer. Figure 3.7 shows that the optimal amplification was achieved using Red Taq polymerase (lane 2) with little non-specific amplification of the 1,000 bp product. Therefore, the SOE-PCR was repeated using Red Taq polymerase with various DMSO concentrations to improve amplification. The purified VH and VL fragments were successfully ligated together using a glycine-serine linker (Figure 3.8) after considerable optimisation to yield an 800 bp product. 112 Figure 3.8: SOE-PCR using different DMSO concentrations to optimise the product yield. A 1 Kb+ DNA molecular weight marker was added to lane 1. The final concentration of DMSO in each PCR reaction was 3%, 5%, 8% and 12%, respectively, as represented by lanes 2,3,4 and 5. The optimal DMSO concentration for the SOE-PCR was 3% (lane 2), as the highest yielding product was obtained. After large-scale amplification of the desired 800 bp PCR fragment, this DNA fragment was gel purified, precipitated in ethanol, quantified and digested using the SfiI restriction enzyme (Section 2.2.10). The scFv fragment was ligated into a digested pAK 100 vector and then transformed into high efficiency electrocompetent E. coli XL-1 blue cells (developed in-house), rescued and the library size calculated. The mouse antifPSA scFv library size was 2.02 x 106 cfu/mL. The library size is limited by the transformation efficiency of electrocompetent cells and the concentration of DNA available for electroporation (Lin et al., 2002). Large libraries sizes can be generated and transformants rescued to yield a large panel of positive clones. In contrast, 500– 2,000 clones would be expected after a typical hybridoma fusion (Schmitz et al., 2000). PSA-specific antibody fragments were selected on fPSA immobilised on the surface of an immunotube, as described in Section 2.2.15. 113 3.6 Selection of fPSA-specific phage-scFv particles by ‘biopanning’ Five rounds of panning were performed and input and output titres for each round of selection determined (Table 3.1). During the selection process, the concentration of fPSA was decreased and the number of washes were increased as described in Section 2.2.15. This increase in stringency after each successive round of panning is important to ensure positive clones are selected and reduces the levels of clones which have no specificity to the target antigen. After each round of selection, the infected E. coli XL-1 Blue cells were spread uniformly across the entire surface of chloramphenicolcontaining agar plates. The plating of the bio-panned library allows every clone the opportunity to be screened. This is important as certain clones may grow more slowly than other clones due to the antibody toxicity within the cell. Therefore, by plating the cells on agar plates there is an increased chance of selecting high affinity antibody fragments. This affinity selection process allows for enrichment of recombinant phagescFv particles against the immobilised fPSA (Oh et al., 2007). Table 3.1: Selection of murine phage displaying anti-fPSA-specific antibody fragments. A phage-titre, before and after selection on fPSA-immobilised immunotubes, expressed as colony forming units (cfu), is shown. Round (cfu/mL) Round 1 Input 4.9 x 1011 Round 1 Output 5.0 x 106 Round 2 Input 2.0 x 1012 Round 2 Output 5.2 x 107 Round 3 Input 1.3 x 1012 Round 3 Output 3.9 x 104 Round 4 Input 1.6 x 1012 Round 4 Challenge remaining bound 3.0 x 102 Round 4 Challenge dissociated 1.2 x 105 Round 5 Input 2.4 x 1012 Round 5 Output 1.3 x 106 114 3.7 Murine polyclonal phage ELISA Five rounds of selection were successfully completed and the eluted phage from each round analysed by polyclonal phage ELISA described fully in Section 2.2.16 (Figure 3.9). The results of the polyclonal phage ELISA give an indication whether or not positive clones were successfully selected from the panned library. The ELISA signal towards the antigen of interest should increase during the course of the selection process if positive anti-fPSA phage scFv fragments were recovered from the generated mouse anti-fPSA library. The pooled phage contains a collection of recombinant antibody fragments with a range of affinities for fPSA and, therefore, individual clones were analysed by ELISA to determine the binding patterns of the clones. Figure 3.9: Phage pools from the unpanned mouse anti-fPSA scFv glycerol library stock and the phage pool obtained after subsequent rounds of panning were tested for binding to fPSA by ELISA. Bound phages were detected with an anti-M13-HRP-conjugated antibody and the absorbance read at 450 nm. A challenge was performed which involves incubating the immunotube with free antigen to remove any weak binding phage-scFv particles. High affinity antibodies remain attached to the fPSA-coated surface and are subsequently propagated for the next stage of selection. A significant absorbance signal increase in pan five suggests the presence of anti-fPSA scFv-phage fragments. 115 Sixty colonies from round 4 and 5 of selection were picked from the bio-panned stock plates, grown in sterile 96 well microtitre plates, infected with helper phage and shaken overnight at 30oC after the addition of kanamycin. Phage were analysed for binding to fPSA-coated ELISA plates and detected with an HRP-conjugated anti-M13 antibody. Approximately 8% of the selected clones showed binding to fPSA coated on the surface of an ELISA plate from Pan 4 output and 60% were positive from the colonies selected from Pan 5 output (Figures 3.10 and 3.11). 3.8 Murine monoclonal phage ELISA Figure 3.10: Sixty individual colonies were randomly selected and analysed by ELISA. Five of the expressed clones showed significant binding to a fPSA-coated ELISA plate suggesting positive selection of phage-scFv particles from the fourth round of ‘biopanning’. Two of the five positive clones were expressed at high levels with a ‘cut-off’ of twice the background (red dotted line). Figure 3.10 shows the binding of individually selected phage-scFv fragments to fPSA following four rounds of panning. Five of the sixty individual colonies selected were 116 shown to be positive for binding to fPSA by ELISA. A ‗cut-off‘ of twice the background was selected for positive fPSA-specific clones (red dotted line). The monoclonal phage ELISA performed on sixty colonies selected from the fifth round of biopanning showed that 60% of the selected clones positively bound fPSA. The expression levels of the positive phage-scFv particles were different with varying degrees of binding to the fPSA-coated ELISA wells. Figure 3.11: Sixty individual colonies were randomly selected and analysed by ELISA from the fifth round of ‘biopanning’. Sixty percent of the recombinant clones showed significant binding to a fPSA-coated ELISA plate. The expression levels were different for each selected clone with a selected cut-off of twice the background (red dotted line). Three individual clones were selected at random from the positive anti-fPSA scFv monoclonal phage ELISA plates and an inhibition phage ELISA performed using 3 fPSA concentrations (5 µg/mL, 1 µg/mL and 200 ng/mL) to investigate if the selected clones were displaced when mixed with fPSA in solution i.e. that the antibodies demonstrated competitive inhibition (Figure 3.12). 117 Figure 3.12: A monoclonal phage inhibition ELISA was performed on 3 random murine antifPSA scFv clones. The phage were incubated for 1 hour with fPSA (5 µg/mL, 1 µg/mL and 200 ng/mL) and added to a fPSA-coated plate. Binding phage-scFv were detected using an antiM13 HRP-labelled antibody. The absorbance readings were normalised whereby the absorbance for each fPSA concentration incubated with antibody (A) was divided by the maximum absorbance for each selected antibody without antigen (Ao). The three random clones were competitive at the selected concentrations of fPSA as there was a decrease in absorbance with increasing fPSA concentration. Two of the clones showed similar binding patterns to fPSA and could potentially be the same clone while the third clone has a completely different binding profile. Figure 3.12 showed that the selected antibodies were competitive inhibitors and were displayed with fPSA in solution (Section 2.2.17). 118 3.9 Soluble expression of mouse anti-fPSA scAb fragments Soluble expression of the clones selected for binding to fPSA from the fifth round of biopanning involved subcloning the scFv fragment into an appropriate expression vector. The Krebber system uses pAK 400 expression vector for soluble expression of mouse scFv‘s. However, it has been widely accepted that murine scFv express poorly, have problems with folding and tend to aggregate in the periplasm (Hayhurst et al., 2003). These issues have been raised in our laboratory and after extensively reviewing of the literature, papers were sourced where these problems were overcome by reformatting the scFv into a scAb. A paper by Hayhurst et al., (2003) addresses the common pitfalls with murine scFv‘s (poor expression levels and solubility). These scientists revealed that reformatting the recombinant antibody to a scAb significantly improves expression levels and overall stability. The pMoPAC vectors are loosely based on the pAK 400 Krebber vector for soluble expression of mouse scFv. The vector has an additional human constant kappa domain upstream of the scFv insert cloning sites. This allows for expression of a scAb format and the presence of a downstream Skp chaperone also improves scAb solubility. The pMoPAC DNA was kindly donated by Dr. Andrew Hayhurst after setting up collaboration with his group. It was transformed in electrocompetent E. coli XL-1 Blue cells and stored at -80oC. The scFv was amplified by PCR from a plasmid preparation on E. coli XL-1 Blue cellscontaining biopanning output round 5 of selection. In parallel, a plasmid purification was performed on the pMoPAC vectors. The scFv fragment was digested using SfiI restriction enzyme and ligated into pMoPAC 16 and pMoPAC 53 restricted vectors. These vectors express the scFv as a single chain Ab (scAb) with a human constant kappa domain which gives extra stability to the expressed antibody fragment and improves overall protein expression. Electrocompetent Tuner cells were prepared and transformed with the pRARE plasmid. Following sub-cloning the scFv into the pMoPAC vectors, electrocompetent Tuner cells containing the pRARE plasmid were transformed with the ligation mixture and individual clones expressed. The pRARE plasmid codes for 6 rare tRNA codons than enhance expression of eukaryotic proteins in prokaryotic systems. Tuner cells allow uniform entry of IPTG into the cell population and expression can be easily regulated. One hundred and ninety two clones were grown in 96 microwell plates and analysed by direct ELISA. Sixty percent of the selected 119 clones bound to fPSA passively immobilised on the surface of the ELISA plate (Figure 3.13). Figure 3.13 : One hundred and ninety two individual mouse anti-fPSA scAb clones were functionally expressed in 2xTY media supplemented with 1 x 505 (0.5% (v/v) glycerol, 0.05% (w/v) glucose final concentration) containing 100 g/mL carbenicillin and 25 µg/mL chloramphenicol overnight at 30oC after induction with 1 mM IPTG. A 1 µg/mL fPSA -coated ELISA plate was blocked with 200 l of 3% (w/v) BSA in PBS. The lysates were diluted 2 fold with 1% (w/v) BSA in PBST and added to the ELISA plate. The scAb clones were detected with an anti-human kappa light chain antibody labelled with alkaline phosphatase. The substrate, pNPP, was added for 30 minutes at 37oC and the resulting absorbance read at 405 nm. Sixty percent of the expressed recombinant antibody fragments successfully bound a fPSA-coated ELISA plate. A cut-off of twice the background (red dotted line) was selected for positive antifPSA-secreting clones. 120 3.10 Differential expression profile of an anti-PSA scFv and reformatted scAb A comparative analysis of the expression profile between the murine anti-fPSA scFv and its corresponding scAb was performed. A scAb with high expression levels was selected for this study and the aim was to evaluate the expression levels of the recombinant antibody fragments (scFv and scAb) in the same cell strain (Tuner) containing the pRARE plasmid, induced at the same time and expressed overnight at 30oC. An ELISA was performed on the extracted recombinant antibody constructs (scFv and scAb) to evaluate the improvement in expression of the scAb over its scFv counterpart. The recombinant antibody fragments were expressed in 3 mL 2xTY media overnight and analysed by ELISA (Figure 3.14). Figure 3.14: A direct ELISA was performed where a plate was coated with 1 µg/mL fPSA, blocked with 3 % (w/v) BSA and serial dilutions (1:2, 1:4, 1:8, 1:16, 1:32, 1:64, 1:128, 1:256 and 1:512 dilution) of the antibody lysate incubated for 1 hour at 37oC. The lysate was detected using an anti-His-HRP-labelled antibody with TMB as the substrate of choice. The wells were read at 450 nm. The expression level of the murine anti-fPSA scAb was considerable higher than its murine anti-fPSA scFv counterpart. 121 This result confirms the low level of expression of the murine scFv. This is mainly due to improper folding and protein aggregation of the scFv. Importantly, the problem of poor expression was addressed by sub-cloning the scFv into pMoPAC series of vectors for scAb expression. The result in Figure 3.14 confirms that by altering the recombinant antibody format, protein expression levels can be significantly enhanced. Another important aspect is the scAb is expressed with a human constant Kappa domain which facilitates the detection of the expressed antibody by ELISA. This was also utilised later in Section 4.2.3 for Biacore ranking of the selected mouse clones. The key result from Figure 3.14 is the significant improvement of expression which allows for further analysis of selected antibody fragments. 3.11 Selection of chicken anti-fPSA scFv fragments Another animal model used to generate recombinant antibodies to fPSA was chickens. Chickens were immunised with fPSA and a large library generated (3.10 x 108 cfu/mL). The objective was to compare the selected antibody fragments from the anti-fPSA chicken and mouse library and to determine whether or not the chicken antibodies recognised different epitopes than the mouse antibodies on the fPSA antigen and to use them in a sandwich assay format. Chickens are a valuable source of antibodies and have been used extensively in biotechnology for the production of polyclonal antibodies. It has been shown that these animals can be immunised easily with a mixture of multiple antigens and recombinant antibodies selected, using phage display, with little or no cross-reactivity. This section describes the selection of chicken anti-fPSA scFv fragments from a chicken anti-fPSA library. The avian immune system is phylogenetically distant from mammals therefore making chickens a very good source of antibodies against some highly conserved targets, like PSA in mammals. Large diverse chicken recombinant antibody libraries have been generated and high affinity antibodies selected through phage display (Finlay et al., 2005). It is important to note that one chicken can be immunised with a number of target antigens and produce high affinity antibodies to each target with little or no crossreactivity (Finlay et al., 2006). Both the VH and VL chain loci in chickens consist of single functional V-region genes and J-region genes. The VH has an additional D 122 segment for diversification purposes. Further diversification is achieved due to the presence of upstream pseudogenes in both the VH and VL chains. The relative ease of amplification of VH and VL chains due to only 2 primer sets for library production is another advantage for using this source for recombinant antibody production. The eggs from immunised hens are a source of large quantities of polyclonal antibodies which can be extracted and subsequently purified. IgY antibodies have important diagnostic and therapeutic applications particularly in detecting antigens in blood and serum (Greunke et al., 2006). A chicken was immunised with two antigens (PSA and BNP) for recombinant antibody library generation. The spleen and bone marrow were removed from an immunised chicken and the RNA extracted as previously described in the mouse antibody library generation section. After reverse transcription the cDNA template was amplified by PCR to generate the VH and VL chain genes. These were resolved on a 1% (w/v) agarose gel and purified by electroelution. The purified VH and VL DNA were amplified by PCR to generate the scFv. The Fab was generated by doing two separate SOE-PCR amplifications and performing a final overlap to link the two constructs. The variable heavy construct was amplified using the VH and CH chain genes and the variable light construct was amplified using the VL and CL chain genes. Both these fragments were gel purified by electroelution and used in the final SOE-PCR to generate the Fab. The scFv and Fab were SfiI digested with the pComb3XSS for 5 hours at 37oC. The DNA was purified by electroelution and the DNA concentration determined. A ligation was performed overnight and the ligation mixture was ethanol-precipitated before the electroporation. The transformants were rescued on carbenicillin-supplemented agar plates at 37oC. 3.12 Optimisation of avian VH and VL chain genes amplification for Fab library construction The avian anti-fPSA scFv and Fab libraries were generated in conjunction with Dr. William Finlay. The generation of the avian scFv library involved amplifying the VH and VL chain genes separately and joining the gel-purified fragments with a glycineserine linker, as described fully in the murine anti-fPSA scFv library Section 2.3. The 123 generation of a Fab library is different as the VH and VL genes are joined to their respective CH and CL chain genes and the Fab construct is amplified by SOE-PCR joining the heavy variable constant domain with the light variable constant domain. A MgCl2 gradient (3.0, 2.5, 2.0 and 1.5 mM) PCR was used for the amplification of the avian VH and VL (kappa and lambda chain) genes. The genes were resolved on a 1% (w/v) agarose gel (Figure 3.15) and gel-purified. Figure 3.15: Amplification of the avian variable genes using a MgCl 2 gradient. Lanes 1 and 14:1 kb+ DNA molecular weight marker; Lanes 2–5: amplification of the variable heavy chain gene using 3.0, 2.5, 2.0 and 1.5 mM MgCl2, respectively. Lanes 6–9: amplification of the variable light kappa chain gene using 3.0, 2.5, 2.0 and 1.5 mM MgCl 2, respectively. Lanes 1013: amplification of the variable light lambda chain gene using 3.0, 2.5, 2.0 and 1.5 mM MgCl 2, respectively. The PCR amplification for each chain was successful with optimal concentrations amplified using 3.0 mM MgCl2. The first PCR overlap involved joining the variable chains with their counterpart human constant genes to give an 800 bp product (Figure 3.16). It is important to ensure the construct is amplified specifically as incorrect amplification would affect the final SOEPCR overlap. 124 Figure 3.16: The first SOE-PCR using the avian variable genes and the constant human chain genes. Lane 1: 1 kb+ DNA molecular weight marker; lane 2: heavy chain construct (VH and CH), lane 3: kappa light chain construct (VL and CL) and lane 4: lambda light chain construct (VL and CL). The amplification of each construct was successful using the gel-purified VH and VL fragments. After large scale amplification these fragments were gel purified and the Fab construct amplified by SOE-PCR. The Fab generated consisted of the heavy/kappa chain construct and the heavy/lambda chain construct joined using a glycine-serine linker region. These were separately resolved on a 1% (w/v) agarose gel (Figure 3.17), purified, pooled and ligated into the pComb 3XSS vector, ethanol precipitated and transformed in electrocompetent E. coli XL-1 Blue cells for selection. 125 Figure 3.17: The final overlap for the generation of the Fab construct. Lane 1: 1kb+ DNA molecular weight marker; lane 2: heavy/kappa light chain Fab and lane 3: heavy/lambda chain Fab. The Fab fragments were successfully amplified without any non-specific amplification. 3.13 Chicken anti-fPSA scFv selection on fPSA-coated ELISA wells The avian anti-fPSA Fab library was screened and the chicken anti-fPSA scFv library (3.10 x 108 cfu/mL) was bio-panned against fPSA immobilised on the surface of an ELISA plate, as described in Section 2.3.4. The input and output titres were measured and are shown in Table 3.2. It is important to ensure no cross contamination between libraries. If a scFv-phage particle infected the generated Fab library it would be preferentially be packaged quicker than the Fab as it is a smaller fragment and could potentially be selected from the library over the Fab. This would render the library useless and cause unnecessary time loss. 126 Table 3.2: Selection of avian anti-fPSA-specific antibody fragments by phage-display. Input and output reflect the number of phage at the start of each round of biopanning and those recovered after each subsequent selection round, expressed as colony forming units (cfu). Round (cfu/mL) Round 1 Input 3.2 x 1012 Round 1 Output 5.0 x 106 Round 2 Input 3.5 x 1012 Round 2 Output 7.0 x 107 Round 3 Input 2.4 x 1011 Round 3 Output 1.5 x 107 Three rounds of selection were completed and the eluted phage from each round analysed by polyclonal phage ELISA as shown in Figure 3.18. The avian anti-fPSA scFv library required 3 rounds of biopanning in comparison to 5 rounds for the murine anti-fPSA scFv library because the avian library was 100-fold bigger and provided an increased opportunity to select positive clones in fewer rounds of selection. After selection the resultant phage pool was tested in an ELISA to evaluate the success of the biopanning process. To monitor the selection process, phage pools acquired in each round of selection were tested in parallel with phage from the unpanned library. The ELISA signal for the antigen, fPSA, should increase during the biopanning process if specific binders have been selected. 127 Figure 3.18: The eluted phage from the chicken anti-fPSA scFv library were analysed in triplicate by ELISA. Three rounds of selection were performed on fPSA-coated ELISA wells and the phage pool obtained after subsequent rounds of biopanning were tested for binding to fPSA. Any binding phage particles were detected with a HRP-labelled anti-M13 antibody. The signal increased from the 1st to the 3rd round of selection indicated that positive clones were recovered from the library. The next stage of the selection process involved soluble expression of these avian anti-fPSA scFv clones. The pComb3XSS phagemid vector contains an amber stop codon (TAG) between the scFv and g3p which is suppressed in E. coli XL-1 Blue cells. However, if the phage is directly infected into non-suppressor TOP10F' cells the scFv is transcribed without the g3p fusion tag due to the amber stop codon. Therefore, E. coli TOP10F' cells were infected with the eluted phage pool from round three of selection and spread uniformly over the surface of agar plates supplemented with carbenicillin (phagemid antibiotic resistance gene). The E. coli TOP10F' cells will grow on the carbenicillin agar plates only if they have been infected with the phagemid. Uninfected TOP10F' cells will not grow as they do not have the appropriate antibiotic resistance gene. The plates were incubated overnight at 37oC 128 and one hundred and ninety two colonies picked and functionally expressed in sterile 96 well plates. The protein was extracted using a freeze (-80oC x 3) - thaw (37oC x 3) method and each clone analysed by ELISA (Figure 3.19) to evaluate binding to fPSA. The ELISA result revealed forty five clones (23%) were positively binding fPSA. 3.14 Soluble expression of chicken anti-fPSA scFv and analysis by ELISA Figure 3.19: One hundred and ninety two individual chicken anti-fPSA scFv clones were solubly expressed and analysed by ELISA. An ELISA plate coated with 100 L of 1 g/mL fPSA was blocked with 200 L of 3% (w/v) BSA in PBS. The lysates were diluted two fold with 1 % BSA (w/v) in PBST and added to the ELISA plate. Any binding scFv clones were detected with a HRP-conjugated anti-hemagglutinin (HA) antibody. The avian Fab library was bio-panned, E. coli TOP10F' cells infected with eluted phage and 60 individual colonies screened. The expressed clones were analysed by ELISA 129 (Figure 3.20) to determine whether the selected clones were kappa or lambda light chain-specific. Figure 3.20: The avian anti-fPSA Fab lysates were analysed by ELISA to determine if they were predominantly lambda or kappa light chains. The Fabs were analysed using an anti-lambdaHRP-labelled antibody and detected with TMB substrate. The majority selected were lambda chain-specific (4 selected clones were not lambda chain-specific). The majority of the selected Fabs (56) were lambda chain specific while 4 clones were kappa chain specific (results not shown) as determined by ELISA. The clones showed a high degree of antibody expression when grown in small scale (96-well sterile plate). The expression levels have different patterns and could potentially be different recombinant antibody fragments so the library diversity was checked. Individual Fab clones from the generated library (5 clones) and from the final round of biopanning (5 130 clones) were subjected to an AluI digest. This enzyme is a high frequency cutting restriction enzyme that recognises the sequence AG/CT and generates blunt ends. The Fabs were resolved on a 2% (w/v) agarose gel (Figure 3.21) to visualise the diversity in the library and the similarity in the Fabs selected from panning as described in Section 2.3.4. Figure 3.21: AluI fragment analysis of selected avian anti-fPSA Fab clones to investigate the diversity between the clones. Lane 1: 100 bp ladder (Invitrogen); lanes 2–6: selected clones from the library. Lanes 7–11: clones selected from the final round of selection. The 5 analysed clones from the new library have no similar fragment patterns confirming a diverse selection of individual clones. Two of the 5 selected Fab clones from the final round of selection have identical DNA sequence patterns (9 and 11). This would suggest that these clones are potentially the same antibody fragment and are specific for a certain epitope of the fPSA molecule. The other selected clones are less diverse than the selected clones from the original library but are different. 131 3.15 Discussion This chapter gives a detailed description on the generation of three recombinant antibody fragment libraries developed from the spleens of mice and chickens immunised with fPSA. Positively-binding anti-fPSA recombinant antibodies were selected from the generated libraries and preliminary studies by ELISA indicated a range of positive clones. Separate approaches were performed for the generation of the murine and avian recombinant antibody libraries. The murine anti-fPSA scFv library was constructed using a method described by Krebber et al. (1997). The spleen, a rich source of antibody-producing cells (lymphocytes) was isolated from the immunised mouse after confirming anti-fPSA-specific antibodies were present in the serum by ELISA. The RNA was extracted and reversed transcribed to cDNA. The variable antibody genes (VH and VL) were successfully amplified using the primer set given in Section 2.2.5. Problems with amplifying the SOE-PCR were encountered as a 1000 bp non-specific band was observed initially. The SOE-PCR was optimised using DMSO in the PCR mix and by using a touchdown PCR. The DMSO inhibits secondary structure by disrupting base pair in GC rich templates and improves the efficiency of amplification. The touchdown PCR enables amplification of the desired product at a high initial annealing temperature with a decrease in annealing temperature after each successive cycle. A range of polymerases were investigated in the PCR amplification to determine the optimal amplification. The desired single chain Fv fragments were assembled with a glycine-serine interface in a VL-(G4S)4-VH orientation. The flexible linker region (20 amino acids) allows folding of the generated scFv. After optimisation, the SOE-PCR product was cloned into pAK 100 Krebber phagemid (phage plasmid) for g3p fusion protein display. The phagemid carries a plasmid origin of replication and a phage origin of replication. After cloning E. coli XL-1 Blue electrocompetent cells were transformed with the DNA and the library selected on agar plates supplemented with chloramphenicol (antibiotic resistance gene). The size of the generated library was 2.2 x 106 cfu/mL. The generated library size indicates a large number of possible clones which could potentially bind fPSA. However, these needed to be selected from the generated murine anti-fPSA recombinant antibody library. Krebber et al. (1997) yielded 132 a library size of 106 after electroporation. The library size is dependent on the concentration of DNA and the efficiency of the E. coli cells. Antigen-specific antibodies were selected from the generated library by ‗biopanning‘. After five rounds of selection on fPSA-coated immunotubes a polyclonal ELISA revealed the presence of positively binding anti-fPSA phage-scFv clones. Krebber et al. (1997) reported 3 rounds of selection to be optimal for the murine scFv generated library. The murine anti-fPSA library required 5 rounds of ‗biopanning‘ for the selection of positive phage-scFv particles. Individual colonies were analysed by monoclonal phage ELISA and a number of clones successfully bound fPSA-coated wells. In the laboratory problems, where poor expression levels of murine scFv were observed and after extensively searching of the literature, Dr. Andrew Hayhurst was contacted about the pMoPAC vectors. The plan was to investigate if improvements in expression levels and stability could be achieved by reformatting the recombinant antibody. The scFv was amplified by PCR from round 5 output of selection and subcloned into the pMoPAC series of vector for soluble expression of a scAb. This construct greatly enhanced soluble protein expression and overall protein stability. The ligated product was transformed in E. coli Tuner cells containing the pRARE plasmid. Tuner cells are lacZY deletion mutants of BL21 and allow uniform entry of IPTG into the cell population ensuring homogenous protein levels of induction. The pRARE plasmid codes for mammalian tRNAs rarely occur in E. coli yet enhance protein expression levels in bacterial cells restricted by codon usage. Individual colonies were analysed for expression by ELISA confirming solubly expression of murine anti-fPSA scAb clones. A differential expression profile of an anti-fPSA scFv and reformatted scAb was performed by ELISA to show the significant expression levels between the two antibody formats (Section 3.10 Figure 3.14). Chickens were immunised with fPSA and the spleen extracted for recombinant antibody generation. Avian anti-fPSA scFv and Fab libraries were constructed using a method described by Andris-Widhopf and co-workers (2000). A library size of 3.10 x 108 cfu/mL for the avian anti-fPSA scFv was bio-panned against fPSA passively immobilised on the surface of ELISA wells (Section 2.3.4). Three rounds of selection were performed and a polyclonal phage ELISA confirmed the presence of anti-fPSA133 specific antibodies. E. coli TOP10F' cells were infected with the phage and individual clones selected and expressed in sterile 96 wells plates. Twenty three of the 192 selected clones were positive by ELISA. The avian anti-fPSA Fab library was bio-panned, individual colonies selected, expressed and screened by ELISA. ELISA showed that the Fab lysates were predominantly lambda light chain domain fragments. The Fab construct contains the human constant domain regions and these allow for extra stability when expressed in E.coli cells. Overall, antigen-specific recombinant antibody fragments were successfully selected from the generated immune libraries by phage display. A large panel of soluble clones were individually expressed and analysed by ELISA. These selected recombinant antibodies fragments were further characterised as described in the next chapter. Both Krebber et al. (1997) and Andris-Widhopf et al. (2000) reported the generation of recombinant antibody libraries from different animal sources (mice and chickens, respectively). Krebber et al. (1997) generated 2 libraries (spleen cells fused to a tumour cell line and B cells) of 4 x 106 and 6 x 106 transformants. Three rounds of ‗biopanning‘ were required for selection of positive binders. Andris-Widhopf and colleagues generated 3 large chicken libraries (approximately 107 transformants), 2 scFvs (long and short glycine-serine linker for monomeric and dimeric expression, respectively) and 1 Fab. Four rounds of ‗biopanning‘ using phage display were performed for each library and individual antibody fragments expressed and screened with a high proportion of positive antibody fragments selected from the library. Three rounds of selection were required for the selection of positive chicken anti-fPSA recombinant antibody fragments. Therefore, highly sensitive recombinant antibody fragments were selected earlier than the expected number of rounds of ‗biopanning‘. Rojas et al. (2002) generated a PSA recombinant antibody library size of 1.51 x 105 transformants. They used a mouse heavy variable chain with a human light chain and successfully selected 2 scFvs that recognised PSA after 5 rounds of biopanning. Both the murine and avian anti-fPSA libraries generated in this research were 10-fold bigger than the libraries reported by Rojas and Andris-Widhopf (2002; 2000). The library size is very important for selection of positive antibody fragments. The larger the library size diversity the greater the chance of recovering high affinity antibody fragments towards the antigen. 134 Chapter 4 Screening and characterisation of putative anti-fPSA scFv and Fab antibodies 135 4.1 Introduction This chapter describes the strategy for selecting high affinity recombinant antibody fragments from a panel of clones using the Biacore system. Ninety four murine antifPSA scAb-secreting clones were evaluated using a custom designed wizard on the Biacore 3000 instrument. A capture assay was employed where the individual scAbs were captured by anti-human C kappa mAb immobilised on the sensor surface and fPSA was subsequently passed over the captured scAb lysates. The scAbs were ranked according to their percentage binding stability to fPSA (30 nM) after 5 min. Preliminary kinetic analysis was performed on 11 clones with good performance characteristics and sequencing of the individual antibodies confirmed that they had different amino acid sequences with variation in the complementary determining region (CDR). The best murine anti-fPSA scAb was selected on the basis of an inhibition ELISA as the affinities of the selected antibodies were similar. The selected clone was expressed in large quantities and successfully purified by IMAC. The selected avian anti-fPSA scFv and Fab clones (Section 3.11, Figures 3.19 and 3.20) were subjected to further analysis and their affinities determined by Dr. Paul Leonard on the Biacore A100 in Uppsala, Sweden. The same capture approach for analysing the murine anti-fPSA scAb antibodies was pursued using two different immobilised surfaces (anti-lambda (Fab) and anti-HA (scFv)) on the Biacore A100. After pre-concentration studies an antilambda antibody was immobilised onto the surface of a CM5 dextran chip and the Fab lysates passed over the surface. The fPSA (30 nM) molecule was passed over the captured Fab, the percentage binding stability calculated and the clones ranked accordingly. Ninety six clones were evaluated using both a single fPSA concentration injection and multiple fPSA concentration injections to determine if the association and dissociation rate constants changed significantly due to the number of fPSA injections. The effect of temperature (37 and 25oC) on assay performance was evaluated. Temperature can cause shifts in the binding abilities of antibody and, therefore, is an important parameter in assay design (Zedar-Lutz et al., 1997). The avian anti-fPSA scFvs were examined using the capture assay format on an anti-HA immobilised surface. Nineteen of the selected clones showed high affinities towards fPSA. 136 4.2 Biacore capture assay design A Biacore capture assay was employed to analyse the selected antibody fragments and to rank them based on their binding abilities. 4.2.1 Preconcentration studies of anti-human C kappa mAb Preconcentration involves determining the optimal buffer conditions for maximum binding of the ligand to the surface of the chip (Section 2.4.4). The protein G-purified anti-human C kappa mAb was diluted in a low ionic strength buffer (10 mM sodium acetate) with different pH values. The principle behind using an array of pH buffers is that the protein carries a ‗net‘ positive charge at pH values less than its isoelectric point causing an electrostatic interaction between the negatively charged dextran surface and the positively charged protein. This results in a higher coupling efficiency with a lower requirement for protein. The study is performed to ensure maximum binding levels of the protein to the chip surface thus, ensuring a reusable assay format. The anti-human C kappa mAb diluted in 10 mM sodium acetate buffer at various pH increments was passed over a blank CM5 dextran surface and the response recorded. The selected pH should be the highest pH that provides adequate immobilisation levels (Murphy et al., 2006). The maximum potential antigen binding was observed in sodium acetate buffer, pH 4.0 (Figure 4.1), and this was used for immobilisation of anti-human C kappa mAb to the surface of all 4 flow cells. 4.2.2 Immobilisation of anti-human C kappa mAb on to the CM5 dextran sensor chip surface The immobilisation of a ligand to a CM5 dextran chip involves a number of steps described in Section 2.4.5. The chip is firstly activated by (N-ethyl-N‘(dimethylaminopropyl) carbodiimide (EDC) and (N-hydroxysuccinimide) (NHS), the ligand is subsequently passed over the surface and the chip surface is then deactivated to block and eliminate any potential non-specific binding (NSB) to the cell surface. The most utilised method of immobilisation involves the activation of the dextran surface using EDC and NHS chemistry. The EDC solution, in the presence of NHS, activates 137 the CM dextran carboxyl groups into functional ester groups. The surface NHS esters react with primary amino groups such as lysine residues on the protein causing covalent binding of the protein to the surface (Murphy et al., 2006). An injection time of 7 minutes for the EDC/NHS solution activates approximately 40 % of the carboxylated groups on the chips surface. The purity of the ligand is of vital importance for immobilisation and should be at least 95 % as this avoids the introduction of NSB or assay interference. Any remaining available unbound functional ester groups are deactivated by passing 1M ethanolamine hydrochloride (pH 8.5) over the chip surface (Casper et al., 2004). Immobilisation of the anti-human C kappa mAb to the dextran surface was performed by passing 60 g/mL of anti-human kappa mAb in 10 mM sodium acetate buffer, pH 4.0, over EDC/NHS activated flow cells 1, 2, 3 and 4 for 30 minutes at a flow rate of 10 l/min. After 30 minutes any remaining active groups on the surface and non-covalently bound material were deactivated and blocked using 1M ethanolamine hydrochloride (pH 8.5) with a 10 minute injection at a flow rate of 10 l/min. The surface was regenerated using 10 mM NaOH to remove non-covalently attached proteins from the surface. Approximately 12,500 RU was immobilised on the dextran surface of each flow cell (Figure 4.2). For characterisation of antibodies from clones with varying expression levels a high capacity surface area was essential. 138 Figure 4.1: The protein G-purified anti-human C kappa mAb, diluted in 10 mM sodium acetate solutions of pH 4.0, 4.2, 4.4, 4.6, 4.8 and 5.0 was passed over the surface of a CM 5 dextran chip at a flow rate of 10 µL/min. During pre-concentrations studies there is an electrostatic interaction between the positive charged protein (due to its charge below the isoelectric point) and the negatively charged surface carboxylated groups. The ionic strength (150 mM) of the HBS running buffer sufficiently removed the electrostatic charged antibody from the surface. The optimal pH for the immobilisation of the anti-human C kappa mAb to the CM5 dextran surface was pH 4.0 as the highest levels of apparent binding was observed. Immobilisation of anti-human C Kappa 139 Figure 4.2: The surface of a CM5 dextran chip was activated with an EDC/NHS solution and the anti-human C kappa mAb passed over the activated carboxyl group surface. The sample was injected for 30 minutes until the surface was fully saturated. Binding of the ligand is via the amino groups on the protein with the activated carboxyl groups on the dextran surface chip. After sufficient capturing of the ligand had been achieved the remaining activated groups were deactivated with ethanolamine-HCl solution. The surface was regenerated using a 10 mM solution of NaOH injected over the newly covalently coupled surface. The levels of anti-human C kappa mAb immobilised onto the surface of each flow cell were 12556, 12640, 12663 and 12467 RU for flow cells 1–4, respectively. Ninety four scAb fragments were further evaluated after preliminary studies showed a number of positive clones as described in Section 3.8 (Figure 3.13). These were solubly expressed in 20 mL cultures (2xTY media) and high throughput analysis performed using a custom wizard application. After successfully immobilising anti-human C kappa mAb on the surface of all four flow cells of a CM 5 dextran chip (Figure 4.2), a capture format assay was performed by passing the crude bacterial scAb protein-containing lysate over the surface of the immobilised anti-human C kappa mAb. Subtractive referencing was implemented whereby the scAb lysate was captured on three flow cells (2, 3 and 4) leaving flow cell 1 as a negative reference blank with no captured scAb 140 fragment (Figure 4.3). HBS and 1 g/mL fPSA (30 nM) were individually passed over the captured scAb fragments. The refractive indexes from flow cell 1 (blank reference) and the binding response from HBS buffer injected over each captured scAb was subtracted from the refractive index recorded for fPSA binding to the captured scAb. The scAbs were ranked according to the antibody-antigen complex percentage stability remaining after 5 minute dissociation and the fragments with the highest percentage complex stability were subjected to preliminary kinetic analysis (Section 2.4.6). Figure 4.3: Anti-human C kappa mAb was immobilised on the surface of a CM5 dextran sensor chip. A high throughput capture assay format was performed where the bacterial lysatecontaining scAb fragments were passed over the surface of the immobilised anti-kappa mAb (flow cells 2, 3 and 4). A set concentration of fPSA (30 nM) was subsequently injected over all 4 flow cells and the antibody fragments ranked (double referencing) according to their complex stability levels. 141 4.2.3 Designed custom kinetic wizard After consulting representatives from Biacore I designed a custom wizard for the simultaneous analysis of recombinant antibody binding clones (Figure 4.4). The designed wizard allowed for the quick analysis and ranking of antibody binding fragments to fPSA. Figure 4.4: The custom designed kinetic wizard allowed the analysis of 3 murine anti-fPSA scAbs simultaneously using double referencing (HBS over the captured murine anti-fPSA scAb (flow cells 2, 3 and 4) and fPSA over flow cell 1 (blank – no captured scAb)). This screen snapshot shows the list of commands used for the assay. The command icons are arranged across the top of the file. The written file determines the flow cells for analysis (2-1, 3-1, and 41 subtractive referencing), flow rate (50 µL/min), baseline report point before sample application, flow path for capturing the murine anti-fPSA scAb (100 µL) at a flow rate of 20 µL/min for 5 minutes, a data report point to confirm the levels of capture, injection of 30 nM fPSA after the 3 murine anti-fPSA scAbs were captured and report points 5 seconds and 5 minutes after dissociation of the fPSA molecule. The extracted lysates were captured on the immobilised anti-human C kappa mAb surface and 30 nM fPSA passed over the lysates. A report point was recorded at the start 142 of dissociation (stability early) and 5 min (stability late) after the injection and the clones ranked on their binding abilities. The percentage stability remaining after a 5 min dissociation period gave a good overall indication of the antibody/antigen complex with the highest affinities (Canziani et al., 2004). The analysis of 94 murine anti-fPSA scAbs took approximately 7 days. The data were analysed using BIAevaluation software and the data report points exported into Excel, the graph of each binding profile for each analysed scAb were plotted (Figure 4.5). Figure 4.5: Ninety four scAb antibody fragments were analysed and ranked according to their binding to a set concentration of fPSA (30 nM). Each curve represents binding of fPSA to a captured scAb fragment. The degree of association and dissociation of fPSA to the scAb represents the apparent binding to the antibody fragment. Reports points were recorded 5 seconds after the end of the injection and 295 seconds after injection and the percentage of stability remaining calculated and the scAb clones ranked (Table 4.1). Each scAb was ranked according to its percentage stability after a 5 min dissociation period. The association and dissociation rates for each selected scAb were significantly different (Figure 4.5). Some clones have quick on rates and quick off rates while some clones have slow on rates and slow off rates suggesting potential differences in binding affinities for fPSA. The results were tabulated (Table 4.1) and the percentage stability calculated (RU). 143 Table 4.1: This table represents the calculated complex stability of each scAb fragment analysed on the Biacore 3000. A range of stability percentage values for the scAb fragments were recorded in Excel. Binding responses less than 5 RU indicates little or no binding to 30 nM fPSA (R<5RU) and such clones were subsequently discarded. scAb % scAb % scAb % scAb % Clone Stability Clone Stability Clone Stability Clone Stability Number fPSA Number fPSA Number fPSA Number fPSA 1 N/A 25 52.92 49 N/A 73 N/A 2 53.43 26 68.20 50 N/A 74 48.30 3 N/A 27 64.29 51 N/A 75 N/A 4 N/A 28 N/A 52 N/A 76 N/A 5 70.17 29 62.35 53 N/A 77 N/A 6 76.13 30 65.67 54 N/A 78 N/A 7 N/A 31 69.01 55 N/A 79 N/A 8 N/A 32 N/A 56 N/A 80 N/A 9 82.04 33 49.36 57 N/A 81 N/A 10 62.00 34 N/A 58 N/A 82 N/A 11 60.00 35 33.79 59 N/A 83 49.84 12 N/A 36 31.31 60 60.74 84 N/A 13 N/A 37 12.94 61 N/A 85 N/A 14 60.41 38 N/A 62 61.85 86 52.70 15 57.94 39 N/A 63 61.96 87 68.39 16 34.17 40 20.34 64 N/A 88 N/A 17 71.04 41 58.31 65 N/A 89 N/A 18 69.22 42 10.48 66 N/A 90 15.13 19 63.40 43 54.36 67 N/A 91 N/A 20 N/A 44 70.73 68 N/A 92 N/A 21 N/A 45 67.55 69 75.47 93 55.76 22 33.97 46 N/A 70 67.08 94 47.21 23 N/A 47 69.70 71 6.45 24 55.31 48 59.49 72 85.00 144 Antibodies with a 68% stability or greater were selected and subjected to preliminary kinetic analysis. This ‗cut-off‘ was chosen to reduce the risk of potentially missing high binder scAbs. Eleven clones with percentages stabilities above 68%, were selected and preliminary evidence demonstrated low nanomolar to picomolar range affinities using a 1:1 Langmuir model (Table 4.2). Table 4.2: The affinities of each murine anti-fPSA scAb analysed were determined and ranked accordingly. The association and dissociation rate constants of each scAb fragment were recorded and preliminary affinities calculated using BIAevaluation software. The percentage stability after 5 minute dissociation (Table 4.1) obtained for each selected scAb is presented with the scAb name. Clone Name ka (1/Ms) kd (1/s) KD (M) scAb F5 (68 %) 2.88 x 106 1.04 x 10-3 3.62 x 10-10 M scAb A1 (70 %) 2.51 x 106 1.09 x 10-3 4.33 x 10-10 M scAb F6 (82 %) 2.51 x 106 1.09 x 10-3 4.33 x 10-10 M scAb D8 (69 %) 3.48 x 106 1.55 x 10-3 4.46 x 10-10 M scAb E7 (70 %) 1.17 x 106 5.99 x 10-4 5.13 x 10-10 M scAb B6 (76 %) 1.67 x 106 9.87 x 10-4 5.91 x 10-10 M scAb C8 (69 %) 1.55 x 106 9.71 x 10-4 6.28 x 10-10 M scAb A8 (71 %) 2.76 x 106 2.63 x 10-3 9.58 x 10-10 M scAb B5 (69 %) 3.08 x 104 7.04 x 10-5 2.29 x 10-9 M scAb E4 (85 %) 3.75 x 106 1.32 x 10-3 3.51 x 10-9 M scAb C2 (75 %) 6.70 x 106 2.67 x 10-3 3.99 x 10-9 M 145 Figure 4.6: Preliminary kinetic analysis of a crude lysate from scAb F5 using a capture approach. A number of concentrations of fPSA were passed over the captured scAb and the curves plotted using a drifting baseline 1:1 Langmuir model in BIAevaluation. The residual plot shows the ‘goodness’ of the fit to the line. The curve (Figure 4.6) was fitted using a 1:1 Langmuir model with drifting baseline with a Chi2 value of 0.172. This value gives information about the ‗goodness‘ of the curve fit and for good qualitative data this value should be as low as possible. The residual plot shows the data points are distributed randomly above and below the zero line but are within acceptable limits. Initial evidence suggests that the 11 selected clones had similar affinities for fPSA as determined using BIAevalution software on the Biacore 3000 (Section 2.4.7). Therefore, the most suitable clone for assay development was chosen by inhibition ELISA based on its binding ability to compete for fPSA in solution or fPSA adsorbed onto the surface of an ELISA well. This involved expressing each individual scAb and analysing the crude protein lysate by ELISA to determine the 146 optimal working dilution for each fragment. Inhibition studies were performed using the appropriate working dilution for each antibody. These dilutions were incubated with various concentrations of fPSA for 1 hour at 37oC and added to a fPSA-coated plate. Bound murine anti-fPSA scAb was detected using anti-human C kappa AP-labelled antibody. The substrate, pNPP, was added for 30 minutes at 37oC and the absorbance of the wells read at 405 nm (Figure 4.7). Figure 4.7: An inhibition ELISA using the selected murine anti-fPSA scAbs. The murine antifPSA scAbs were incubated with a range of fPSA concentrations (500, 333, 222, 148, 98, 66, 44, 29, 19.5, 13 and 8.67 ng/mL) for 1 hour at 37oC and added to a fPSA-coated plate. Bound antifPSA scAb was detected using anti-kappa alkaline phosphatase (AP)-labelled antibody. The substrate, pNPP, was incubated for 30 minutes at 37oC and the absorbance of the wells read at 405 nm. The ELISA analysis showed varying degrees of binding to fPSA. The majority of the scAbs have different binding patterns as observed by ELISA but the scAb with the best potential for immunoassay development was scAb B5. Figure 4.7 would suggest that scAb B5 has an IC50 value less than 10 ng/mL. This selected murine anti-fPSA scAb was subjected to further characterisation and analysis. The amino acid sequences of each of the selected murine anti-fPSA scAbs (A1 to E4) were determined to investigate similarities and differences in the fragments. 147 4.2.4 Alignment of scAb clones from murine anti-fPSA library Bacterial stocks containing the murine anti-fPSA scAb clones were sent for sequencing to Eurofins MWG as agar stab cultures in 1.5 mL centrifugation tubes supplemented with carbenicillin (Section 2.5). The DNA sequences were received and subsequently translated into their amino acid sequence using ExPASy translation tool. The amino acid sequences were aligned and the complementary determining binding (CDR) regions of the antibody evaluated using ClustalW2 software. The CDR regions of the antibodies were highlighted according to the Kabat scheme. The glycine-serine linker region of the clones was highlighted in green and scAb E7 is missing a glycine residue (indicated by a dash) in the linker region but this should have no impact on binding of the clone. The diversity between the clones is evident from the sequence variation between the heavy and light chains which are highlighted in red (Figure 4.8). The CDR L2, L3, H1 have slight differences in amino acid residues while CDR H2 is highly conserved between the analysed fragments. There is more diversity observed between the scAbs in CDR L1 and H3. The scAbs have 2 predominant amino acid sequences in the CDR L1 region with the exception of scAb E4. It has a hydrophobic proline amino acid residue in the CDR L1 region which is not present in any of the other sequences. Despite its hydrophobicity prolines are usually found on protein surfaces and play an important role in molecular recognition. Many serine residues are observed in the CDR L1 region and these are normally common within tight turns of proteins allowing for flexible movement and binding in the antigen pocket. A positively charged arginine was found in scAbs B5 and E4 which frequently plays a role in protein structure and prefers to be on the outside of proteins. However, this residue can cause non-specific binding to negatively charged surfaces (Birtalan et al., 2008). Asparagine was present in scAbs B6, A1 and E7 and tends to be frequently found in the active site of the protein. ScAb E7 has an isoleucine residue which is probably buried deep within the proteins hydrophobic core. ScAbs A1, A8 and E7 have the greatest diversity in the CDR H3 region. Ippolite et al. (2006) revealed that 40% of the amino acid residues in the CDR H3 are tyrosine or glycine. Interestingly, scAb A1 has 5 tyrosine residues in CDR H3 which is an important amino acid residue in creating interactions at protein-protein interfaces and has a dominant role in antigen recognition (Koide and Sidhu, 2009). ScAb A8 has a proline residue at the end of the CDR H3 region while scAb E7 sequence was unclear as 148 a few amino acid residues were missing (indicated by a dash). Tyrosine in combination with glycine and serine residues have been shown to mediate high affinity antigen recognition (Biratalan et al., 2008) and the sequenced scAb fragments possessed an abundant level of these residues. CDR L1 Framework 1 F6 B6 E4 A1 A7 A8 B5 E7 -XXLHGRRTQDIVMETTQSPASLPVSLGQRATISCRASEGVDLYGYSFIQ--WYQQKTGQ -XXLHGRRTKDIVMETTQSPASLTVSLGQRATISCRASESVDSYGNSFMETHWYQQKPGQ XMETHGRRTQDIVMETTQSPASLPVSLGQRATISCRAREGVDLYGYSFIP--WYQQKTGQ ------------RIVITQSPASLAVSLGQRATISCRASESVDSYGNSFMETHWYQQKPGQ -XXLHGRRTQDIVMETTQSPASLPVSLGQRATISCRASEGVDLYGYSFIQ--WYQQKTGQ XMETHGRRTQDIVMETTQSPASLPVSLGQRATISCRASEGVDLYGYSFIQ--WYQQKTGQ XCHRHGG-IKDIVMETTPSPASLPVSLGQRATISCRAREGVDLYGYSFIQ--WYQQKTGQ -XXCTGWRTQDI--ELTQSPASLAVSLGQRATISCRASESVDNYGISFMETNWFQQKPGQ work 2 CDR L2 F6 B6 E4 A1 A7 A8 B5 E7 177 179 178 168 177 178 177 174 Framework 3 YWMETHWIKQRPGQGLEWIGYINPSTGYTEYSQKFKDKATLTADKSSSTAYMETQLSSLT YWMETHWIKQRPGQGLEWIGYINPSTGYTEYSQKFKDKATLTADKSSSTAYMETQLSSLT YWMETHWIKQRPGQGLEWIGYINPSTGYTEYSQKFKDKATLTADKSSSTALHA--LSSLT YWMETHWIKQRPGQGLEWIGYINPSTGYTEYNQKFKDKATLTADKSSSTAYMETQLSSLT YWMETHWIKQRPGQGLEWIGYINPSTGYTEYSQKFKDKATLTADKSSSTAYMETQLSSLT YWMETHWIKQRPGQGLEWIGYINPSTGYTEYSQKFKDKATLTADKSSSTAYMETQLSSLT YWMETHWIKQRPGQGLEWIGYINPSTGYTEYSQKFKDKATLTADKSSSTAYMETQLSSLT YWMETHWIKQRPGQGLEWIGYINPSTGYTEYSQKFKDKATLTADKSSNTAYMETQLNSLT 237 239 236 228 237 238 237 234 Framework 4 CDR H3 F6 B6 E4 A1 A7 A8 B5 E7 117 119 118 108 117 118 117 117 CDR H1 KLELKRGGGGSGGGGSGGGGGSGGGGSEVQLQQSGTELAKPGASVKMETSCEASGYTFTN KLELKRGGGGSGGGGSGGGGGSGGGGSEVQLQQSGTELAKPGASVKMETSCEASGYTFTN KLELKRGGGGSGGGGSGGGGGSGGGGSEVQLQQSGTELAKPGASVKMETSCEASGYTFTN KLELKRGGGGSGGGGSGGGGGSGGGGSEVQLQQSGTELAKPGASVKMETSCEASGYTFTN KLELKRGGGGSGGGGSGGGGGSGGGGSEVQLQQSGTELAKPGASVKMETSCEASGYTFTN KLELKRGGGGSGGGGSGGGGGSGGGGSEVQLQQSGTELAKPGASVKMETSCEASGYTFTN KLELKRGGGGSGGGGSGGGGGSGGGGSEVQLQQSGTELAKPGASVKMETSCEASGYTFTN KLELKRGGGGSGGGGSGGGG-SGGGGSQVQLQQSGAELARPGASVKLS--CKASGYTFTD CDR H2 57 59 58 48 57 58 57 57 CDR L3 Frame- Framework 1 Linker region Framework 2 F6 B6 E4 A1 A7 A8 B5 E7 Framework 3 PPKLLISRASNLESGIPARFSGSGSRTDFTLTINPVEADDVATYYCQQSRDDPWTFGGGS PPKLLIYRASNLESGIPARFSGSGSRTDFTLTINPVEADDVATYYCQQSRDDPWTFGGGS PPKLLISRASNLESGIPARFSGSGSRTDFTLTINPVEADDVATYYCQQSRDDPWTFGGGS PPKLLIYRASNLESGIPARFSGSGSRTDFTLTINPVEADDVATYYCQQSRDDPWTFGGGS PPKLLISRASNLESGIPARFSGSGSRTDFTLTINPVEADDVATYYCQQSRDDPWTFGGGS PPKLLISRASNLESGIPARFSGSGSRTDFTLTINPVEADDVATYYCQQSRDDPWTFGGGS PPKLLISRASNLESGIPARFSGSGSRTDFTLTINPVEADDVATYYCQQSRDDPWTFGGGS PPKLLIYASSNLKSGIPARFSGSGSGTDFTLNIHPVEEEDAATYYCQQSNEDPWTFGGGT work 4 F6 B6 E4 A1 A7 A8 B5 E7 Frame- SEDSAVYYCTRSRKLAMETDHWGQGTSVTVSS-ASGAEFAAAAPSVFIFPPSDEQLKSG 295 SEDSAVYYCTRSRKLAMETDHWGQGTSVTVSS-ASGAEFAAAAPSVFIFPPSDEQLKSG 297 SEDSAVYYCTRSRKLAMETDHWGQGTSVTVLLGLGGPNSRPPAHLSFLFPPS------- 288 SEDSAVYYCASYYGSSYATDYXGQGTSVTVSR-PRGPNSRPLHHLSSSSAH-------- 278 SEDSAVYYCTRSRKLAMETDHWGQGTSVTVSSASGAEFAAAAPSVFIFPPSDEQLKSG-- 295 SEDSAVYYCTRSRKLAMETDPWGQGNSVTVSSASGAEFAAAAPSVFTFPPSDEQL----- 293 SEDSAVYYCTRSRKLAMETDHWGQGTSVTVFSASGAEFAAAAPSVFIFPPSDEQLKSG-- 295 SEDSAVYFCAR------RLYVWGAGTTVTVSSASGAEFAAAAPSVFIFPPSDEQLKSGTASV 290 149 Figure 4.8: Murine anti-fPSA scAb gene sequences from the immunised murine anti-fPSA library were translated into amino acid sequences using Expasy Translate tool and aligned using ClustalW2 sequence alignment program. The highlighted sequences in red show the CDRs (heavy and light chain) of the recombinant antibody fragments. The framework regions are highlighted in blue and the glycine-serine linker region is green. 4.2.5 Protein expression, extraction, purification and analysis Large-scale expression of the murine anti-fPSA scAb B5 was performed in 500 mL culture as outlined in Section 2.7. The protein was extracted by sonication and the bacterial pellet removed by centrifugation. The protein lysate was passed through a 0.2 µM filter to reduce clogging of the nickel resin. It was then applied to the resin and any histidine-tagged expressed proteins captured by the nickel resin. Nickel has a high affinity for histidine residues and other proteins pass through the column. Generally, histidine residues are uncommon in proteins with only 2% present in globular proteins and only half are exposed on the protein surface (Ueda et al., 2003). Material binding non-specifically was removed by washing the resin with 20 mL of running buffer (300 mM NaCl, 20 mM imidazole and 0.5% (v/v) Tween) and the protein successfully eluted using a low pH buffer (10 mM sodium acetate pH 4.4) (Section 2.7). An immunodot blot visualised using an AP-labelled anti-human kappa antibody was used to confirm the presence of the murine anti-fPSA scAb in collected fractions (Figure 4.9). The IMAC-purified containing scAb fractions were pooled, centrifuged and analysed by SDS-PAGE and Western blotting (Figures 4.10 and 4.11). Bacterial lysate preparation is a critical step because the main purpose is to maximise protein extraction while minimising unwanted protein oxidation, proteolysis and possible contamination with genomic DNA (Structural Genomics Consortium, 2008). During IMAC purification it is important to resuspend the protein in high ionic strength buffer to ensure protein solubility and stability. Modest levels of imidazole in the resuspension buffer bind proteins with less histidine residues, thus, reducing non-specific binding to the IMAC resin (Structural Genomics Consortium, 2008). 150 Figure 4.9: Each fraction was analysed to confirm elution of the murine anti-fPSA scAb from the nickel resin column. The individual fractions spotted (2.5 µL) on nitrocellulose paper were unpurified lysate, flowthrough, wash and each IMAC-eluted fractions The paper was blocked using 3% (w/v) BSA and detected using an anti-human C kappa AP-labelled antibody. The signal intensity increases significantly after the fifth elution fraction and the intensity of the signal is fairly uniform for all eluted fractions thereafter. The eluted fractions were pooled and concentrated by centrifugation at 4,000 rpm at 4oC using a 5,000 molecular weight cut-off spin column. It is important to select a molecular weight cut-off (MWCO) below (approximately 4-fold lower) the protein size to ensure retention of sample. The protein was buffer exchanged to remove any residual sodium acetate solution and to ensure a high protein concentration of purified sample for analysis. Ultrafiltration is a common technique in separating proteins in size graded fractions. The MWCO of the vivaspin column denote the size of the protein for retention. The fractions (lysate, flow-through, wash and elution) were analysed by SDSPAGE (Figure 4.10) and Western blotting (Figure 4.11) to determine the degree of purity and the functionality of the murine anti-fPSA scAb (Section 2.7). 151 Figure 4.10: Lane 1 and 6 represents the Sigma colorburst marker, lane 2 contains unpurified sonicated lysate with all the proteins expressed in the bacterial cell lysate; lane 3 contains the flow-through with all unbound proteins passing through the resin; lane 4 contains any impure non-specific hydrophobic proteins which may weakly bind to the resin and are removed using an increased concentration of Tween 20 and lane 5 contains the purified eluted murine antifPSA scAb with few contaminating proteins. Figure 4.11: Lanes 1 and 6 contain the Sigma colorburst marker; lanes 2, 3 and 4 contain the unpurified lysate, flow-through and wash fraction from the purification. Lane 5 represents the purified protein with a molecular size of 42 kDa’s. The membrane was probed using anti-human kappa AP-labelled antibody and developed using BCIP liquid substrate. It was quenched using 100 mM EDTA. 152 The protein samples were resolved on SDS-PAGE gels to verify that the scAb exhibited the correct molecular weight (42 kDa) and to assess the purity of the isolated protein. While one gel was stained with Coomassie blue to visualise individual proteins expressed within the bacterial cell, the proteins on the other gel were transferred to a nitrocellulose membrane and subjected to Western blot analysis using an anti-kappa AP-labelled antibody. A large number of proteins were clearly visible in the lysate and flow-through with little or no proteins in the wash fraction for the Coomassie bluestained SDS-PAGE gel. A single band at the correct molecular weight for the expressed scAb fragment was evident in Figure 4.11 indicating its relative purity. 4.2.6 Time course expression profile analysis for scAb expression An expression time course experiment was performed on the murine anti-fPSA scAb described in Section 2.8. Before induction with IPTG (1 mM final concentration) a 1 mL sample was removed and every hour after induction 1 mL samples were removed from the culture and stored at -20oC. In addition, a 1 mL sample was removed from the expressed overnight culture. The samples were sonicated, the bacterial pellet centrifuged and the protein analysed by SDS-PAGE (Figure 4.12), Western blotting (Figure 4.13) and ELISA (Figure 4.14). The highest level of murine anti-fPSA scAb expression was observed in the overnight sample. 153 Figure 4.12: Time course experiment for the expression of the murine anti-fPSA scAb product. Lane 1 represents the fermentas pagerulerTM prestained marker for protein size determination while lanes 2 – 8 represent 1 mL sample fractions harvested at 0, 1, 2, 3, 4, 5, 6 hours and overnight post-induction, respectively. Figure 4.13: The individual samples were analysed to investigate the levels of protein expression over time. The proteins were transferred to a nitrocellulose membrane and detected with an AP-labelled anti-human C kappa antibody. The level of murine antifPSA scAb increased after 3 hours of induction with the greatest level expressed in the overnight sample. 154 Figure 4.14: The individual 1 mL samples (0, 1, 2, 3, 4, 5, 6 hours and overnight) were analysed for expression by ELISA. The lysates were detected using an anti-human kappa APlabelled antibody. The substrate, pNPP was incubated for 30 minutes at 37oC and the absorbances read at 405 nm. The SDS-PAGE (Figure 4.12), Western blotting (Figure 4.13) and ELISA results (Figure 4.14) confirmed the maximum expression levels of protein expression for the murine anti-fPSA scAb was observed after incubating the culture overnight at 30oC following induction with 1 mM IPTG. It is important to induce the culture at mid-to-late log phase of growth for maximum levels of expression with a lower temperature for induction to allow proper folding of the newly transcribed recombinant protein (Structural genomics Consortium, 2008). They report that cells should be grown to high cell density overnight for sufficient levels of protein expression. Large scale expression, protein extraction and IMAC purification of the murine anti-fPSA scAb were performed for downstream assay development. 155 4.3 Avian clone analysis The selected avian anti-fPSA recombinant antibody fragments from panning were subjected to further characterisation on the Biacore A100. The assay format employed was similar to the custom design method for ranking the murine anti-fPSA scAb. However, analysis was performed at a significantly faster rate due major advancements in the software and hardware of the A100 compared to the Biacore 3000. The A100 is ideal for high sample throughput analysis of proteins with high quality data analysis and an integrated evaluation software for rapid interpretation of results. 4.3.1 Analysis of the avian anti-fPSA Fab and scFv fragments on the Biacore A100 instrument The avian anti-fPSA scFv and Fab fragments were analysed by Dr. Paul Leonard in Uppsala, Sweden using the A100 Biacore (Section 2.11). The avian anti-fPSA Fabs were predominantly expressed with a constant lambda (Cλ) chain as determined by ELISA as described in the previous, section 3.11, (Figure 3.20). Therefore, all four flow cells were immobilised with an anti-Cλ antibody using amine coupling chemistry. The 96 avian anti-fPSA Fabs were screened against three concentrations of fPSA (30, 5 and 0 nM). The recombinant antibody was captured and fPSA injected for 2 minutes with a 10 minute dissociation time. The surface was finally regenerated with a 30 second injection of 10 mM NaOH. The complete analysis time of the 96 anti-fPSA Fabs was 26 hours. All sensorgrams were referenced subtracted against an unmodified CM5 surface (spot 3). The zero concentration was then subtracted from the 30 and 5 nM sensorgrams (double referencing) to remove any systematic instrument disturbances. The data were evaluated using a 1:1 Langmuir equation with drifting baseline and global fit. Twenty six Fabs were classified as ‗OK‘ and therefore, the kinetic rate constant values can be trusted. Seventeen were classified as ‗inspect‘ but the reason may be largely attributed to low signal to noise ratio. The avian anti-fPSA scFv were analysed using a similar capture assay approach with an anti-HA immobilised surface and kinetic analysis performed on 7 of the 48 tested fragments. 156 The percentage stability was calculated by dividing the stability late dissociation response (after 10 min) by the stability early response (5 seconds after the end of the fPSA injection) and multiplying by one hundred. Subtractive referencing was implemented whereby the refractive index from the blank flow cell and the binding response from HBS buffer injected over the captured antibody fragments were subtracted from the fPSA binding response 4.3.1.1 Avian anti-fPSA Fab clone analysis The selected avian anti-fPSA Fabs were ranked according to the percentage stability of binding to 30 nM fPSA after a 10 minute dissociation. The percentage stability for each Fab fragment was calculated and a bar chart plotted in Excel (Figure 4.16). Figure 4.16: Percentage stability remaining during dissociation for all Fab samples calculated using double reference data for 30 nM on spot 1 (high immobilisation level). X-axis scale does not contain all sample names due to space constraints. Samples that due to drift were above 100% were set to 100%. 157 Figure 4.17: The percentage stability remaining plotted against the calculated ‘off-rates’. The two calculated numbers show high correlation especially for the slower ‘off-rates’. The majority of the Fabs analysed had high percentage stability after 10 minute dissociation. The clone ranking patterns were different suggesting diverse affinities for fPSA. An important consideration when designing the Biacore screening assay was to determine the most productive approach that can be used for screening large panels of antibodies in the shortest time-frame. Ideally, it would be acceptable to screen each antibody once and determine the best clones from the data. To evaluate this, association and dissociation rates of each clone were analysed against PSA at multiple analyte concentrations and at 30 nM (Figure 4.18 and 4.19). 158 Figure 4.18: The association rate of nineteen of the analysed Fabs against single and multiple fPSA concentrations were compared. The ‘on-rate’ plot shows that the majority of the Fabs gave good correlation between single and multiple analyte analysis. However, there are Fabs, such as 2D3, that show large variation in ‘on-rate’. In this assay setup with a very low Rmax the system can measure ‘on-rates’ as fast as 1x107 and therefore, the data fitting for clone 2D3 is not correct and cannot be trusted. However, using multiple analyte concentrations the on-rate is still quite fast and would be included for further analysis. Fourteen of the 19 clones analysed had similar ‗on-rates‘ when analysed using single and multiple injections of fPSA. Two (1A4 and 2F8) of the remaining 5 had faster ‗onrates‘ when analysing multiple injections while 3 Fabs (1D1, 2D3 and 2E7) had faster ‗on-rates‘ with a single fPSA injection. 159 Figure 4.19: The dissociation rate of the nineteen analysed Fabs against single and multiple fPSA concentrations. It shows that the majority of these antibodies have practically the same ‘off-rate’. However, we do see some outliers with up to 10 fold difference in ‘off-rate’ for Fab 1D5. This difference is probably due to sensorgram artefacts which can occur and either decrease or increase ‘off-rate’ values. Sixteen of the 19 Fabs analysed had similar off rates when analysed using single and multiple injections of fPSA. Two (1D5 and 2F8) of the remaining 3 had quicker ‗offrates‘ when analysing multiple injections while clone 1D1 had quicker off rates with a single fPSA injection. Kinetic analysis was performed on 26 of the best selected avian anti-fPSA Fabs. These were selected on the basis of their percentage complex stability (Figure 4.16). The individual Fabs were captured and then fPSA injected. The data was interpreted using BIAevaluation software using a 1:1 Langmuir model with a drifting baseline and a global fit. The curve was fitted with a drifting baseline as the avian anti-fPSA Fab was dissociating from the surface and this needs to be taken into final consideration in the evaluation of the data. A global fit was applied to the data as the response levels of the captured avian anti-fPSA Fab lysate vary due to expression level differences. 160 Figure 4.20: The Biacore A100 instrument software comes with the added advantage of automated data evaluation analysis allowing for quality control of data. The software ranks the curve fitting into four categories, GOOD, ATTENTION, INSPECT and DISCARD. The four sensorgrams shown here are examples of good fits irrespective of capture level. The curves on the right demonstrate that the capture level is very low but the fitting is still reasonably good. The capture levels of fPSA are higher for Fabs 1C1 and 1C6 than 2D8 and 2D3 (Figure 4.20). However, the data curve fitting for all of the sensorgrams was acceptable. All these clones were analysed at room temperature and temperature can have a huge effect on binding of the antibody to the protein and this needs to be considered when designing an immunoassay. Changes in temperature affect the binding abilities of antibodies as well as stability. For commercialization purposes it is important to determine which antibodies are suited for the proposed assay. The clones were analyzed at two temperatures (25 and 37oC). Nineteen of the antibodies were classified as either ‗OK‘ or ‗inspect‘ at both temperatures. The differences in kinetics for these antibodies were compared in two bar graphs (Figures 4.21 and 4.22) 161 Figure 4.21: Dissociation rates for the 19 antibodies with well determined kinetics at both 25 and 37oC. Most antibodies change by about a factor of 5 in ‘off-rate’. 1A5 changes very significantly whereas 1D5 seems relatively unaffected. Figure 4.22: Association rates for the 19 antibodies with well determined kinetics at both 25 and 37oC. Sixteen of the screened clones showed a faster on-rate at 37oC with three having a slower association (1A4, 1D6 and 2F8). 162 Figure 4.23: ‘On/off-rate’ map of the analysed clones at 37 and 25oC. There is a general shift to the top right corner when analysing the fragments at a higher temperature (37oC) and the association and dissociation rates are faster but the general spread of affinities are the same. Interestingly temperature can have a major effect on affinity as seen in clone 1B5 with an affinity change from 170 nm at 30oC to 5 nm at 37oC. The antibodies of major interest are those that do not change in affinity with respect to temperature change (e.g. 1D5). Assay performance can be hugely influenced by temperature (Figure 4.23). Clones with unfavourable shifts in performance at different temperatures (clone 1B5) are unsuitable for certain assays. Clones (1D5) with similar affinities over a range of temperatures are desirable for assay design. 4.3.1.2 Avian anti-fPSA scFv analysis Forty eight avian anti-fPSA scFv were analysed using an anti-HA immobilised chip. Four fPSA concentrations, one in duplicate and a negative (0 nM fPSA) were passed over each captured scFv and the interaction studied (Figure 4.24). Among the 48 scFvs only 13 showed any binding at 10 nM, which was the highest analyte concentration included. Binding kinetics could be determined for 7 of the 13 binders. The remaining 163 scFvs showed too low binding level characteristics preventing a good assessment of the kinetics. Figure 4.24: Anti-HA-labelled antibody was immobilised onto 4 spots within a flow cell using one spot as a blank reference. Two scFv fragments are captured per flow cell and fPSA injected over every spot. The binding response from the blank flow cell and the reference spot were subtracted from the binding response from the capture scFvs. 164 Figure 4.25: ‘On’ and ‘off-rates’ for the 7 scFv’s for which the kinetics could be determined. The ‘off-rates’ are generally faster than for the Fab antibodies against fPSA. Series 32 (B7), 39 (C4), 22 (D6) are the only three with affinities below 1 nM (Figure 4.25). They have calculated affinities from 50 to 500 nM. Overall, the avian anti-fPSA recombinant antibody fragments were successfully evaluated using the Biacore A100 and avian anti-fPSA scFv clone D6 was shown to have the best affinity of all the avian antibodies analysed. 4.4 Determination of antibody pairs for sandwich assay development This type of assay is the one of the most common immunoassays performed in which 2 antibodies recognise different epitopes of the same antigen. The analyte is sandwiched between the immobilised capture antibody on the surface and probed using a detection antibody. An important consideration when designing a sandwich assay is to categorically confirm both antibodies do not bind overlapping antigenic epitopes. Therefore, a Biacore sandwich assay was completed to investigate antibody pairs which 165 could be utilised in a sandwich assay format. The purified murine anti-fPSA scAb was captured using an anti-human C kappa mAb-immobilised-CM5 dextran surface chip. The fPSA molecule (1 µg/mL) was passed over the captured antibody and probed using avian anti-fPSA Fab and scFv lysates (Table 4.3). The assay was repeated without the presence of the fPSA molecule to confirm no NSB between the capture and detection antibody (Section 2.12). Table 4.3: Epitope mapping was performed by capturing the purified murine anti-fPSA clone B5 scAb on the anti-human kappa mAb immobilised surface, passing over 1 µg/mL fPSA and injecting the avian clone lysates. A number of avian antibodies could be considered for a sandwich assay format as they seem to bind different epitopes of fPSA. Avian Clone Captured scAb (RU) Captured fPSA (RU) Captured Avian (RU) 2F8 Fab 93.8 110.5 135.1 1C1 Fab 100.4 118.4 109.3 1A5 Fab 114.2 131.3 132.5 1D5 Fab 97.5 110.5 135.1 1D6 scFv 95.8 115.5 98.1 The epitope mapping Biacore assay identified a number of potential avian antibody fragments (2F8 and 1D5 Fabs) which could be utilised in a sandwich immunoassay format. These Fabs recognise and bind different epitopes on the fPSA molecule than the captured murine anti-fPSA scAb. 166 4.5 Discussion This chapter describes the screening and characterisation of anti-fPSA scAb, scFv and Fab antibody fragments. These antibody fragments were expressed in bacteria with different epitope tags (kappa domain for scAb, HA domain for scFv and lambda domain for Fab) which do not interfere with the antigen/antibody binding complex formation and seldom affect biological activity (Structural Genomics Consortium, 2008). These tags facilitated high throughput analysis of positive clones by immobilising anti-kappa, anti-lambda or anti-HA antibodies onto the surface of a CM5 dextran chip and subsequent Biacore-based analysis. In the previous chapter the selection of a large panel of positive recombinant antibody clones from immunised animals by phage display was outlined. This created a positive dilemma as each clone needed to be evaluated to determine the highest affinity antibody. The analysis process can be extremely time-consuming and expensive depending on the selection format (e.g. ELISA). Also, important information like dynamic complex formation cannot be determined from ELISA-based assays. The Biacore offers a rapid high-throughput ‗real-time‘ analysis format for ranking individual clones. Leonard et al., (2007) reported screening and ranking of avian recombinant antibody fragments using crude bacterial lysates on the Biacore A100. They developed a capture assay format which could potentially screen 400 antibody clones per day. The purity of the immobilising ligand is critical to prevent NSB and ensure good quality data for interpretation. The anti-human C kappa antibody was supplied in ascites fluid so it had to be purified using protein G, passed through a size exclusion PD-10 column and eluted in molecular grade H2O. Preconcentration studies of the protein G-purified anti-kappa antibody diluted in 10 mM sodium acetate buffer at different pH values (Figure 4.1) were evaluated. The maximum levels of apparent binding were observed in 10 mM sodium acetate, pH 4.0, which was used for the immobilisation. The purified anti-human C kappa antibody was immobilised on all four flow cell surfaces using EDC/NHS chemistry. A custom wizard was designed on the Biacore 3000 instrument for ranking of the murine anti-fPSA scAb clones. The wizard enabled three scAb clones to be captured simultaneously leaving flow cell one as a blank reference. Therefore, a capture assay format using a drifting baseline was implemented. One µg/mL fPSA (30 167 nM) was passed over the surface of all four flow cells and the percentage stability (stability late/stability early x 100 %) for 94 antibody fragments measured over a five minute dissociation period. The 94 fragments were ranked on the basis of their binding to 30 nM fPSA (Table 4.1) and the best selected with a ‗cut-off‘ of over 68 % stability after 5 minute dissociation. Kinetic analysis was performed with initial affinities ranging from 3.99 x 10-9 M to 3.62 x 10-10 M (Table 4.2). The murine anti-fPSA B5 scAb was selected as it was found to be more inhibitive in ELISA than the other clones even though their affinities were very similar (Figure 4.7). The murine anti-fPSA B5 scAb was expressed in large scale (500 mL) and the protein extracted by sonication. The protein was purified by IMAC and each eluted fraction confirmed the presence of the murine anti-fPSA scAb by immunodot blot (Figure 4.9). The fractions containing the purified scAb were buffer exchanged and stored at -20oC. The fractions (lysate, flowthrough, wash and elution) were analysed by SDS-PAGE and Western blot (Figures 4.10 and 4.11). A time course experiment confirmed that the maximum level of expression of the murine anti-fPSA scAb was achieved overnight at 30oC. Individual samples were analysed by SDS-PAGE, Western blotting and ELISA (Figures 4.12, 4.13 and 4.14, respectively). The murine anti-fPSA scAbs were sent to MWG for sequencing and differences in the amino acid residues in binding regions of the CDR confirmed the selection of a range of antibody fragments (Figure 4.8). ScAb A1 has 3 tyrosine amino acid residues in CDR H3 which has shown to be an important residue in protein-protein interactions. A high proportion of the tested scAb fragments had serine residues in the CDR L1 which allows flexible movement of the antibody fragment which is important for binding in the antigen active site. Two scAb fragments (B5 and E5) had a positive arginine residue in the CDR L1 domain which prefers to be positioned on the outside of the protein and is involved in protein structure. A recent report by Sidhu et al., (2009) suggests that tyrosine, tryptophan and arginine are common interface amino acid residues for protein-protein recognition. The avian anti-fPSA scFvs and Fabs were analysed on the Biacore A100 instrument by Dr. Paul Leonard in Uppsala, Sweden. They were ranked based on their percentage stability after 10 minute dissociation. Multiple and single injection of fPSA were examined to determine similarities or differences in binding with respect to 168 concentration. The binding abilities of the Fabs were analysed under different temperatures (25 and 37oC) to determine the effect of temperature on each clone. The temperature increases the association rate of 18 of the 19 clones and the dissociation rate of 16 of the 19 analysed. The affinity of one of the avian anti-fPSA Fab (1D5) seemed unaffected by the change in temperature, therefore making it a good antibody candidate for assay development. The avian anti-fPSA scFvs were captured on an anti-HA immobilised chip and individually ranked. Out of the 48 scFvs analysed 3 scFv fragments had affinities of less than 1 nm (B7, C4 and D6). A significant advantage of using the Biacore A100 over the 3000 instrument is the time reduction in high throughput analysis. Using the Biacore 3000 instrument, 94 murine anti-fPSA scAbs were analysed in 7 days while on the Biacore A100 96 avian anti-fPSA Fabs were analysed in 26 hours. This is due to the number of flow cells available for analysis. The Biacore 3000 has 4 flow cells while the A100 has 4 flow cells with 5 spots in each flow cell allowing for 20 interactions per cycle. The instrument has a built-in software system that selects the data and evaluates it with confidence for good kinetic and affinity values. On the Biacore 3000 data interpretation and analysis of results can be a very time consuming but are reliable and reproducible. A comparative study reported by Katsamba involved 22 investigators exploring assay reliability on the Biacore. The assay design involved immobilising the monoclonal antibody onto the chip surface and analysing a series of PSA concentrations. Systematic artefacts were removed from the curves for good quality data and kinetic analysis determination using BIAevaluation software. The antibody showed an overall affinity of 1.1 x 10-9 nM (Katsamba et al., 2006). Some of the selected murine and avian clones show better affinity than the tested monoclonal antibody in this study. Both the murine and avian recombinant antibody fragments have low picomolar affinity as determined by Biacore. Currently, few papers describing murine and avian anti-fPSA recombinant antibody fragments have been published as most researchers have generated monoclonal antibodies. Rojas et al. (2002) constructed a phage antibody fragment library by combining a synthetic human light chain variable region (VL) with a diverse set of heavy chain variable regions selected from a mouse immunised with PSA. The apparent 169 affinities for 2 clones selected were 3.5 x 10-9 M and 1.5 x 10-9 M as determined by ELISA. Leinonen et al. (2004) investigated the development of monoclonal antibodies against PSA by genetic immunisation. Only five monoclonal antibodies were successfully obtained and their affinity constants were between 3.2 x 10-8 and 7.0 x 10-9 M. Popkov et al. (2004) developed rabbit monoclonal antibodies that recognise cell surface tumour-associated antigens expressed in PCa. Their study involved using phage display technology for the selection of human prostate cancer cell reactive antibodies, namely, Fab antibody fragments. New Zealand white rabbits were immunised with human prostate cancer cell lines (LNCap or DU145). Chimeric rabbit/human Fab libraries were generated and selected by a positive/negative selection strategy. Acevedo et al. (2002) developed a quantitative ELISA for the measurement of PSA concentration. Mouse monoclonal antibodies with affinities of 3.7 x 10-9 M and 4.7 x 10-10 M were used to develop this assay. The assay can detect both free and complex PSA in the sera of patients. The monoclonal antibodies discussed in this paper have similar affinities to the generated anti-fPSA scAbs (3.99 x 10-9 M to 3.62 x 10-10 M). However, 4 of the selected murine scAb fragments (F5, A1, F6 and D8) show better affinity determined by Biacore. A number of the analysed avian antibody fragments (1A5 Fab and D6 scFv) have better affinities over the reported monoclonal antibodies. Several surface-plasmon resonance (SPR)-based assays for the detection of PSA have been reported. Huang et al. (2005) have recently reported the development of an immunosensor using a commercial SPR biosensor (Biacore 2000) for detection of PSA concentrations as low as 1 ng/mL. Biacore SPR chips are usually composed of a carboxymethyl dextran matrix linked to a gold substrate on the sensor surface. However, self-assembled monolayers (SAMs) have been used as the immobilisation substrate and have been shown to give better sensitivities than dextran layers (Frederix et al., 2003). The PSA assay reported by Huang and co-workers used camel anti-PSA antibody fragment attached to a mixed SAM of thiols for PSA capture, and a biotinylated mouse monoclonal anti-PSA antibody and streptavidin-modified gold nanoparticles for detection. Yu et al. (2004) reported the use of a CM5 Biacore chip as a dextran surface onto which a sandwich assay for fPSA was located. The assay combined SPR and fluorescent labelling, i.e. surface plasmon field-enhanced fluorescence spectroscopy (SPFS). This technology uses surface plasmons to excite the 170 fluorescent label and produce the signal. The limit of detection observed was 80 fM for a 40 minute contact time, many orders of magnitude lower than detection limits of PSA assays commercially available today. The murine and avian recombinant antibody fragments were analysed using the Biacore 3000 and A100, respectively. Capture assays were developed with low picomolar affinity antibodies selected for both species. Thaxton et al. (2009) reported an ultrasensitive nanoparticle-based bio-barcode assay for the detection of PSA. The assay is approximately 300 times more sensitive than commercially available immunoassays and has the ability to detect 330 fg/mL. It is a promising diagnostic tool capable of detecting extremely low levels of PSA in men after radical prostatectomy which would go unnoticed using commercial immunoassays. Researchers are aiming to develop point-of-care testing (POCT) which will undoubtedly help in PCa detection. Currently, the turnaround time for patient results could be a number of weeks as samples are analysed at dedicated clinical centres. There is a progression towards the use of label-free biosensors formats for PSA detection and the success of these biosensor formats in future POCT devices depends on the ability to integrate the format into devices that include simple and efficient sample handling and readout systems (Healy et al., 2007). 171 Chapter 5 Assay development and immunohistochemistry studies for fPSA detection 172 5.1 Introduction This chapter focuses primarily on the development of a sensitive assay for the detection of fPSA by Biacore and ELISA. Inhibition and sandwich-based assays were performed using the murine anti-fPSA scAb and avian anti-fPSA scFv. A subtractive Biacore inhibition assay was developed using a fPSA-immobilised CM5 dextran surface chip. The purified murine anti-fPSA scAb was incubated with varying concentrations of fPSA, injected over the fPSA-immobilised chip surface and the binding response recorded. A decrease in the binding levels of antibody (reported in RU) to the surface was observed with increasing fPSA concentration. The results were normalised by dividing the binding response for each concentration by the maximum binding response (Ro, scAb incubated with no fPSA). The Biacore inhibition assay confirmed that the murine anti-fPSA scAb was displaced in the presence of fPSA. Inter-day and intra-day assays were performed to verify the accuracy and precision of the immunoassay format. The coefficients of variation (CV‘s) were determined for each concentration and calibration curves generated from the data using a four-parameter equation for immunoassays using BIAevalution 3.1 softwareTM and the percentage accuracies for each sample back-calculated from the curve. An inhibition ELISA using the murine anti-fPSA scAb was also evaluated to compare the level of sensitivity to the Biacore inhibition assay. Inhibition studies showed that the ELISA assay format was not as sensitive as the Biacore assay. The assay sensitivity was significantly improved by performing a sandwich-based ELISA assay. The murine anti-fPSA scAb was successfully biotinylated and subsequently captured using neutravidin with the avian anti-fPSA clones used as the detection antibodies. The five avian (4 Fabs/1 scFv) antibody fragments were retested by ELISA to determine whether or not they bind non-overlapping epitopes of fPSA. Intra-day and inter-day assay calibration curves were generated with CVs and percentage accuracies for each analyte. A Biacore-based sandwich assay was investigated but showed no improvement in assay performance over the direct murine anti-fPSA scAb Biacore inhibition assay. Precision assays were evaluated using 20 blanks and 20 replicates of fPSA at 2 concentrations (2.5 and 1.8 ng/mL in PBST) to determine the limit of detection of the assay. The sandwich ELISA assay approach was 173 repeated using serum from females spiked with fPSA to determine sensitivity levels in the serum matrix. The serum of individuals presenting with PCa and with known tPSA values were analysed using the sandwich ELISA to determine the performance of the assay on clinical samples. Immunohistochemistry studies were performed on prostate tissue samples using both murine and avian anti-fPSA recombinant antibody fragments. A key parameter for the generated recombinant antibodies is that they recognise PSA in the context of the secreted protein in tissue samples. Therefore, studies involving normal and PCa tissues samples were undertaken in Beaumont Hospital. It is vital that the protein is not altered by subtle downstream changes (e.g. proteolysis) that could minimise binding of the recombinant antibody fragments. It was confirmed that both murine and avian anti-fPSA antibodies bound successfully to the tissue samples analysed. However, the degree of tissue staining intensity was less than the positive control but this was expected since the control antibody was a polyclonal and detects tPSA while both the avian and murine recombinant antibody fragments bind fPSA which is approximately 25 % of tPSA. This study showed that recombinant antibodies can be successfully employed in detecting secreted proteins in prostate tissue samples and can be used in the diagnosis of prostate cancer. 174 5.2 Development of a subtractive inhibition Biacore assay for fPSA The purified murine anti-fPSA scAb B5 was chosen based on its overall ability to bind fPSA in solution, as determined by ELISA in Section 4.2.4 (Figure 4.7). The fPSA molecule was successfully immobilised onto the surface of a CM 5 dextran chip and, following optimisation of the conditions for assay performance an inter-day study was completed. The range of assay sensitivity as determined by Biacore was 31.3 – 6.3 ng/mL (results not shown). Biacore-based PSA assays have been reported in the literature. Huang et al. (2005) reported a PSA SPR immunoassay using camel antibodies immobilised on the chip surface. The concentration detected using the direct approach was 10 ng/mL PSA. However, sensitivity was improved to the sub ng/mL by employing a sandwich assay approach. Cuong et al. (2005) fabricated a sandwich SPR strategy for the detection of PSA-ACT in both HBS buffer and human serum samples with detection concentrations of 10.2 and 18.1 ng/mL, respectively. 5.3 Murine anti-fPSA inhibition ELISA assay with inter-day and intra-day studies A solid phase inhibition-based ELISA was performed using the purified murine antifPSA scAb to compare levels of assay sensitivity with the Biacore assay format, described in Section 5.2. After optimising the antibody working dilution for assay development inter-day and intra-day inhibition studies were performed. Calibration curves were generated with percentage CV and accuracy values. The range of sensitivity for the ELISA-based inhibition assay was 100 to 3.9 ng/mL fPSA (results not shown). This ELISA assay format showed similar levels of sensitivity as the Biacore assay. Therefore, the assay format was modified to investigate if sensitivity could be improved. As reported in the literature by both Huang et al. (2005) and Cuong et al. (2005) assay sensitivity could be further improved by implementing a sandwich-based assay. They reported using a biotinylated antibody for sandwich assay development. Therefore, the murine anti-fPSA scAb was biotinylated and utilised in ELISA and Biacore-based immunoassays. 175 5.4 Biotinylation of the purified murine anti-fPSA scAb and assay development NHS-Biotin is a 244 Da water soluble vitamin which can bind primarily to lysine residues of proteins. The murine anti-fPSA scAb was biotinylated and employed in a lateral flow immunoassay. Naturally biotin has a high affinity for streptavidin/neutravidin so a sandwich assay was investigated. The sandwich assay format involved capturing the biotinylated murine anti-fPSA scAb on a neutravidincoated plate and using an avian anti-fPSA scFv as the detection antibody. Precision assays were performed to determine the limit of detection of the assay with 95% confidence. Female serum samples were spiked and assay sensitivity investigated and serum samples from patients presenting with PCa analysed. PCa tissue samples were analysed using the generated recombinant antibody fragments to confirm specific binding to the secreted protein. 5.4.1 Biotinylated of the murine anti-fPSA scAb The purified murine anti-fPSA scAb was successfully biotinylated using the Pierce EZlink biotinylation kit. During the biotinylation procedure biotin is covalently attached to the primary amines (lysines) of the recombinant antibody. The degree of biotinylation was determined using the HABA/Avidin test, as described in section 2.16. A sandwich assay format was investigated with the aim of improving assay sensitivity. 5.4.2 Sandwich ELISA-based assay After confirming by ELISA the anti-fPSA secondary antibodies bound different epitopes of the captured fPSA molecule as described in Section 2.17 (Figure 5.1), the optimal coating concentration of neutravidin was determined (5 µg/mL) and the TMB substrate incubation time was investigated to compare assay sensitivity between 10 and 20 min incubation periods. A twenty minute incubation period was optimal with no increase in background signal observed. 176 Figure 5.1: Sandwich ELISA using the captured biotinylated murine anti-fPSA scAb and antitPSA mAb, anti-fPSA mAb, anti-cPSA mAb and purified anti-fPSA scFv as detection antibodies. Each of the primary anti-PSA antibodies were detected using the appropriate enzyme-labelled secondary antibodies. Both commercial (anti-tPSA/fPSA mAbs) and the avian anti-fPSA scFv successfully recognised different epitopes on the fPSA molecule than the neutravidin captured biotinylated murine anti-fPSA scAb. The commercial anti-cPSA mAb did not bind to the captured fPSA molecule and no background signal was observed (Ao). 5.4.3 Pre-study validation of sandwich ELISA assay To ensure an accurate, reliable working assay format the sandwich ELISA was repeated over three days (inter-day assay) as decribed in Section 2.23. The sample batches were made fresh on the day of analysis. The dilutions, incubation times and washing steps were performed accurately each day to guarantee a reproducible assay format (Figure 5.2). The % CV and % accuracies were calculated for each concentration assessed (Table 5.1). An intra-day sandwich ELISA was performed 4 times using the same dilution batches over a period of one day to evaluate the overall precision of the assay. 177 The data was evaluated using a 4-parameter fit equation using BIAevaluation (Table 5.2). Figure 5.2: Inter-day assay calibration curve for the determination of fPSA in PBST. A plate was coated with 5 g/mL neutravidin and blocked using 3% (w/v) BSA in PBS. The biotinylated murine anti-fPSA scAb was captured and a number of fPSA concentrations (25, 18.8, 14, 10.5, 8.0, 6.0, 4.5, 3.3, 2.5, 1.8, 1.4, 1.0, 0.8 and 0.6 ng/mL) added to the plate. The purified avian anti-fPSA scFv was detected using an anti-HA-HRP-labelled antibody. The data is a result of three replicate measurements over a three day period. Using BIAevaluation 3.1 software the data was fitted with a 4 parameter equation and the original data points for each concentration added to the curve. The error bars represent the standard deviation for each concentration analysed in triplicate over a three day interval. 178 Table 5.1: Inter-day studies using a sandwich assay format with various fPSA concentrations and determination of the coefficient of variation. Concentration Back-calculated fPSA (ng/mL) concentrations % CV’s % Accuracies (ng/mL) 25.0 25.6 6.3 97.5 18.8 18.0 4.6 104.2 14.0 14.5 5.1 96.5 10.5 9.7 9.6 108.8 8.0 7.8 2.2 102.0 6.0 6.6 5.2 89.8 4.5 4.6 3.7 97.5 3.3 3.2 6.0 103.3 2.5 2.5 9.9 99.9 1.8 1.9 16.6 93.7 1.4 1.2 33.7 115.7 1.0 0.9 26.6 102.9 0.8 0.7 50.6 106.1 0.6 0.8 64.0 88.2 The sandwich assay can detect low levels of fPSA (1.8 ng/mL) with acceptable % CV (16.6%) and % accuracy (93.7%) values. However, at the lower fPSA concentrations (1.4, 1.0, 0.8 and 0.6 ng/mL) the % CVs are outside the suggested 20% limit and the assay is unreliable at these concentrations. The range of assay sensitivity is between 25 and 1.8 ng/mL fPSA. 179 Table 5.2: Intra-day studies using nine fPSA concentrations and determination of the coefficient of variation. Concentration Back-calculated fPSA (ng/mL) concentrations % CV’s % Accuracies (ng/mL) 10.5 10.5 2.9 100.4 8.0 7.9 2.6 99.6 6.0 5.9 10.6 100.3 4.5 4.5 4.7 98.5 3.3 3.2 5.4 103.9 2.5 2.6 4.5 98.0 1.8 1.9 13.6 95.8 1.4 1.5 6.5 94.4 1.0 0.7 9.5 138.0 The intra-day study suggested that the assay could detect 1.4 ng/mL as the values were well inside the acceptable limits. At 1.0 ng/mL the percentage accuracy of 138.02 showed that the assay was unreliable at this concentration. The inter-day and intra-day studies show the proposed assay had good precision with CV‘s ranging from 16.61% to 2.22% between 1.8 ng/mL and 25 ng/mL fPSA and 13.55% to 2.56% between 1 ng/mL and 10.5 ng/mL fPSA, respectively. The percentage accuracies determined in both assay formats provide an accurate assessment of the measured fPSA concentrations. Both assays had acceptable concentration values back-calculated from the curves. 5.4.4 Determining the limit of detection (LOD) of the sandwich ELISA assay The LOD for any assay can be regarded as the lowest concentration which can be reliably differentiated with 95% confidence from the limit of the blank (LOB) (Section 2.24). The LOB was determine using equation 2.3.2 and a precision assay performed with 20 positive and negative samples using the sandwich ELISA assay approach to investigate the accuracy of the test and to verify the LOD (Figure 5.3). 180 Figure 5.3: Twenty replicates of 2.5 ng/mL (blue diamonds) and 1.8 ng/mL (red square) fPSA were analysed with 20 blank samples to determine the lowest concentration of analyte that can be detected with 95 % confidence. The cut-off (LOB) was determined by getting the mean and adding 1.645 standard deviation of the 20 negative samples (red dotted line). The value 1.645 is related to Gaussian curve where 90 % of the distribution falls between -1.645 and 1.645. The limit of detection determined using the precision assay (Figure 5.3) was 1.8 ng/mL fPSA with 95% confidence using 20 blanks and 20 sample replicates. After successfully determining the limit of detection of the sandwich assay spiked female serum samples were analysed. The serum was spiked with fPSA and a sandwich ELISA performed to investigate assay sensitivity (Figure 5.4). 181 5.4.5 Spiked female serum samples An inter-day study using serum samples spiked with different concentrations of fPSA was performed to evaluate assay sensitivity (Figure 5.4) over a 3 day period (Section 2.25). Figure 5.4: An inter-day calibration curve using female spiked serum using a 4-parameter equation fit. The decrease in assay sensitivity is due to serum matrix which interferes with assay sensitivity. Female serum was spiked with fPSA and analysed using a sandwich assay approach with the biotinylated murine anti-fPSA scAb as the capture antibody and the avian anti-fPSA scFv as the detection antibody. 182 Table 5.3: Inter-day studies using female serum spiked with different fPSA concentrations and determination of the coefficient of variation. Concentration Back-calculated fPSA (ng/mL) concentration % CV’s % Accuracies (ng/mL) 25.0 25.3 8.5 99.0 18.8 18.5 12.1 101.4 14 13.5 4.2 103.6 10.5 11.2 13.5 94.0 7.9 7.7 11.4 103.0 5.9 6.4 0.8 91.9 4.5 3.8 4.5 115.8 3.3 3.5 5.6 94.0 2.5 2.9 10.9 87.1 1.8 1.4 146.7 131.4 1.4 1.0 284.8 138.7 1.0 1.0 232.7 105.3 0.8 0.9 290.2 89.4 0.6 1.0 143.3 61.9 The female serum spiked samples (Table 5.3) had range of CV‘s (0.80% to 12.08%) between 2.5 ng/mL and 25 ng/mL fPSA while the lower concentrations had values well outside the acceptable limit. The calculated % accuracies were 87.08% and 115.80% between 2.5 ng/mL and 25 ng/mL fPSA. Therefore, the LOD for the sandwich assay in spiked female serum samples is 2.5 ng/mL fPSA. At the lower fPSA concentrations (1.8, 1.4, 1.0, 0.79 and 0.59 ng/mL), the assay is very unreliable and does not display any accurate values. The next stage of assay validation involved determining levels of fPSA in clinical patient serum samples presenting with PCa symptoms. 5.4.6 Real patient serum sample analysis by sandwich ELISA Serum samples from patients presenting with PCa were kindly donated by Professor William Watson, Conway Institute, UCD, Belfield, Dublin. These samples were stored 183 at -80oC and analysed using the sandwich assay format using the biotinylated murine anti-fPSA scAb as the capture antibody and the avian anti-fPSA scFv as the detection antibody. The serum samples were added in duplicate and absorbances recorded. The unknown fPSA concentrations were back-calculated from the curve and correlated to the known tPSA values obtained from each sample analysed (Table 5.4). Table 5.4: Serum samples analysed by sandwich ELISA. Five patients presenting with PCa were tested to determine the levels of fPSA in each sample and to correlate the levels of fPSA to the tPSA serum levels analysed using the commercially available Abbott Diagnostic AxSYM test at Beaumont Hospital. Sample ID Serum analysis Calculated fPSA tPSA concentration concentration (ng/mL) (ng/mL) MMH/05/0121 Yes 1.8 6.0 MMH/04/0068 Yes 1.9 6.2 MMH/04/0086 Yes 1.8 6.1 MMH/05/0104 Yes 2.2 6.4 MMH/05/0111 Yes 2.5 7.0 The levels of tPSA in the patient serum samples were previously tested in Beaumont Hospital using the Abbott Diagnostic AxSYM tPSA test. The fPSA levels were analysed using the sandwich assay approach and the unknown concentrations evaluated from the calibration curve. The levels of fPSA in the sample contribute to approximately 25% of tPSA as detected by ELISA. 5.4.7 Immunohistochemistry study using purified murine anti-fPSA scAb and avian antifPSA scFv antibodies on prostate tissue samples This study was performed on normal and cancerous prostate tissue samples and involved detecting fPSA localised within the tissue microarrays (TMAs). The purified recombinant antibodies were sent to Beaumont Hospital and the assay performed by Gillian O‘Hurley on TMAs using a Bond Max Automated Machine with a standard diagnostic commercially available antibody as an internal positive control. Both normal 184 (BPH) and tumour (PCa) prostate tissue cells were investigated using the murine and avian recombinant antibodies and the staining intensity evaluated. Since normal epithelial cells express PSA it was not a surprise to observe a higher staining intensity than tumour cells as PSA is released in the bloodstream when the architecture of the gland is disrupted and normal function is altered. The cells were visualised under high magnification and overall both recombinant antibody fragments bound the PSA in prostate cells albeit with less intensity compared to the positive control polyclonal antibody (Section 2.27 Figures 5.5, 5.6 and 5.7). The polyclonal antibody is a mixture of antibodies that recognise many epitopes of the PSA molecule whereas the recombinant antibody fragments are monospecific and bind to a single epitope. Therefore, this would corroborate the stronger staining intensity observed. Figure 5.5: Normal prostate tissue cells were analysed with the purified avian anti-fPSA scFv using a positive control. The tissue sections were incubated with HRP-labelled secondary antibodies using DAB chromagen as substrate. The staining intensity was greater using the positive control antibody. However, no surrounding background stromal tissue binding was observed with the avian anti-fPSA scFv. 185 Figure 5.6: The tumour cells have a smaller lumen than normal prostate cells and are clustered together with little differentiation. The tissue section was screened using the purified avian anti-fPSA scFv and incubated with HRP-labelled secondary antibodies using DAB chromagen as substrate. The staining pattern is similar to the positive control (polyclonal antibody) but with less intensity. Figure 5.7: The purified murine anti-fPSA scAb demonstrated excellent staining identifying both epithelium and secretary PSA levels in the prostate gland but is not as intensity as the positive control test antibody (polyclonal antibody). This could be attributed to the fact that the polyclonal antibody was raised against tPSA while the recombinant antibody fragments were against fPSA. The lumen and epithelium of the prostate gland are clearly defined and recognisable when stained. 186 Figure 5.8: BPH glands (non-cancerous growth of prostate) were analysed using the murine and avian anti-fPSA recombinant antibody fragments. PSA is expressed within the gland, however, no staining was observed. The primary antibody (murine scAb or avian scFv) was not incubated on the tissue sections and the secondary-labelled antibody did not bind nonspecifically to the tissue sample. The slight blue staining of the nuclei is due to the destaining solution (Haematoxylin) which causes the cells to stain brown when positive. Both primary and secondary antibodies are required for staining of the tissue samples. Figure 5.8 demonstrates no staining due to the fact that the primary antibody (murine or avian recombinant antibody) is omitted from the experiment. 187 5.5 Discussion The aim of this chapter was to assess the potential of employing SPR biosensor, ELISAbased and immunohistochemical assays for the detection of fPSA. The purified recombinant antibody fragments described in Chapter 4 were involved in immunoassay development and patient sample analysis. Initial studies using a Biacore-based inhibition assay showed a working range of 31.16.3 ng/mL. This result was mirrored by an ELISA-based inhibition assay with poor reliability at low fPSA concentrations. This was a major concern as the FDA approved range of detection for PSA is 2-4 ng/mL. Therefore, the detection levels of fPSA were not acceptable using the inhibition-based immunoassays. Huang et al. (2005) developed a novel commercial PSA SPR biosensor immunoassay with detection limits of 10 ng/mL using camel antibodies. The assay involved covalently attaching the recombinant antibody onto a gold surface and injecting PSA over the surface. They noticed that the limit of detection could be further improved to sub ng/mL range by constructing a sandwich assay involving a biotinylated antibody. Saerens et al. (2005) reported a Biacore sandwich PSA assay involving capturing camel single-domain antibodies and detecting clinical levels of different PSA in 15 minutes. Acevedo et al. (2002) demonstrated a sandwich ELISA-based immunoassay with a limit of detection of 0.1 ng/mL using mouse monoclonal antibodies. Their ELISA-based sandwich immunoassay incorporated highly sensitive mAbs with a detection range of 25–0.12 ng/mL which was comparable to commercially available immunoassays. This assay format was different to the inhibition-based immunoassays. Therefore, a sandwich assay approach was investigated. The underlying achievement was the development of a sandwich assay format with high levels of sensitivity. Sandwich-based immunoassays were investigated, however, no improvement in assay sensitivity was observed over the inhibition-based assays using a Biacore assay format. It involved capturing fPSA using a biotinylated scAb and detecting bound fPSA with the purified avian scFv. The fPSA capture levels by the biotinylated scAb especially at the lower fPSA concentrations were statistically unreliable for accurate data analysis on the Biacore. A sandwich ELISA-based assay 188 demonstrated acceptable levels of sensitivity. Inter-day and intra-day studies confirmed a reliable working assay with a limit of detect of 1.8 ng/mL (Figure 5.2). A precision assay confirmed the detection limit with 95% confidence (Figure 5.3). Butch et al. (2001) investigated the analytical performance of Roche total and free PSA sandwich assays. They reported linear ranges of 0.01–103 ng/mL and 0.02–25 ng/mL, respectively. The CV‘s for both assays were < 5% for intra-day studies with inter-day study values of < 13.6%. The sandwich inter-day study showed CV‘s between 2.2– 16.6% (Table 5.1) at a linear range of 25–1.8 ng/mL (Figure 5.2). The intra-day study reported CV‘s between 2.6–13.6% (Table 5.2) at a linear range of 10.5–1.4 ng/mL. The developed sandwich assay showed acceptable levels of detection. Commercially available immunoassays (e.g. Roche) require expensive large automated machines to analyse samples. These assays are performed at dedicated centres throughout the world with trained personnel with prolonged waiting times for results (Healy et al., 2007). The aim was to develop an economical sensitive sandwich ELISA-based immunoassay which could be executed in a diagnostic environment using standard laboratory equipment. To the author‘s best knowledge, this is the first reported fPSA sandwich immunoassay using murine and avian recombinant antibody fragments. Zundel et al. (1990) reported a sandwich-based immunoassay using 2 monoclonal antibodies with detection levels below 2 ng/mL. In their study they confirmed no PSA in female serum and evaluated 79 clinical male samples with known PSA concentrations. Acevedo et al. (2002) also confirmed no PSA levels in the serum of healthy females. Therefore, spiked female samples were evaluated using the sandwich assay format (Figure 5.4) and a shift in sensitivity to 2.5 ng/mL was observed. Serum matrix interference can affect the binding ability of the antibodies and impact the limit of detection. After confirming low detection levels in spiked female serum samples real patient samples were analysed using the sandwich ELISA format. The concentrations of the unknown samples were calculated from the generated calibration curve (Table 5.4). The back-calculated fPSA concentrations were approximately 25% of the tPSA concentrations as reported using the Abbott Diagnostic AxSYM tPSA test in Beaumont Hospital. This would suggest that the assay could potentially be used for the detection of fPSA in clinical samples. 189 Surface-enhanced Raman scattering (SERS), ‗real-time‘ Immuno-PCR and SPR sandwich-based assay have shown pg/mL detection levels (Healy et al., 2007). The format severely influences the performance of the assay and sensitivity can be improved considerably. Assays such as ‗real-time‘ immuno PCR require careful sample preparation to prevent sample contamination, dedicated equipment and data interpretation. Sensitivity is limited by background signal due to non-specific adsorption of assay reagents to the walls of the tube (Lind et al., 2005). There are a number of commercial tests available that use monoclonal antibodies for the detection of fPSA in serum. Abbott Diagnostics have 2 commercial assays available (Architect and AxSYM), with detection limits of 0.3 ng/mL and 0.1 ng/mL, respectively. Diagnostic Products Corporation have the Immulite assay with detection limits of 0.25 ng/mL. Roche supply the Elecsys assay with a detection limit of 0.5 ng/mL and Beckman Coulter assay has limits of 0.2 ng/mL. These commercially available immunoassays require large expensive equipment at dedicated centres. Many other novel PSA assay formats have been reported in the literature. (Grubisha et al., 2003; Soukka et al., 2001; Fernandez-Sanchez et al., 2005; Escamilla-Gomez et al., 2009). Grubisha et al. (2003) reported sensitivity levels as low as 1 pg/mL using a sandwich assay on gold nanoparticles coated with a Raman scatterer. Soukka et al. (2001) reported sensitivity levels in the 0.04 pg/mL range using fluorescently-labelled nanoparticles. These assays are ultrasensitive and could be employed in detecting levels of PSA after radical prostatectomy but would not be suitable for early detection of PCa. Fernandez-Sanchez et al. (2005) reported sensitivities in the region of 1 ng/mL fPSA using a disposable one-step lateral flow assay but these immunoassays can have significant drawbacks such as sensitivity. Escamilla-Gomez et al. (2009) described a method for the detection of fPSA and tPSA using a voltammetric electrochemical sensor using screen-printed electrodes. The reported sandwich assay format had a range of detection between 1–10 ng/mL and required a level of expertise in electrochemical immunoassay development. A major disadvantage when designing electrochemical immunoassays is the diffusion of the electroactive indicator where the electrodes are in close proximity resulting in negative results (Escamilla-Gomez et al., 2009). These reported assays have better sensitivity levels than the sandwich ELISA-based immunoassay described here, however, there are certain disadvantages associated with 190 using these assays, as previously mentioned. Assay sensitivity could be improved by performing mutagenesis on the selected antibody fragments (murine and avian). This would involve site-directed or random mutagenesis improving the affinities and the assay performance of the antibodies. Assay sensitivity could be further improved by incorporating the recombinant antibody fragments into an immuno-PCR (Lind et al., 2005) using a capture antibody with a DNA label for product amplification or nanoparticle-based immunoassays using a bio-barcode assay for ultrasensitive levels of detection (Thaxton et al., 2009). These would be suitable immunoassays for detecting very low levels of PSA after radical prostatectomy where commercial immunoassays fail to detect PSA levels. The murine and avian recombinant antibody fragments were analysed on both normal and tumour prostate cancer cells. BPH is a non-cancerous growth of the prostate gland. As men get older the prostate grows as a result of proliferation of epithelial cells within the basal cell layer. BPH is characterized histologically by the presence of discrete nodules in the periurethral zone of the prostate gland (Edwards, 2008). The immunohistochemical study showed that both murine and avian recombinant antibody fragments can be utilised in TMAs for detection of the secreted PSA protein within the tissue sample. Both antibody fragments bound with less intensity than a polyclonal positive control but this may be due to the fact that the polyclonal antibody binds many epitopes on the PSA molecule while the recombinant antibodies have one specific epitope for binding. This would confirm and support the TMAs results. Varma and coworkers (2002) analysed PCa tissue sections using immunohistochemistry staining and noticed that polyclonal anti-PSA antibodies were more sensitive than monoclonal antibodies. It would also be possible to use a cocktail of recombinant antibodies to different discrete epitopes on PSA. Expression of recombinant antibody fragments in bacterial cells offer an unlimited supply of protein at low costs. These are seen as advantageous characteristics and could substitute polyclonal/monoclonal antibodies for analysing PCa tissue samples. 191 Chapter 6 Overall Conclusions 192 6.1 Overall conclusions The aim of the work presented in this thesis was the development of immunoassays for the detection of PCa. The strategy used involved the generation and characterisation of recombinant antibody fragments (scFv, scAb and Fab) to fPSA. Individual fragments were selected by phage display and characterised by ELISA, SDS-PAGE and Western blotting. Biacore and ELISA-based immunoassays were developed after significant optimisation for the detection of fPSA. Chapter 3 describes the methodologies used for the generation of recombinant antibody fragment libraries. Three antibody libraries were constructed using systems described by Krebber et al. (1997) and Barbas et al. (2001) from the spleens of immunised mice and chickens, respectively. Two scFv libraries and one Fab library were constructed and individual antibody fragments selected by panning. After polyclonal and monoclonal phage ELISAs confirmed positively-binding antibody fragments individual colonies were screened by ELISA to determine expression levels of selected antibody fragments. Comparative analysis between a murine scAb and its scFv counterpart showed significant differences in expression levels between antibody formats. Avian recombinant antibody fragments (scFvs and Fabs) were expressed and individual colonies screened for protein expression by ELISA. The diversity of the original avian Fab library was clearly observed (Figure 3.21) and colonies selected from round three of the selection panning process showed that two out of the five analysed antibody fragments had similar DNA patterns suggesting they could be the same antibody fragment. After identification of positive antibody fragments binding to fPSA the next stage involved additional screening and characterisation of the generated antibody fragments. A Biacore capture assay was employed to rank 94 expressed scAbs on the basis of their percentage stability after 5 min dissociation. Eleven antibody fragments with high percentage stability (above 68%) were selected and subjected to further analysis. The affinities were calculated using BIAevaluation software and the values were extremely similar. The most suitable antibody for immunoassay development was selected on a basis of its competitiveness in an inhibition ELISA. The selected antibody fragment 193 (scAb B5) was expressed in large-scale, purified using IMAC resin and the purity verified by SDS-PAGE and Western blotting. The avian antibody fragments were evaluated using the same capture assay approach on the Biacore A100 which allows for rapid data analysis and interpretation. Chapter 5 describes the generation of Biacore and ELISA-based immunoassays using the recombinant antibody fragments selected from Chapter 4. The murine anti-fPSA scAb was used to develop a subtractive inhibition Biacore assay. The fPSA antigen was immobilised on the surface of a CM5 sensor chip and the scAb concentration optimised for assay development using an appropriate flow rate. Decreasing concentrations of fPSA were incubated with anti-fPSA scAb and passed over the sensor surface. A decrease in binding was observed with increasing fPSA concentration. Regeneration studies proved the sensor surface to be stable for over 40 binding cycles with the assay format demonstrating reproducible and accurate inter-day coefficients of variation values of less than 20%. Overall, the assay showed good sensitivity and the sensor chip surface was very stable. A solid phase inhibition-based ELISA assay was also developed with similar sensitivity to the Biacore-based assay. The sensitivity was investigated using a sandwich assay approach to determine whether or not it could be improved. The purified scAb was biotinylated with the biotin covalently attached to the primary lysine residues of the antibody. This was captured on the chip using a neutravidin-coated surface and different concentrations of fPSA captured. The avian anti-fPSA recombinant antibody fragments (scFv/Fab) were injected over the captured fPSA and binding levels recorded. Suitable antibody pairs were selected for immunoassay development. A Biacore sandwich assay proved unsuccessful with no binding observed at low fPSA concentrations (15 ng/mL). The ELISA-based immunoassay showed a significant improvement in the limit of detection (1.8 ng/mL). Spiked female serum samples were analysed to investigate any change in sensitivity using a serum matrix. A slight shift is sensitivity was observed (2.5 ng/mL) but this was expected as the performance of the antibody fragment may be altered in a complex serum matrix. Samples from individuals presenting with PCa were analysed by sandwich ELISA and the unknown concentrations determined using a calibration curve and compared to the values obtained in Beaumont Hospital. The levels 194 of fPSA in each of the samples were in line with reported levels of fPSA. This would suggest that the generated sandwich immunoassay could potentially be used to detect fPSA levels in serum. Improvements in assay sensitivity could be achieved using mutagenesis or employing a novel assay platform as described in Section 5.5. The immunohistochemistry studies proved that the generated recombinant antibody fragments (murine scAb and avian scFv) recognised secreted PSA in PCa tissue samples. This proves that the selected recombinant antibody fragments can be successfully utilised in TMAs and demonstrates clearly the possibility of using recombinant antibody fragments to detect markers of disease in tissue samples. 195 Chapter 7 Bibliography 196 Acevedo, B., Perera, Y., Ruiz, M., Rojas, Benetiz, J., Ayala,M. and Gavilondo, J. (2002). Development and validation of a quantitative ELISA for the measurement of PSA concentrations. Clin. Chim. Acta, 317, 55-63. Allard, W. J., Zhou, Z. and Yeung, K. K. (1998). Novel immunoassay for the measurement of complexed prostate-specific antigen in serum. Clin Chem., 44, 121623. Anderson, J. (2002). Does PSA testing influence the natural history of prostate cancer? Eur. Urol. Supp., 1, 3-10. Andriole, G. L., Levin, D. L. Crawford, E. D., Gelmann, E. P., Pinsky, P. F., Chia, D., Kramer, B. S., Reding, D., Church, T. R. Grubb, R. L., Izmirlian, G., Ragard, L. R., Clapp, J. D., Prorok, P. C. and Gohagan, J. K. (2005). Prostate cancer screening in the prostate, lung, colorectal and ovarian (PLCO) cancer screening trial: findings from the initial screening round of a randomized trial. J. Natl. Cancer Inst., 97, 433-438. Andris-Widhopf, J., Rader, C., Steinberger, P., Fuller, R. and Barbas, C. F. (2000). Methods for the generation of chicken monoclonal antibody fragments by phage display. J. Immunol. Methods, 242, 159-181. Aus, G., Abbou, C. C., Bolla, M., Heidenreich, A., Schmid, H. P., van Poppel, H., Wolff, J. and Zattoni, F. (2005). EAU guidelines on prostate cancer. Eur. Urol., 48, 546-551. Bartal, A. H., Hirshaut, Y. (1987). Methods for hybridoma formation. The humana press Inc. Barbas, C. F., Burton, D. R., Scott, J. K., Silvermann, G. J. (2001). Phage display: A laboratory manual. Cold spring harbour laboratory press, Cold spring, New York. 197 Besselink, G. A., Kooyman, R. P., van Os, P. J., Engbers, G. H. and Schasfoort, R. B. (2004). Signal amplification on planar and gel-type sensor surfaces in surface plasmon resonance-based detection of prostate-specific antigen. Anal Biochem., 333, 165-173. Birtalan, S., Zhang, Y., Fellouse, F. A., Shao, L., Schaefer, G. and. Sidhu, S. S. (2008). The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J. Mol. Biol., 377, 1518-1528. Bradbury, A. R. M. and Marks, J.D. (2004). Antibodies from phage antibody libraries. J. Immunol. Methods, 290, 29-49. Brawer, M. K., Cheli, C. D., Neaman, I. E., Goldblatt, J., Smith, C., Schwartz, M. K., Bruzek, D. J., Morris, D. L., Sokoll, L. J., Chan, D. W., Yeung, K. K., Partin, A. W. and Allard, W. J. (2000). Complexed prostate specific antigen provides significant enhancement of specificity compared with total prostate specific antigen for detecting prostate cancer. J. Urol., 163, 1476-1480. Breul, J., Pickl, U. and Hartung, R. (1994). Prostate-specific antigen in urine. Eur. Urol., 26, 18-21. Bruckler, D. R., Park, A., Viswanathan, M., Hoet, R. M. and Ladner, R. C. (2008). Screening isolates from antibody phage-display libraries. Drug Discovery Today, 13, 318-323. Butch, A. W., Crary, D. and Yee, M. (2001). Analytical performance of the Roche toal and free PSA assays on the Elecsys 2010 immunoanalyzer. Clin. Biochem., 35, 143145. Cao, C., Kim, J. P., Kim, B. W., Chae, H., Yoon, H. C., Yang, S. S. and Sim, S. J. (2006). A strategy for sensitivity and specificity enhancements in prostate specific antigen-alpha (1)-antichymotrypsin detection based on surface plasmon resonance. Biosens. Bioelectron., 21, 2106-2113. 198 Canziani, G. A., Klakamp, S. and Myszka, D. G. (2004). Kinetic screening of antibodies from crude hybridoma samples using Biacore. Anal. Biochem., 325, 301-307. Carvalho A. L., Sanz, L., Barettino, D., Romero, A., Calvete, J. J., and Roma, M. J. (2002). Crystal structure of a prostate kallikrein isolated from stallion seminal plasma: A homologue of human PSA. J. Mol. Biol., 322, 325-337. Casper, D., Bukhtiyarova, M. and Springman, E. B. (2004). Comparison of p38 kinase inhibitor binding affinity and kinetics using Biacore. Anal. Biochem., 325, 126-136. Catalona, W. J., Bartsch, G., Rittenhouse, H. G., Evans, C. L., Linton, H. J., Amirkhan, A., Horninger, W., Klocker, H. and Mikolajczyk, S. D. (2003). Serum pro prostate specific antigen improves cancer detection compared to free and complexed prostate specific antigen in men with prostate specific antigen 2 to 4 ng/ml. J. Urol., 170, 2181-2195. Catalona, W. J., Partin, A. W., Slawin, K. M., Brawer, M. K., Flanigan, R. C., Patel, A., Richie, J. P., deKernion, J. B., Walsh, P. C., Scardino, P. T., Lange, P. H., Subong, E. N., Parson, R. E., Gasior, G. H., Loveland, K. G. and Southwick, P. C. (1998). Use of the percentage of free prostate-specific antigen to enhance differentiation of prostate cancer from benign prostatic disease: a prospective multicenter clinical trial. JAMA., 279, 1542-1547. Catalona, W. J., Smith, D. S., Wolfert, R. L., Wang, T. J., Rittenhouse, H. G., Ratliff, T. L. and Nadler, R. B. (1995). Evaluation of percentage of free serum prostate-specific antigen to improve specificity of prostate cancer screening. JAMA ., 274, 1214-20. Catalona, W. J., Richie, J. P., Ahmann, F. R., Hudson, M. A., Scardino, P. T., Flanigan, R. C., deKernion, J. B., Ratliff, T. L., Kavoussi, L. R., Dalkin, B. L., et al. (1994). Comparison of digital rectal examination and serum prostate specific antigen in the early detection of prostate cancer: results of a multicenter clinical trial of 6,630 men. J. Urol., 151, 1283-1290. 199 Catalona, W. J., Smith, D. S., Ratliff, T.L., Dodds, K. M., Coplen, D. E., Yuan, J. J., Petros, J. A. and Andriole, G. L. (1991). Measurement of prostate-specific antigen in serum as a screening test for prostate cancer. N. Engl. J. Med., 324, 1156-1161. Charnow, J. (2008). Statins raise PCa odds in obese men. Amer. J. Epidemiol., 168, 250-260. Chen, Z., Chen, H., and Stamey, T. A. (1997). Prostate-specific antigen in benign prostatic hyperplasia: purification and characterization. J. Urol. 157, 2166-2170. Chen, L., Stacewicz-Sapuntzakis, M., Duncan, C., Sharifi, R., Ghosh, L., van Breemen, R., Ashton, D. and Bowen, P. E. (2001). Oxidative DNA damage in prostate cancer patients consuming tomato sauce-based entrees as a whole-food intervention. J. Natl. Cancer. Inst., 93, 1872-1879. Cherian, S., Gupta, R. K., Mullin, B. C. and Thundat, T. (2003). Detection of heavy metal ions using protein-functionalized microcantilever sensors. Biosens. Bioelectron., 19, 411-416. Cuong, C., Kim, J. P., Kim, B. W., Chae, H. C., Yang, S. S. and Sang, S. J. (2005). A strategy for sensitivity and specificity enhancements in prostate specific antigen-α1antichymotrypsin detection based on surface plasmon resonance. Biosens. Bioelectron., 21, 2106-2113. Darson, M. F., Pacelli, A., Roche, P., Rittenhouse, H. G., Wolfert, R. L., Saeid, M. S., Young, C. Y., Klee, G. G., Tindall, D. J., and Bostwick, D. G. (1999). Human glandular kallikrein 2 expression in prostate adenocarcinoma and lymph node metastases. Urol., 53, 939-944. Delves, P. J., Lund, T. and Roitt, I. M. (1997). Can epitope-focused vaccines select advantageous immune responses? Mol. Med. Today., 3, 55-60. 200 Denmeade, S. R. and Isaacs, J. T. (2004). Development of prostate cancer treatment: the good news. Prostate, 58, 211-224. Dennis, L. K., Lynch, C. F. and Torner, J.C. (2002). Epidemiologic association between prostatitis and prostate cancer. Urol., 60, 78-83. Diamandis, E. P., Okui, A., Mitsui, S., Luo, L. Y., Soosaipillai, A., Grass, L., Nakamura, T., Howarth, D. J. and Yamaguchi, N. (2002). Human Kallikrein 11: A new biomarker of prostate and ovarian carcinoma. Cancer Res., 62, 295-300. Diamandis, E. P. and Yousef, G. M. (2002). Human tissue kallikreins: a family of new cancer biomarkers. Clin. Chem., 48, 1198-1205. Dillon, P. P., Manning, B. M., Daly, S. J., Killard, A. J. and O'Kennedy, R. (2003). Production of a recombinant anti-morphine-3-glucuronide single-chain variable fragment (scFv) antibody for the development of a "real-time" biosensor-based immunoassay. J. Immunol. Methods, 276, 151-61. Djavan, B., Fong, Y. K., Remzi, M., Fakhari, M. and Marberger, M. (2004). New serum and urinary markers for prostate cancer detection in the new millennium. Eur. Urol. Supp., 3, 25-32. Don, R. H., Cox, P. T., Wainwright, B. J., Baker, K. and Mattick, J. S. (1991). 'Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res., 19, 4008. Drukier, A. K., Ossetrova, N., Schors, E., Brown, L. R., Tomaszewski, J., Sainsbury, R and Godovac-Zimmermann, J. (2005). Ultra-sensitive immunoassays using multiphoton-detection in diagnostic proteomics of blood. J. Proteome Res., 4, 2375-2378. Edwards, J.L. (2008). Diagnosis and management of benign prostatic hyperplasia. Amer. Family Phys., 77, 1403-1410. 201 Escamilla-Gomez, V., Hernandez-Santos, D., Gonzalez-Garcia, M. B., PingarronCarrazon, J. M. and Costa-Garcia, A. (2009). Simultaneous detection of free and total prostate specific antigen on a screen-printed electrochemical dual sensor. Biosens. Bioelectron., 24, 2678-2683. Fernandez-Sanchez, C., McNeil, C. J., Rawson, K., Nilsson, O., Leung, H. Y. and Gnanapragasam, V. (2005). One-step immunostrip test for the simultaneous detection of free and total prostate specific antigen in serum. J. Immunol. Methods, 307, 1-12. Finlay, W. J. J., Shaw, I., Reilly, J. P. and Kane, M. (2006). Generation of high-affinity chicken single-chain fv antibody fragments for measurement of the Pseudonitzschia pungens toxin Domoic acid. Appl. Environ. Microbiol. 72, 3343-49. Finne, P., Zhang, W. M., Auvinen, A., Leinonen, J., Maattanen, L., Rannikko, S., Tammela, T. L. and Stenman, U. H. (2000). Use for the complex between prostate specific antigen and alpha-1-protease inhibitor for screening prostate cancer. J. Urol. 164, 1956–1960. Frederix, F., Friedt, J. M., Choi, K. H., Laureyn, W., Campitelli, A., Mondelaers, D., Maes, G. and Borghs, G. (2003). Biosensing based on light absorption of nanoscaled gold and silver particles. Anal. Chem., 75, 6894-6900. Gann, P. and Giovannucci, E. (2005) Nutrition, exercise and prostate cancer http://www.prostatecancerfoundation.org/atf/cf/%7B705B3273-F2EF-4EF6-A653E15C5D8BB6B1%7D/Nutrition_Guide.pdf. Gann, P. H., Ma, J., Giovannucci, E., Willett, W., Sacks, F. M., Hennekens, C. H. and Stampfer, M. J. (1999). Lower prostate cancer risk in men with elevated plasma lycopene levels: results of a prospective analysis. Cancer Res., 59, 1225-30. Garattini, S. and Shore, P. A. (1966). Advances in pharmacology. Academic Press Inc., New York. 202 Giovannucci, E., Rimm, E. B., Colditz, G. A., Stampfer, M. J., Ascherio, A., Chute, C. C. and Willett, W. C. (1993). A prospective study of dietary fat and risk of prostate cancer. J. Natl. Cancer Inst ., 85, 1538-1540. Gram, H., Marconi, L. A., Barbas, C. F., Collet, T. A., Lerner, R. A. and Kang, A. S. (1992). In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc. Natl. Acad. Sci. USA, 89, 3576-3580. Graves, H. C., Sensabaugh, G. F. and Blake, E. T. (1985). Postcoital detection of a male-specific semen protein. Application for the investigation of rape. N. Engl. J. Med., 312, 338-343. Gregorakis, A. K., Malovrouvas, D., Stefanakis, S., Petrakic, K., and Scorilas, A. (2005). Free/Total PSA (F/T ratio) kinetics in patients with clinically localized prostate cancer undergoing radical prostatectomy. Eur. Urol. Supp., 5, 472-478. Greunke, K., Spillner, E., Braren, I., Seismann, H., Kainz, S., Hahna, U., Grunwald, T. and Bredehorst, R. (2006).Bivalent monoclonal IgY antibody formats by conversion of recombinant antibody fragments. J. Biotech., 124, 446–456. Grubisha, D. S., Lipert, R. J., Park, H. Y., Driskell, J. and Porter, M. D. (2003). Femtomolar detection of prostate-specific antigen: an immunoassay based on surfaceenhanced Raman scattering and immunogold labels. Anal. Chem., 75, 5936-5943. Haese, A., Graefen, M., Steuber, T., Becker, C., Pettersson, K., Piironen, T., Noldus, J., Huland, H. and Lilja, H. (2001). Human glandular kallikrein 2 levels in serum for discrimination of pathologically organ-confined from locally-advanced prostate cancer in total PSA-levels below 10 ng/ml. Prostate, 49, 101-109. Hayhurst, A. (2000). Improved expression characteristics of single-chain Fv fragments when fused downstream of the Escherichia coli maltose-binding protein or upstream of a single immunoglobulin-constant domain. Protein Exp. Purif. 18, 1-10. 203 Hayhurst, A., Happe, S., Mabry, R., Koch, Z., Iverson, B.L. and Georgiou, G. (2003). Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. Immunol. Methods, 276: 185-196. Hayward, S. W. and Cunha, G. R. (2000). The prostate: development and physiology. Radiol. Clin. North Am., 38, 1-14. Healy, D. A., Hayes, C. J., Leonard, P., McKenna, L. and O‘Kennedy, R. (2007). Biosensor developments: application to prostate-specific antigen detection. Trends in Biotech., 3, 125-131. Hernandez, V. Q., Juárez-González, V. R., Ortíz-León, M., Sánchez, R., Possani, L. D. and Becerril, B. (2006). The change of the scFv into the Fab format improves the stability and in vivo toxin neutralization capacity of recombinant antibodies. Mol. Immunol., 44, 1307-1315. Hessels, D., Verhaegh, G. W., Schalken, J. A. and Witjes, J. A. (2004). Applicability of biomarkers in the early diagnosis of prostate cancer. Expert Rev. Mol. Diagn., 4, 513526. Hilz, H., Noldus, J., Hammerer, P., Buck, F., Luck, M. and Huland, H. (1999). Molecular heterogeneity of free PSA in sera of patients with benign and malignant prostate tumors. Eur. Urol., 36, 286-292. Hochmeister, M. N., Borer, U., Budowle, B., Dirnhofer, R., Gehrig, C., Rudin, O., and Thali, M. (1999). Evaluation of prostate-specific antigen (PSA) membrane test assays for the forensic identification of seminal fluid, J. Forensic Sci., 44, 1057-1060. Hoffman, R.M., Gilliland, F.D., Eley, J.W., Harlan, L.C., Stephenson, R.A., Stanford, J.L., Albertson, P.C., Hamilton, A.S., Hunt, W.C. and Potosky, A.L. (2001). Racial and ethnic differences in advanced-stage prostate cancer: the Prostate Cancer Outcomes Study. J. Natl. Cancer Inst., 93, 388-395. 204 Hoogenboom, H. R., Henderikx, P. and de Haard, H. (1998). Creating and engineering human antibodies for immunotherapy. Adv. Drug Deliv. Rev., 31, 5-31. http://www.mamashealth.com/organs/prostate.asp. http://www.psa-rising.com/prostatecancer/prostate.htm Hsing, A. W. and Chokkalingam, A. P. (2006). Prostate cancer epidemiology. Frontiers Biosci., 11, 1388-1413. Hsing, A. W., Tsao, L. and Devesa, S. S. (2000). International trends and patterns of prostate cancer incidence and mortality. Int. J. Cancer, 85, 60-67. Huang, L., Reekmans, G., Saerens, D., Friedt, J. M., Frederix, F., Francis, L., Muyldermans, S., Campitelli, A. and Van Hoof, C. (2005). Prostate-specific antigen immunosensing based on mixed self-assembled monolayers, camel antibodies and colloidal gold enhanced sandwich assays. Biosens. Bioelectron., 21, 483-490. Huhtinen, P., Soukka, T., Lovgren, T. and Harma, H. (2004). Immunoassay of total prostate-specific antigen using europium(III) nanoparticle labels and streptavidin-biotin technology. J. Immunol. Methods, 294, 111-122. Hutchinson, L. M., Chang, E. L., Becker, C. M., Shih, M. C., Brice, M., DeWolf, W. C., Gaston, S. M. and Zetter, B. R. (2005a). Use of thymosin beta15 as a urinary biomarker in human prostate cancer. Prostate, 64, 116-127. Hutchinson, L. M., Chang, E. L., Becker, C. M., Ushiyama, N., Behonick, D., Shih, M. C., DeWolf, W. C., Gaston, S. M. and Zetter, B. R. (2005b). Development of a sensitive and specific enzyme-linked immunosorbent assay for thymosin beta15, a urinary biomarker of human prostate cancer. Clin. Biochem., 38, 558-571. Hwang, K. S., Lee, J. H., Park, J., Yoon, D. S., Park, J. H. and Kim, T. S. (2004). Insitu quantitative analysis of a prostate-specific antigen (PSA) using a nanomechanical PZT cantilever. Lab Chip, 4, 547-552. 205 Ippolito, G. C., Schelonka, R. L., Zemlin, M., Ivanov, I. I., Kobayashi, R., Zemlin, Gartland, G. L., Nitschke, L., Pelkonen, J., Fujihashi, K., Rajewsky, K. and Schroeder Jr. H. W. (2006). Forced usage of positively charged amino acids in immunoglobulin CDR-H3 impairs B cell development and antibody production. J. Experim. Med., 6, 1567-1578. Irving, R. A., Korett, A. A. and Hudson, P. J. (1996). Affinity maturation of recombinant antibodies using E. coli mutator cells. Immunotechnol., 2, 127-143. Irani, J., Salomon, L., Soulie, M., Zlotta, A., de la Taille, A., Dore, B. and Millet, C. (2005). Urinary/serum prostate-specific antigen ratio: comparison with free/total serum prostate-specific antigen ratio in improving prostate cancer detection. Urol., 65, 533537. Ismail, H. A., Pollak, M., Behlouli, H., Tanguay, S., Begin, L. R. and Aprikian, A. G. (2003). Serum insulin-like growth factor (IGF)-1 and IGF-binding protein-3 do not correlate with Gleason score or quantity of prostate cancer in biopsy samples. BJU Int., 92, 699-702. Jacobs, E. J., Rodriguez, C., Mondul, A. M., Connell, C. J., Henley, S. J., Calle, E. E. and Thun, M. J. (2005). A large cohort study of aspirin and other nonsteroidal antiinflammatory drugs and prostate cancer incidence. J. Natl. Cancer Inst., 97, 975-980. Janeway, C., Travers, P., Walport, M., Capra, J. D. (1999). In immunology: the immune system in health and disease. 4th edition New York, Garland Publishing. Johnson, E. D. and Kotowski, T. M. (1993). Detection of prostate-specific antigen by ELISA. J. Forensic Sci., 38, 250-258. Jimenez-Verdejo, A., Osuna, E., Garcia-Olivares, E. and Luna, A. (1994). Study of the enzymatic activity of GGT, LDH, PAP and PSA in semen stains: application to age calculation. Forensic Sci. Int., 68, 7-15. 206 Jung, K., Brux, B., Lein, M., Rudolph, B., Kristiansen, G., Hauptmann, S., Schnorr, D., Loening, S. A. and Sinha, P. (2000a). Molecular forms of prostate-specific antigen in malignant and benign prostatic tissue: biochemical and diagnostic implications. Clin. Chem., 46, 47-54. Jung, K., Elgeti, U., Lein, M., Brux, B., Sinha, P., Rudolph, B., Hauptmann, S., Schnorr, D., Loening, S. A. (2000b). Ratio of free or complexed prostate-specific antigen (PSA) to total PSA: which ratio improves differentiation between benign prostatic hyperplasia and prostate cancer? Clin. Chem., 46, 55-62. Katsamba, P. S., Navratilova, I., Calderon-Carcia, M., Fan, L., Thornton, K., Zhu, M., Vandel Bos, T., Forte, C., Friend, D., Laird-Offringa, I., Tavares, G., Whatley, J., Shi, E., Widom, A., Lindquist, K. S., Klakamp, S., Drake, A., Bohmann, D., Roell, M., Rose, L., Dorocke, J., Roth, B., Luginbuhl, B. and Myszka, D. G. (2006). Kinetic analysis of a high-affinity antibody/antigen interaction performed by multiple Biacore users. Anal. Biochem., 352, 208-221. Khaldi, N., Miras, A., Botti, K., Benali, L. and Gromb, S. (2004). Evaluation of three rapid detection methods for the forensic identification of seminal fluid in rape cases. J. Forensic Sci., 49, 749-753. Khan, M. A., Partin, A. W., Rittenhouse, H. G., Mikolajczyk, S. D., Sokoll, L. J., Chan, D. W. and Veltri, R. W. (2003). Evaluation of pro-PSA for the early detection of prostate cancer in men with a total PSA range of 4.0-10 ng/ml. J. Urol., 170, 723-726. Klassen, A. C. and Platz, E. A. (2006).What can geography tell us about prostate cancer? Am. J. Prev. Med., 30, S7-15. Klotz, L. and Teahan, S. (2006). Current role of PSA kinetics in the management of patients with prostate cancer. Eur. Urol. Supp., 5, 472-478. Kohler, G. and Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature (London), 256, 495-497. 207 Koide, S. and Sidhu, S. S. (2009). The importance of being tyrosine: lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem. Biol., 4, 325-334. Khosravi, M. J., Papanastasiou-Diamandi, A. and Mistry, J. (1995). An ultrasensitive immunoassay for prostate-specific antigen based on conventional colorimetric detection. Clin. Biochem., 28, 407-414. Krebber, A., Bornhauser, S., Burmester, J., Honegger, A., Willuda, J., Bosshard, H. R. and Pluckthun, A. (1997). Reliable cloning of functional antibody variable domains from hybridomas and spleen cell repertoires employing a reengineered phage display system. J. Immunol. Methods, 201, 35-55. Krumholtz, J. S., Carvalhal, G. F., Ramos, C. G., Smith, D. S., Thorson, P., Yan, Y., Humphrey, P. A., Roehl, K. A. and Catalona, W. J. (2002). Prostate-specific antigen cutoff of 2.6 ng/mL for prostate cancer screening is associated with favorable pathologic tumor features. Urol., 60, 469-473. Kumar, A., Mikolajczyk, S. D., Goel, A. S., Millar, L. S. and Saedi, M. S. (1997). Expression of pro form of prostate-specific antigen by mammalian cells and its conversion to mature, active form by human kallikrein 2. Cancer Res., 57, 3111-3114. Kuriyama, M., Wang, M. C., Papsidero, L. D., Killian, C. S., Shimano, T., Valenzuela, L., Nishiura, T., Murphy, G. P. and Chu, T. M. (1980). Quantitation of prostate-specific antigen in serum by a sensitive enzyme immunoassay. Cancer Res., 40, 4658-4662. Kwiatkowski, M. K., Recker, F., Piironen, T., Pettersson, K., Otto, T., Wernli, M. and Tscholl, R. (1998). In prostatism patients the ratio of human glandular kallikrein to free PSA improves the discrimination between prostate cancer and benign hyperplasia within the diagnostic 'grey zone' of total PSA 4 to 10 ng/ml. Urol., 52, 360-365. Labrie, F., Candas, B., Dupont, A., Cusan, L., Gomez, J. L., Suburu, R. E., Diamond, P., Levesque, J. And Belanger, A. (1999). Screening decreases prostate cancer death: 208 first analysis of the 1988 Quebec prospective randomized controlled trial. Prostate, 38, 83-91. Laux, D. L. and Custis, S. E. (2003). Forensic Detection of Semen III. Detection of PSA Using Membrane Based Tests: Sensitivity Issues with Regards to the Presence of PSA in Other Body Fluids. Submitted MAFS newsletter. Lee, J. H., Hwang, K. S., Park, J., Yoon, K. H., Yoon, D. S. and Kim, T. S. (2005). Immunoassay of prostate-specific antigen (PSA) using resonant frequency shift of piezoelectric nanomechanical microcantilever. Biosens. Bioelectron., 20, 2157-2162.# Leinonen, J., Niemelä, P., Lövgren, J., Bocchi, L. Pettersson, K., Nevanlinna, H. and Stenman, U. H. (2004). Characterization of monoclonal antibodies against prostate specific antigen produced by genetic immunization. J. Immunol. Methods, 289, 157167. Levesque, M., Hu, H., D'Costa, M. and Diamandis, E. P. (1995). Prostate-specific antigen expression by various tumors. J. Clin. Lab. Anal., 9, 123-128. Leonard, P., Safsten, P., Hearty, S., McDonnell, B., Finlay, W. and O‘Kennedy, R. (2007). High throughput ranking of recombinant avian scFv antibody fragments from crude lysates using Biacore A100. J. Immunol. Methods, 323, 172-179. Lichtenstein, P., Holm, N. V., Verkasalo, P. K., Iliadou, A., Kaprio, J., Koskenvuo, M., Pukkala, E., Skytthe, A. and Hemminki, K. (2000). Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med., 343, 78-85. Lilja, H., Ulmert, D. and Vickers, A. J. (2008). Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nature Reviews Cancer, 8, 268-278. 209 Lilja, H., Christensson, A., Dahlen, U., Matikainen, M. T., Nilsson, O., Pettersson, K. and Lovgren, T. (1991). Prostate-specific antigen in serum occurs predominantly in complex with alpha 1-antichymotrypsin. Clin. Chem., 37, 1618-1625. Lin, H., and Cornish, V. W. (2002). Screening and selection methods for large-scale analysis of protein function. Angew. Chem, Int. Ed., 41, 4402-4425. Lind, K. and Kubista, M. (2005). Development and evaluation of three real-time immuno-PCR assemblages for quantification of PSA. J. Immunol. Methods, 304, 107116. Linton, H. J., Marks, L. S., Millar, L. S., Knott, C. L., Rittenhouse, H. G. and Mikolajczyk, S. D. (2003). Benign prostate-specific antigen (BPSA) in serum is increased in benign prostate disease. Clin. Chem., 49, 253-259. Little M., Kipriyanov S. M., Le Gall F. and Moldenhauer G. (2000). Of mice and men: hybridoma and recombinant antibodies. Immunol. Today, 21, 364-370. Low, N. M., Holliger, P. H. and Winter G. (1996). Mimicking somatic hypermutation: Affinity maturation of antibodies displayed on bacteriophage using a bacterial mutator strain. J. Mol. Biol., 260, 359-368. Lutz, G. L., Zuber, E., Jean Witz, J. and Regenmortel, M. H. V. (1997). Thermodynamic analysis of antigen–antibody binding using biosensor measurements at different temperatures. Anal. Biochem., 246, 123-132. Lytton, B. (2001). Prostate cancer: a brief history and the discovery of hormonal ablation treatment. J. Urol., 165, 1859-1862. Magee, J. A., Araki, T., Patil, S., Ehrig, T., True, L., Humphrey, P. A., Catalona, W. J., Watson, M. A. and Milbrandt, J. (2001). Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res., 61, 5692-5696. 210 Mahabeer, R. and Bhoola, K. D. (2000). Kallikrein and kinin receptor genes. Pharmacol. Ther., 88, 77-89. Maher, J., Vintiner, S., Elliot, D. and Melia, L. (2002). Evaluation of the BioSign PSA membrane test for the identification of semen stains in forensic casework. N. Z. Med. J., 115, 48-49. Marks, J. D., Griffiths, A. D., Malmqvist, M., Clackson, T. P., Bye, J. M. and Winter, G. (1992). By-passing immunisation: Building high affinity human antibodies by chain shuffling. Nature Biotech., 10, 779-783. Matsumoto, K., Konishi, N., Hiasa, Y., Kimura, E., Takahashi, Y., Shinohara, K. and Samori, T. (1999). A highly sensitive enzyme-linked immunoassay for serum free prostate-specific antigen (f-PSA). Clin. Chim. Acta ., 281, 57-69. Matsumoto, K., Konishi, N., Samori, T., Kimura, E., Doi, M., Kato, S. and Yuki, Y. (2000). ELISA for a complexed antigen with a monoclonal antibody blocking reaction with the free antigen—assay-specific for complexed prostate-specific antigen. J. Immunol. Methods, 234, 99-106. McCafferty J, Griffiths, A. D, Winter, G. and Chiswell D. J. (1990). Phage antibodies: filamentous phage displaying antibody variable domains. Nature, 348, 552–554. Mikolajczyk, S. D., Song, Y., Wong, J. R., Matson, R. S. and Rittenhouse, H. G. (2004). Are multiple markers the future of prostate cancer diagnostics? Clin. Biochem., 37, 519-528. Mikolajczyk, S. D. and Rittenhouse, H. G. (2003). Pro PSA: a more cancer-specific form of prostate specific antigen for the early detection of prostate cancer. Keio. J. Med., 52, 86-91. Mikolajczyk, S. D., Marker, K. M., Millar, L. S., Kumar, A., Saedi, M. S., Payne, J. K., Evans, C. L., Gasior, C. L., Linton, H. J., Carpenter, P. and Rittenhouse, H. G. (2001). 211 A truncated precursor form of prostate-specific antigen is a more specific serum marker of prostate cancer. Cancer Res., 61, 6958-6963. Mikolajczyk, S. D., Millar, L. S., Wang, T. J., Rittenhouse, H. G., Wolfert, R. L., Marks, L. S., Song, W., Wheeler, T. M. and Slawin, K. M. (2000). "BPSA," a specific molecular form of free prostate-specific antigen, is found predominantly in the transition zone of patients with nodular benign prostatic hyperplasia. Urol., 55, 41-45. Mikolajczyk, S. D., Grauer, L. S., Millar, L. S., Hill, T. M., Kumar, A., Rittenhouse, H. G., Wolfert, R. L. and Saedi, M. S. (1997). A precursor form of PSA (pPSA) is a component of the free PSA in prostate cancer serum. Urol., 50, 710-714. Miller, D. C., Hafez, K. S., Stewart, A., Montie, J. E. and Wei, J. T. (2003). Prostate carcinoma presentation, diagnosis, and staging: an update form the National Cancer Data Base. Cancer, 98, 1169-1178. Mistry, K., and Cable, G. (2003). Meta-analysis of prostate-specific antigen and Digital rectal examination as screening tests for prostate carcinoma. J. Am. Board Family Pract., 16, 95-101. Mitchell, I. D., Croal, B. L., Dickie, A., Cohen, N. P. and Ross, I. (2001). A prospective study to evaluate the role of complexed prostate specific antigen and free/total prostate specific antigen ratio for the diagnosis of prostate cancer. J. Urol., 165, 1549-1553. Murphy, M., Jason-Moller, L. and Bruno, J. (2006). Using Biacore to measure the binding kinetics of an antibody-antigen interaction. Current Protocols in Protein Science, Chapter 19. 19.14.1-19.4.17 (John Wiley & Sons). Nadler, R. B., Humphrey, P. A., Smith, D. S., Catalona, W. J. and Ratliff, T. L. (1995). Effect of inflammation and benign prostatic hyperplasia on elevated serum prostate specific antigen levels. J. Urol., 154, 407-413. 212 Nakamura, T., Scorilas, A., Stephan, C., Jung, K., Soosaipillai, A. R. and Diamandis, E. P. (2003). The usefulness of serum human kallikrein11 (hK11) for discriminating between prostate cancer and benign prostatic hyperplasia. Cancer Res., 63, 6543-6546. Naya, Y., Fritsche, H. A., Bhadkamkar, V. A., Mikolajczyk, S. D., Rittenhouse, H. G. and Babaian, R. J. (2005). Evaluation of precursor prostate-specific antigen isoform ratios in the detection of prostate cancer. Urol. Oncol., 23, 16-21. Ni, J., Lipert, R. J., Dawson, G. B. and Porter, M. D. (1999). Immunoassay readout method using extrinsic Raman labels adsorbed on immunogold colloids. Anal. Chem., 71, 4903-4908. Noldus, J., Chen, Z. and Stamey, T. A. (1997). Isolation and characterization of free form prostate specific antigen (f-PSA) in sera of men with prostate cancer. J. Urol., 158, 1606-1609. Nurmikko, P., Pettersson, K., Piironen, T., Hugosson, J. and Lilja, H. (2001). Discrimination of prostate cancer from benign disease by plasma measurement of intact, free prostate-specific antigen lacking an internal cleavage site at Lys145-Lys146. Clin. Chem., 47, 1415-1423. Nurmikko, P., Vaisanen, V., Piironen, T., Lindgren, S., Lilja, H. and Pettersson, K. (2000). Production and characterization of novel anti-prostate-specific antigen (PSA) monoclonal antibodies that do not detect internally cleaved Lys145-Lys146 inactive PSA. Clin. Chem., 46, 1610-1618. Oberpenning, F., Weining, C., Brandt, B., De Angelis, G., Heinecke, A., Hamm, M., Stieber, P., Hertle, L., Schmid, H.P. and Semjonow, A. (2002). Combining free and total prostate specific antigen assays from different manufacturers: the pitfalls. Eur. Urol., 42, 577-82. Obiezu, C. V. and Diamandis, E. P. (2005). Human tissue kallikrein gene family: applications in cancer. Cancer Lett., 224, 1-22. 213 Obiezu, C. V., Soosaipillai, A., Jung, K., Stephan, C., Scorilas, A., Howarth, D. H. and Diamandis, E. P. (2002). Detection of human kallikrein 4 in healthy and cancerous prostatic tissues by immunofluorometry and immunohistochemistry. Clin. Chem., 48, 1232-1240. Oh, M. Y., Joo, H. Y., Hur, B. U., Jeong, Y. H. and Cha, S. H. (2007). Enhancing phage display of antibody fragments using gIII-amber suppression. Gene, 386, 81-89. Özena, H., and Sözen, S. (2006). PSA isoforms in prostate cancer detection. Eur. Urol. Supp., 5, 495-499. Pakioures M., Borgono, C. and Diamandis, E. (2007). Human tissue kallikreins: The cancer biomarker family. Cancer letters, 249, 61-79. Papotti, M., Paties, C., Peveri, V., Moscuzza, L. and Bussolati, G. (1989). Immunocytochemical detection of prostate-specific antigen in skin adnexal and breast tissues and tumors. Basic Appl. Histochem., 33, 25-29. Partin, A. W., Catalona, W. J., Finlay, J. A., Darte, C., Tindall, D. J., Young, C. Y., Klee, G. G., Chan, D. W., Rittenhouse, H. G., Wolfert, R. L. and Woodrum, D. L. (1999). Use of human glandular kallikrein 2 for the detection of prostate cancer: preliminary analysis. Urol., 54, 839-845. Peter, J., Unverzagt, C., Krogh, T. N., Vorm, O. and Hoesel, W. (2001). Identification of precursor forms of free prostate-specific antigen in serum of prostate cancer patients by immunosorption and mass spectrometry. Cancer Res., 61, 957-962. Peyromaure, M., Fulla, Y., Debre, B. and Dinh-Xuan, A. T. (2005). Pro PSA: a "pro cancer" form of PSA? Med. Hypotheses, 64, 92-95. Popkov, M., Rader, C. and Barbas, III, C. F. (2004). Isolation of human prostate cancer cell reactive antibodies using phage display technology. J. Immunol. Methods, 291, 137151. 214 Roddam, A. W., Duffy, M. J., Hamdy, F. C., Ward, A. M., Patnick, J., Price, C. P., Rimmer, J., Sturgeon, C., White, P. and Allen, N. E. (2005). Use of prostate-specific antigen (PSA) isoforms for the detection of prostate cancer in men with a PSA level of 2-10 ng/ml: systematic review and meta-analysis. Eur. Urol., 48, 386-99. Rella, R., Spadavecchiaa, J., Manera, M. G., Siciliano, P., Santino, A., and Mita, G. (2004). Liquid phase SPR imaging experiments for biosensors applications. Biosens. Bioelectron., 20, 1140-1148. Rhodes, D. R., Sanda, M. G., Otte, A. P., Chinnaiyan, A. M. and Rubin, M. A. (2003). Multiplex biomarker approach for determining risk of prostate-specific antigen-defined recurrence of prostate cancer. J. Natl. Cancer Inst., 95, 661-668. Roitt, I. M. (2006). Roitt‘s essential immunology. 11th edition. Publishers, Maden mass, Oxford, Blackwell. Rojas, G., Almagro, J. C., Acevedo, B. And Gavilondo, J. V. (2002). Phage antibody fragments library combining a single human light chain variable region with immune mouse heavy chain variable regions. J. Biotechnol., 94, 287-298. Rubin, M. A., Mucci, N. R., Figurski, J., Fecko, A., Pienta, K. J. and Day, M. L. (2001). E-cadherin expression in prostate cancer: a broad survey using high-density tissue microarray technology. Hum. Pathol., 32, 690-697. Rubin, M. A., Zhou, M., Dhanasekaran, S. M., Varambally, S., Barrette, T. R., Sanda, M. G., Pienta, K. J., Ghosh, D., and Chinnaiyan, A. M. (2002). Alpha-methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA., 287, 16621670. Saerens, D., Frederix, F., Reekmans, G., Conrath, K., Jans, K., Brys, L., Huang, L., Bosmans, E., Maes, G., Borghs, G. and Muyldermans, S. (2005). Engineering camel single-domain antibodies and immobilization chemistry for human prostate-specific antigen sensing. Anal. Chem., 77, 7547-7555. 215 Sano, T., Smith, C. L. and Cantor, C. R. (1992). Immuno-PCR: very sensitive antigen detection by means of specific antibody-DNA conjugates. Science, 258, 120-122. Sato, I., Sagi, M., Ishiwari, A., Nishijima, H., Ito, E. and Mukai, T. (2002). Use of the "SMITEST" PSA card to identify the presence of prostate-specific antigen in semen and male urine. Forensic Sci. Int., 127, 71-74. Schier, R., McCall, A., Adams, G. P., Marshall, K. W., Merritt, H., Yim, M., Crawford, R. S., Weiner, L. M., Marks, C. and Marks, J. D. (1996). Isolation of picomolar affinity anti-c-erbB-2 single-chain Fv by molecular evolution of the complementarity determining regions in the center of the antibody binding site. J. Mol. Biol., 263, 551567. Schmitz, U., Versmold, A., Kaufmann, P. and Frank, H. G. (2000). Phage display: a molecular tool for the generation of antibodies-a review. Placenta, 21, 106-112. Schulman, C.C., Ekane, S. and Zlotta, A.R. (2001). Nutrition and prostate cancer: evidence or suspicion? Urol., 58, 318-334. Shannon, J., Tewoderos, S., Garzotto, M., Beer, T. M., Derenick, R., Palma, A. and Farris, P. E. (2005). Statins and prostate cancer risk: a case-control study. Am. J. Epidemiol., 162, 318-325. Shariat, S., Canto, E., Kattan, M. and Slawin, K. (2004). Beyond prostate-specific antigen: new serologic biomarkers for improved diagnosis and management of prostate cancer. Rev. Urol., 6, 58-72. Simich, J. P., Morris, S. L., Klick, R. L. and Rittenhouse-Diakun, K. (1999). Validation of the use of a commercially available kit for the identification of prostate specific antigen (PSA) in semen stains. J. Forensic Sci., 44, 1229-1231. Smith, G. P. (1985). Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science, 14, 1315-1317. 216 Sokoll, L. J., Chan, D. W., Mikolajczyk, S. D., Rittenhouse, H. G., Evans, C. L., Linton, H. J., Mangold, L. A., Mohr, P., Bartsch, G., Klocker, H., Horninger, W. and Partin, A. W. (2003). Proenzyme PSA for the early detection of prostate cancer in the 2.5-4.0 ng/ml total PSA range: preliminary analysis. Urol., 61, 274-276. Soukka, T., Paukkunen, J., Harma, H., Lonnberg, S., Lindroos, H. and Lovgren, T. (2001). Supersensitive time-resolved immunofluorometric assay of free prostatespecific antigen with nanoparticle label technology. Clin. Chem., 47, 1269-1278. Sozen, S., Eskicorapci, S., Kupeli, B., Irkilata, L., Altinel, M., Ozer, G., Uygur, C., Alkibay, T. and Ozen, H. (2005). Complexed prostate specific antigen density is better than the other PSA derivatives for detection of prostate cancer in men with total PSA between 2.5 and 20 ng/ml: results of a prospective multicenter study. Eur. Urol., 47, 302-307. Stangelberger, A., Margreiter, M., Seitz, C. and Djavan, B. (2007). Prostate cancer screening markers. J. Men’s Health, 4, 233-244. Stenman, U. H., Leinonen, J., Zhang, W. M. and Finne, P. (1999). Prostate-specific antigen. Semin. Cancer Biol., 9, 83-93. Stephan C., Cammann, H., Meyer, H. A., Lein, M. and Jung K. (2007). PSA and new biomarkers within multivariate models to improve early detection of prostate cancer. Cancer Letter, 24, 18–29. Stephan, C., Jung, K., Lein, M., and Diamandis, E.P. (2004). PSA and other tissue kallikreins for prostate cancer detection. Eur. J. Cancer, 43, 1918-1926. Steuber, T., Nurmikko, P., Haese, A., Pettersson, K., Graefen, M., Hammerer, P., Huland, H. and Lilja, H. (2002). Discrimination of benign from malignant prostatic disease by selective measurements of single chain, intact free prostate specific antigen. J. Urol., 168, 1917-1922. 217 Storb, U., Shen, H.M., Michael, N., and Kim, N. (2001). Somatic hypermutation of immunoglobulin and non-immunoglobulin genes. Philos. Trans. R. Soc. Lond. B. Biol. Sci., 356, 13-19. Stowell, L. I., Sharman, L. E. and Hamel, K. (1991). An enzyme-linked immunosorbent assay (ELISA) for prostate-specific antigen. Forensic Sci. Int., 50, 125-138. Structural Genomics Consortium (2008). Protein production and purification. Nature Methods, 5, 135-146. Struewing, J.P., Hartge, P., Wacholder, S., Baker, S.M., Berlin, M., McAdams, M., Timmerman, M. M., Brody, L. C. and Tucker, M. A. (1997). The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N. Engl. J. Med., 336, 1401-1408. Studier, F.W. (2005). Protein production by auto-induction in high density shaking cultures. Protein Exp. Purif., 41, 207-234. Thaxton, C. S., Elghanian, R., Thomas, A. D., Stoeva, S. I., Lee, J. S., Smith, N. D., Schaeffer, A. J., Klocker, H., Horninger, W., Bartsch, G and Mirkin, C. A. (2009). Nanoparticle-based bio-barcode assay redefines ―undetectable‖ PSA and biochemical recurrence after radical prostatectomy. PNAS (USA), 10, 1-6. Thompson, I. M. Pauler, D. K., Goodman, P. J., Tangen, C. M., Lucia, M. S., Parnes, H. L., Minasian, L. M., Ford, L. G., Lippman, S. M., Crawford, E. D., Crowley, J. J. and Coltman, C. A., Jr. (2004). Prevalence of Prostate Cancer among Men with a ProstateSpecific Antigen Level 4.0 ng per Milliliter. N. Engl. J. Med., 350, 2239-2246. Trojan, L., Bode, C., Weiss, C., Mayer, D., Grobholz, R., Alken, P. and Michel, M. S. (2006). IGF-II serum levels increase discrimination between benign prostatic hyperplasia and prostate cancer and improve the predictive value of PSA in clinical staging. Eur. Urol., 49, 286-292. 218 Ueda, E. K. M., Gout, P. W. and Morganti, L. (2003). Current and prospective applications of metal ion-protein binding. J. Chromatogr., 988, 1-23. Varambally, S., Dhanasekaran, S. M., Zhou, M., Barrette, T. R., Kumar-Sinha, C., Sanda, M. G., Ghosh, D., Pienta, K. J., Sewalt, R. G., Otte, A. P., Rubin, M. A. and Chinnaiyan, A. M. (2002). The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature, 419, 624-629. Varma, M., Morgan, M., Jasani, B., Tamboli, P. and Amin, M.B. (2002). Polyclonal Anti-PSA Is More Sensitive but Less Specific Than Monoclonal Anti-PSA Implications for Diagnostic Prostatic Pathology. Amer. J. Clin. Path., 118, 202-207. Vihinen, M. (1994). Modeling of prostate specific antigen and human glandular kallikrein structures. Biochem. Biophys. Res. Commun., 204, 1251-1256. Weir, H.K., Thun, M.J., Hankey, B. F., Ries, L. A., Howe, H. L., Wingo, P. A., Jemal, A., Ward, E., Anderson, R. N. and Edwards, B. K. (2003). Annual report to the nation on the status of cancer, 1975–2000, featuring the uses of surveillance data for cancer prevention and control. J. Natl. Cancer Inst., 95, 1276-1299. Whittemore, A. S., Kolonel, L. N., Wu, A. H., John, E. M., Gallagher, R. P., Howe, G. R., Burch, J. D., Hankin, J., Dreon, D. M., West, D. W. and et al. (1995). Prostate cancer in relation to diet, physical activity, and body size in blacks, whites, and Asians in the United States and Canada. J. Natl. Cancer Inst., 87, 652-661. Wilson, W. D. (2002). Analyzing biomolecular interactions. Science, 295, 2103-2105. Woodrum, D. L., Brawer, M. K., Partin, A. W., Catalona, W. J. and Southwick, P. C. (1998). Interpretation of free prostate specific antigen clinical research studies for the detection of prostate cancer. J. Urol., 159, 5-12. 219 Wu, J.T., Zhang, P., Liu, G.H. and Wilson, L. (1998). Development of an immunoassay specific for the PSA-ACT complex without the problem of high background. J. Clin. Lab. Anal., 12, 14-19. Wu, G., Datar, R. H., Hansen, K. M., Thundat, T., Cote, R. J. and Majumdar, A. (2001). Bioassay of Prostate Specific Antigen (PSA) using microcantilevers. Nature Biotechnol., 19, 856-860. Ye, Z., Tan, M., Wang, G. and Yuan, J. (2004). Preparation, characterization, and timeresolved fluorometric application of silica-coated terbium(III) fluorescent nanoparticles. Anal. Chem., 76, 513-518. Ye, Z., Tan, M., Wang, G. and Yuan, J. (2005). Preparation, characterization and application of fluorescent terbium complex-doped zirconia nanoparticles. J. Fluoresc., 15, 499-505. Yokota, M., Mitani, T., Tsujita, H., Kobayashi, T., Higuchi, T., Akane, A. and Nasu, M. (2001). Evaluation of prostate-specific antigen (PSA) membrane test for forensic examination of semen. Leg. Med (Tokyo)., 3, 171-176. Yousef, G. M. and Diamandis, E. P. (2001). The new human tissue kallikrein gene family: Structure, function, and association to disease. Endocr. Rev., 22, 184-204. Yu, F., Persson, B., Lofas, S. and Knoll, W. (2004). Surface plasmon fluorescence immunoassay of free prostate-specific antigen in human plasma at the femtomolar level. Anal. Chem., 76, 6765-6770. Yu, H. and Diamandis, E. P. (1995a). Prostate-specific antigen in milk of lactating women. Clin. Chem., 41, 54-58. Yu, H. and Diamandis, E. P. (1995b). Prostate-specific antigen immunoreactivity in amniotic fluid. Clin. Chem., 41, 204-210. 220 Zeder-Lutz, G., Zuber, E., Witz, J. and Van Regenmortel, M. H. V. (1997). Thermodynamic analysis of antigen-antibody binding using biosensor measurements at different temperatures. Anal. Biochem., 246, 123-132. Zhang, W. M., Finne, P., Leinonen, J., Vesalainen, S., Nordling, S. and Stenman, U. H. (1999). Measurement of the complex between prostate-specific antigen and alpha-1protease inhibitor in serum. Clin. Chem., 45, 814-821. Zhang, W. M., Leinonen, J., Kalkkinen, N., Dowell, B. and Stenman, U. H. (1995). Purification and characterization of different molecular forms of prostate-specific antigen in human seminal fluid. Clin. Chem., 41, 1567-1573. Zheng, G., Patolsky, F., Cui, Y., Wang, W. U., Lieber, C. M. (2005). Multiplexed electrical detection of cancer markers with nanowire sensor arrays. Nature Biotechnol., 23, 1294-1301. Zhu, Y., Williams, S. and Zwiggelaar, R. (2006). Computer technology in detection and staging of prostate carcinoma: a review. Med. Image Anal., 10, 178-199. Zundel , D., Jarry , H., Kestler, D., Holzapfel, G., Bartels, H., Scheit, K. H. and Wuttke W. (1990). Development and evaluation of an enzyme-linked immunoassay for the prostate: specific antigen utilizing two monoclonal antibodies. Urol. Res., 18, 327-330. 221






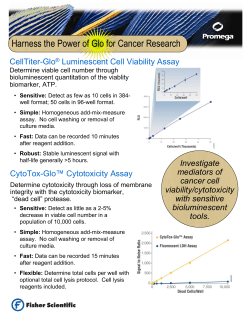



© Copyright 2026