
Document 327026
The Journal of the Pharmaceutical Information & Pharmacovigilance Association Issue 45 • Sept 2014 Contents 2 President’s Report 4 From The Editors Articles and Meeting Reports 5 Citizen Science 6 Pharmacovigilance Compliance and Market Research 8 Why Orange is the New Black 10 Outsourcing Pharmacovigilance - Pro’s, Con’s and Vendor Selection 12 How Will the EU Clinical Trial Affect You? 14 The Role of Clinical Pharmacists in Pharmacovigilance: Some Thoughts From India 17 Meeting Customer Expectations - Is There An App For That 18 How Will The Change in European Device Regulations Affect Vigilance Practises?- A Future without Scandal 20 Risk Management - How Well Are We Doing? 22 PIPA Discussion Forum 23 Diary 24 Online Learning 25 New Members 26 Noticeboard 27 PIPA Committee Members Copyright for any article accepted for publication in PIPELINE is transferred to PIPA once the article is submitted. Copyright covers the exclusive rights to reproduce & distribute the article in any form (such as photocopies or electronic copies) and applies to the complete article and any part within. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form without written permission from PIPA. PIPA will, wherever possible, grant permission to authors to subsequently use their articles, or to others to take limited numbers of copies, provided permission is obtained from the editors in advance. PIPA members are permitted to print a copy of PIPEline and/or save a single copy of the electronic file for their personal use. While all reasonable efforts are made to ensure the accuracy of the information presented in this newsletter, the editors do not accept any liability for loss arising from reliance on the information presented. The opinions expressed in the newsletter are not necessarily those of PIPA, its Committee or companies to which members belong - unless otherwise stated. PIPELINE Editor: Sanjay Motivaras and Giulia Brichetto Designed by: Zebrahouse Design and Print Ltd www.zebrahouseprint.co.uk Click here to go to Contents page 1 PRESIDENT’S REPORT Dear Members, Sarah our President is taking well deserved annual leave – so it has fallen to me to write the president’s report for this issue of PIPELINE. Christine Needham, PIPA Vice President Despite the recent rain the weather has been good and I hope you have all managed to get a few days away to enjoy the Sunshine. The power of Social media has again been illustrated by the success of the ALS Ice Bucket Challenge. The challenge has become a viral internet sensation in the last few weeks and involves dousing yourself with a bucket of icy water, whilst agreeing to donate to the ALS Association. ALS is the most common type of motor neurone disease, which is a progressive neurodegenerative disease that affects nerve cells in the spinal cord and brain. Pharmaceutical news items have featured executives at GlaxoSmithKline, Evotec and Biogen all taking part in this Challenge. Sarah described in her last report how we have also re-organised PIPA. We have revisited our vision continue to redefine our structure to enable us to play to our strengths. We have now developed nine complementary workstreams and will continue to focus on these to deliver a range of services to our members. We have now had our first committee meeting with our new members and it was great to see so many new ideas bought to the table. Vicki Page Clarke has sadly left the committee, her role is now based in Scotland and it has become increasingly difficult for her to attend meetings. Vicki led the Signal Detection working party successfully for some years and we would like to thank her for all her hard work. Moving forward we have replaced this working party with a Pharmacovigilance Regulations and Practice” Workstream lead by Tom Nichols (Cancer Research UK) and deputy John Barber (Dr Reddys). This is a great time for you to get involved with PIPA and help shape its 2 future. If you have feedback or would like to join one of the workstreams, please email me or Sarah. The PIPA Committee has also been busy with arrangements for this Year’s Annual conference. We have moved from a the traditional Summer timeslot and this year will be holding the conference on the 6th and 7th November, in the hope that by avoiding the holiday season more of you will be able to come along. We have analysed your valuable feedback and suggestions from last year and are putting together a full, varied and topical agenda for both Medical Information and Pharmacovigilance. Full details of the agenda will be available shortly, but sessions are likely to include: yy PMCPA Code of Practice changes and their impact yy Medical Information and Pharmacovigilance through acquisition yy Interactive ‘Zinc’ workshop yy Drug driving legislation for pharmaceutical products yy Medical Information and Pharmacovigilance Standards in practice yy Risk Management yy A dedicated half day Managers’ work stream yy Patient reporting of adverse events yy Periodic Benefit-Risk Evaluation Reports (PBRERs) yy Mindfulness in the workplace yy Global Medical Information websites As usual the conference will also provide a fantastic platform for networking opportunities during discussions in the programme sessions and through the more informal evening social event, The power of Social media has again been illustrated by the success of the ALS Ice Bucket Challenge which this year promises to be as entertaining as ever! We look forward to seeing you there. Book early to secure your place and to take advantage of our preferential earlybird booking rates. Please go to the PIPA website for further details and full booking information: http://www.pipaonline.org/ Conference-2014 With Best Wishes Christine Needham – PIPA Vice President [email protected] Click here to go to Contents page … And a brief update from Sarah I hope you all had a great Summer – I certainly did as our family ventured into Asia for the first time, touring and enjoying the diverse culture, landscape and wildlife of China. I’ll never forget the immense Terracotta Army - approx. 8000 unique warriors and horses buried for centuries and rediscovered only 40 years ago. Standing in awe on the Great Wall, I tracked it’s path snaking across the mountain ridge in each direction – a breath-taking landmark. We were also privileged to see five baby panda - three were only 20cm long and being nursed in incubators while two others were sleeping in a large wooden playpen. Thank you Christine for stepping in to prepare the Pipeline report while I was somewhat distracted! Just a few short paragraphs from me this time…. Last Autumn, I shared the Committee concerns regarding PIPA’s cash flow and manpower capacity. Having critically reviewed how we could stabilize and sustain PIPA, we made a range of operational changes, refocused our efforts and announced the EGM to consult with the membership; we were very keen but not totally confident as to what we could achieve in the current climate. Almost twelve months on, I am delighted to update on our status. Our membership base has grown and our cash flow has been restored (a key milestone in August - we held >£60K in our Current Account and decided it was time to transfer £35K to our Reserve Account). New committee members have enriched our core team and the Click here to go to Contents page future looks most promising. As per constitution, Emma and Sarah A have recently filed our accounts for our formal 5-Year Audit with our Accountants. I am particularly delighted with the professional and social programme for our November conference and hope you can join us. While many of us feed in suggestions, it’s the Conference Workstream, notably Christine, Tom and Anne, who bring this all to life. A rich mix of personal and technical learning opportunities through presentations, breakout workshops and our Managers Track, coupled with informal networking opportunities. Please visit the website for more details and to book your place. Sarah Dunnett, PIPA President Many of you have already registered and completed the Module VI and / or the Basic PV training courses launched by our e-Learning Workstream (these are continuously available through our website). Sarah H, Ester and the team have also been busy authoring, uploading and quality checking a number of additional e-learning modules so look out for announcements regarding Module XV, I and X. Kind Regards, Sarah Dunnett (PIPA President) 3 FROM THE EDITORS Dear Readers, Welcome to our third edition of PIPELINE. I am Giulia the new co-editor of PIPELINE and I am delighted to have joined forces with Sanjay to deliver a journal loaded with hot topics that stimulate discussion. We hope you will enjoy reading this edition as summer is a great time to relax, enjoy the milder weather and spend time on our wider interests. In this edition we have the pleasure of providing you with some brilliant articles on a variety of topics, ranging from the use of new technologies in the pharmaceutical industry to a discussion on Pharmacovigilance compliance and marketing regulations. There is also a very handy overview of the MHRA Orange Guide, which we hope many of you will find useful. Giulia Brichetto As always if you would like to read an article on a specific topic not covered by us or showcase your writing skills and become a PIPELINE author, please get in touch. We would love to hear from you. The last couple of weeks we have been very busy organising the PIPA Conference, which is just around the corner, so hurry up and buy your tickets if you haven’t already! I hope to see many of you at the conference so I can introduce myself in person and meet all the new faces. Looking forward to hearing from you all soon Sanjay Motivaras Giulia & Sanjay Contact the editors: [email protected] Become an Author Fellow member, are you a budding author with an interest in writing articles that would be of interest to medical information and pharmacovigilance professionals? Would you like to impart knowledge that you have gained recently through work or training, which you feel the membership would find useful? e-PIPELINE is a journal which is sent to all members electronically and is read by both members and non-members. PIPA welcomes contributions of articles from its membership for e-PIPELINE, which is registered with the British Library. So why not take this opportunity to see your name in print….write for us! Please contact the editorial team with your ideas or article, or for further information, and we would be delighted to discuss the process for contributing an article in more detail: [email protected] 4 Click here to go to Contents page ARTICLES Citizen Science Following the last President’s Report, we thought we would give you more details on Cancer Research UK’s exciting Citizen Science initiative Citizen Science is a world-leading initiative from Cancer Research UK that harnesses the power of the public to help scientists analyse real cancer data, accelerating their research to beat cancer sooner. Why it’s important The most effective tool for analysing cancer data is often the human eye. Computers simply aren’t good enough at understanding patterns or spotting things that look a bit unusual. It can take scientists years to look over the huge volumes of data they need to analyse, which delays the search for new cancer treatments. But what if the public could do this analysis? With enough eyes on the data we could analyse it in a fraction of the time. Citizen scientists from around the world have already joined our cause, saving scientists thousands of hours by helping to analyse genuine research data. How it works Our products visualise real cancer data in engaging ways that make it exciting and enjoyable for the general public to get involved. The more people analysing data, the more accurate the results will be, and the faster new breakthroughs can be discovered. The results of their data analysis have been consistent with the experts. They’ve also shown that crowd sourcing saves time. In just 3 months the public analysed data that would have taken a research team 18 months. Play to Cure™: Genes in Space Play to Cure™: Genes in Space is the world’s first app to incorporate the analysis of genes into an action-packed mobile game. As players traverse space searching for a mysterious and valuable substance and dodging oncoming asteroids, they’re helping us to discover critical clues about the genetic mistakes which underpin cancer. We know that faults in our genes can lead to cancer cells forming. This can be Click here to go to Contents page linked to the amount of genes in our cells - sometimes we have more and sometimes we have less. It‘s critical we recognise these patterns to understand which cancer causing genes to target when developing treatments. With thousands of citizen scientists playing Play to Cure™: Genes in Space, the process is greatly accelerated. Players first plot a galactic route. In the context of the game, they’re choosing their flight path, but these “space coordinates” are actually a visualisation of DNA data, and players are showing our scientists where the genetic variations are which may lead to cancer. Tom Nichols well a patient will respond to different treatments. Users are guided through a tutorial to make sure they know what to do. They are then shown real life breast cancer data (below) and asked a few questions to determine details about the cells in the image. Our scientists use this insight to answer key questions about the type of cancer they are studying; ultimately helping to improve the diagnosis and treatment of cancer. Then they collect Element Alpha. This mist like substance is collected as players fly through space and can be traded for ship upgrades. It actually represents the same DNA data that has just been mapped – which means our scientists have two perspectives on the same sample, from one player. And we’ve added an asteroid field. This makes the gameplay more engaging and challenging, players need to dodge or shoot a multitude of asteroids to complete a stage. Each data sample is analysed multiple times for accuracy. Citizen scientists don’t have to worry about making mistakes. The more people who use Play to Cure™: Genes in Space, the more accurate the results will be and the faster data can be translated into new ways to beat cancer. Cell Slider Cell Slider is the world’s first online platform that harnesses the collective force of the public to help beat cancer sooner. By examining tumour tissue samples and spotting cancerous cells, citizen scientists from all over the world can help us understand how 5 Pharmacovigilance Compliance and Market Research Ejaz Butt MRQA, MICR, Pharmacovigilance Compliance & Risk Management Consultant Ejaz Butt Summary: Adverse event reporting has been closely monitored over the years and has developed into a tighter, regulated process than ever before, not at least with the emergence of the new Legislation. With every update in Pharmacovigilance - bringing along with it new challenges and currently one of those new challenges is presented in the form of Market Research. The message resonating from the Authorities would suggest that drug safety departments are to tighten their grip on marketing and provide similar, if not exact, kind of oversight portrayed with clinical studies. Each company is equipped with a team of experts in the form of scientific/ medical advisors who in turn work closely with product/brand managers to either seek new information regarding a therapeutic area or a particular medicinal product, in order to try and better understand the market or the scientific data involved to help patients and /or healthcare professionals improve the management of disease and optimisation of treatment. Along with that however, also brings the risk of adverse events (AE) being reported. As when a market research programme (MRP) involves direct interaction with patients, there is always the possibility that AEs or product complaints relating to any of the marketing authorization holder’s (MAH) products may be mentioned. And where there are possibilities of AE’s - there are risks of under-reporting - and when the programmes are contracted out to third party entities, the risk increases twofold, as first line safety information is no longer in the domain of the MAH. Market Research Programmes, according to the Good Vigilance Practice Module VI (VI.C.2.2.11Reports from Patient Support Programs (PSP) and MRPs) is defined as a “systematic collection, recording and analysis of data and findings concerning medicinal products, relevant for marketing and business 6 development”. It is often conducted by a third party on behalf of the MAH, and can be conducted face to face, by telephone, online and through mail (be it in the electronic format). It may involve fully structured questionnaires including closed questions, or semistructured, which involve a mix of open and closed questions. Or it could be completely unstructured and therefore all open questions (qualitative). When responding to any of the questions presented to the participant of the research, the possibility for patients or health care practitioners (HCPs) - who are the participants of the research - to report adverse events which could be suspected adverse drug reactions. The GVP Module VI (VI.C.2.2.11 Reports from PSPs and MRPs), outlines the legal requirements in the collection of safety data, and this module also contains requirements applicable to MAHs for the management and reporting of safety data arising from PSPs and MRPs, when made aware of them. Serious and non-serious cases of suspected adverse reactions originating in those programmes are required to be reported by MAHs as solicited reports. A Patient Support Programs (PSPs) is defined by Module VI, as “an organised system where a MAH receives and collects information relating to the use of its medicinal products. Examples are post …where there are possibilities of AE’s - there are risks of underreporting and when the programmes are contracted out to third party entities, the risk increases twofold … Click here to go to Contents page authorisation patient support and disease management programmes, surveys of patients and healthcare providers, information gathering on patient compliance, or compensation/ reimbursement schemes”. The Association of the British Pharmaceutical Industry (ABPI) defines a PSP as “a service for direct patient or patient carer interaction/engagement designed to help management of medication and/ or disease outcomes (e.g. adherence, awareness and education), or to provide healthcare professionals (HCPs) with support for their patients”. A program, therefore, falls into the category of a PSP, if there is direct contact with patients or patient carers. The intention of a PSP is to support patient care provided by the MAH or by a third party on the MAH’s behalf. Thus, PSP research presents itself as a “higher risk” in terms of AEs being reported due to the direct nature of its interactions with participants. Further examples of activities revolving around PSPs include (but the list is not an exhaustive one): helping to manage a patient’s medication and/or disease outcomes (e.g., adherence, awareness, education), providing healthcare professionals with support for their patients, compliance programmes where consenting patients on a medication are contacted to see how they are managing with their medication, etc. Click here to go to Contents page It is important to note that third parties are acting as “delegates” of the MAH, therefore, the AE reporting clock starts when the third party entity is made aware of the AE and not the date the report is passed onto the MAH. It is therefore of paramount importance, that those involved with carrying out activities within the programme are appropriately trained. As a general guidance, there are certain factors which should be covered when meeting your regulatory obligations and providing a PV compliance oversight over MRPs. These can be summarised through a checklist (here enlists a minimum criteria that ought to be covered): 1. Appropriate quality assessment of vendor involved (due diligence) 2. Adequate training (including reporting and/or product information related training if applicable) The guidance provided is rather broad, but the message is quite clear. So where does the responsibilities lie? With different parties involved in such activities, who takes the lead to ensure compliance is met? It is clear that the expertise of the Drug Safety department must be utilised to develop a system where everyone has a precise understanding of the pharmacovigilance requirements and compliance of market research programmes. Drug safety needs to be prepared to work together with the different entities involved, be it Medical Affairs, Marketing, Legal and Commercial areas of the business. They need to encourage transparency and be open to provide advice, training and guidance. Ultimately, the goal is to drive the message and promote a culture that pharmacovigilance is very much part of the business and together can bring quality medicinal products to the patient. 3. A safety data exchange agreement or similar 4. Monitoring of program compliance throughout the lifecycle of the project involved, e.g. periodic reconciliation for the process of data exchange, etc – relevant forms/ templates can be obtained from the BHBIA or ABPI website 7 Why Orange is the New Black A brief overview of MHRA Orange Guide 2014 – a Medical Information perspective Perveen Kashem, Medical Information Scientist, Baxter Healthcare Perveen Kashem We are aware of Good Manufacturing Practice (GMP) and Good Distribution Practice (GDP) as necessary to ensure safe and effective products reach customers. Numerous directives, guidance and regulatory requirements have been developed over the years which cover a diverse range of medicines. So wouldn’t it be useful to have the essential information in one handy book? Well, the good news is a resource known as the Orange Guide, ‘Rules and Guidance for Pharmaceutical Manufacturers and Distributors.’ This rough guide to the latest edition of the Orange Guide gives an overview of the contents, key changes and how information from the guide may be useful from a Medical Information perspective. I hope you will agree with me after this brief overview, that Orange is indeed the new black! History The Orange Guide is divided into four major sections First published in 1971 consisting of just 30 pages, the original Orange Guide was a voluntary guide to help British manufacturers understand the regulatory requirements for the manufacture of pharmaceutical products. Later editions followed with more detailed requirements for manufacturing sites, addition of Good Distribution Practice (GDP) and in the late 90’s it was harmonised to European Legislation. The guide is traditionally orange and has maintained this cover, perhaps for easy identification and to maintain continuity in the Pharmaceutical Industry. The latest edition, produced by the Medicines Health Regulatory Agency (MHRA) contains the changes since the 2007 edition. The Orange Guide is more than a guide to GMP, as it covers both UK and EU legislation on pharmaceuticals, as well as GDP and Active Pharmaceutical Ingredients and a whole lot more. 8 Click here to go to Contents page MHRA The first section of the book is an overview of the MHRA and the work they do. All licensed medicines available in the UK are subjected to rigorous scrutiny by the MHRA before they can be used by patients. The MHRA assess all applications for new medicines so they meet acceptable standards on safety, quality and efficacy. This is then maintained by regular inspections and testing throughout the lifetime of the medicine. Good Manufacturing Practice -GMP (parts I, II and III) GMP ensures products are consistently produced and controlled according to quality standards. GMP covers all aspect of production from the starting materials, premises and equipment to training and personal hygiene of the staff. The GMP section forms a large section of the guide consisting of three parts; Part I is for basic requirements for Medicinal Products, which includes the quality system, personnel, equipment, documentation, outsourcing etc. followed by 20 separate appendices or annexes specific for product type, e.g. sterile medicines, biologic active substances for human use, medicines derived from human plasma. Part II is the basic requirements for active substances used as starting materials and Part III is a new section which clarifies all the regulatory documents required for GMP. Manufacture and Importation The third section is about Manufacture and Importation (includes extracts from Human Medicines Regulation 2012). This gives guidance on the conditions required for Manufacturer’s licence, set out in the Human Medicines Regulation 2012. This section also details the code of practice for a ‘Qualified Person’, the legal and regulatory requirements for imported products. Wholesale Distribution and Brokering (Falsified Medicines Directive) Click here to go to Contents page Manufacturers performing any distribution activities with their own products must comply with Good Distribution Practice (GDP). details on how to prepare a Site Master File, provides a template exporting active substances to the EU and ICH requirements for batch certification. This ensures products are consistently stored, transported and handled under the Marketing Authorisation (MA) holder or product specification. Application within Medical Information There are detailed instructions for the wholesale distribution of medicinal products by manufacturers, importers or other wholesale distributors, or pharmacists supplying medicinal products to the public. The Falsified Medicines directive came into force in Europe in January 2013. This introduces measures to prevent the entry of falsified medicines into the supply chain. This includes guidelines to assist wholesale distributors in conducting their activities and to prevent falsified medicines entering the legal supply chain. The legislation is not just for wholesale distributors but also brokers who are involved in the purchase of medicinal products without selling or purchasing those products themselves and without owning and physically handling the medicinal products. The Orange Guide is most relevant to colleagues in Supply Chain, Quality and Manufacturing however the principles GMP and GDP are important for Medical Information. When questions are raised around manufacturing processes, temperature controlled delivery, product quality, product testing etc., then the Orange Guide may well serve as a useful background resource. It is useful to be able to refer to it as you research standard answers and respond to enquiries. For further information please contact Perveen Kashem at: [email protected] Key Changes There have been many changes to this latest edition, however the key changes have been in the GMP part I ‘Quality Risk Management’ has been harmonised with the ICH guidelines so this is an integral part of the quality system. The section on document retention has been amended to include electronic documents in light of the increasing use of electronic documents in GMP. Guidance on the outsourced GMP regulated activities has also been updated. There have been further clarifications in the individual annexe sections, e.g. in ‘Biologic active substances’ now includes transgenic derived products. The other major change has been the addition of a new chapter in the GMP section, part III. This section was added in December 2010, providing a section dedicated to GMP related documentation and clear guidance to Regulatory expectations. This lists 9 Outsourcing Pharmacovigilance – Pros, Cons and Vendor Selection Jonathan Hart-Smith Historically, outsourcing drug safety and pharmacovigilance was not an option a company would consider. The pressure to keep the expertise and responsibility in-house was powerful, coupled with concerns over data protection, confidentiality and potential non-compliance. Companies were keen to retain control. These reservations are still evident but a track record of successful outsourcing initiatives, together with an increasing number of competent providers, has made it more commonplace for companies to consider the benefits. There are three main options available when considering outsourcing: yy Strategic full service partnership yy Functional Service / Business Process Outsourcing (FSP / BPO) yy Temporary staffing / contracting Each of these options has pros and cons: Option Pros Cons Strategic full service partnership • Simplified arrangement as part of a larger outsourcing alignment • Reduced level of daily oversight required • Reduced fixed costs • Access to expertise • A shift to low cost centres • Reduced pressure on internal staff to recruit/train • Access to SOPs/tools • • • • Functional service provision • Increased flexibility • Cost reductions • Reduced demand on internal staff for management • Access to capabilities/skills • Rapid response to demand • Reduced pressure on internal staff to recruit/train • Fixed costs become variable • Vendor works on the clients systems reducing data integration challenges • External supplier has the skills and they are now “owned” by the company • Investment required in training the vendor in internal systems. Temporary • Flexible • Rapid response to changes staffing • Fixed costs become variable • Control over work allocation and outputs • Pay per use 10 Perceived lack of control/governance Reduced visibility of cost allocation Significant investment in long term relationship Potential “them and us” relationship • Internal staff spend time recruiting and selecting suitable contractors • Increased cost per hour Click here to go to Contents page Even for a company that desires keeping responsibility in house, the demand on pharmacovigilance departments is increasing and the growth in demand for both capacity and skills cannot always be delivered by an existing workforce. Therefore regardless of your company’s chosen arrangement, the question of whether to outsource or not, and how, is likely to be raised. If you do decide to engage a third party vendor, there are a number of questions to ask: yy Is this a long term strategic goal? What are the motivators for this (cost, expertise, location)? yy Is this a long or short term project? yy Is external expertise required? yy Will this form part of a larger outsourcing initiative? yy What level of control will be retained? yy What resources are available for this initiative? Once these questions are answered and the scope of this work defined, it is time to undertake vendor selection. The contracts and outsourcing department will often provide support. This is likely to follow the process of identifying some potential providers, sending a request for information (RFI), a request for quotation (RFQ), supplier meetings, selection and contract negotiation. Throughout this process you should have some core questions to answer: yy Does this supplier have the expertise, track record and capacity to deliver? yy Can you check their references? yy Do they have established process, systems and tools? yy Do they have an established governance structure? yy Can they integrate with your business? yy Will they provide the service to meet your budget? yy What is your budget? yy Will they be responsive to your demands? yy Do you intend to use your SOPs or the vendors? Having selected the right vendor, take some time at the start of the relationship Click here to go to Contents page to make sure you build a shared vision for success and to establish the trust and two-way communication that will be the foundation of your partnership. Summary: Outsourcing pharmacovigilance is becoming far more commonplace within the pharmaceutical industry. With careful vendor selection and a clear definition of the work, outsourcing provides flexibility, expertise and cost control. Ultimately a strong working relationship, collaborative approach and shared common goal will lead to a successful partnership. CK Aspire provides dedicated teams and departments to help businesses do what they do best. We provide a high quality service managing projects, studies or functions for clients within the pharmaceutical science and technology sector. For more information on how CK Aspire can help you, contact us on +44 (0)1438 842 967 or email [email protected] By Jonathan Hart-Smith MD of CK Group 11 How Will The EU Clinical Trial Regulation Affect You? By Tom Nichols Tom Nichols The current rules to regulate the conduct of clinical trials in the EU are set out in the ‘Clinical Trials Directive’ (2001/20/EC). However, this has been criticised by patients, researchers and industry for disproportionate regulatory requirements. The new Regulation aims to cut red-tape and bring patient-orientated research back to Europe. On April 2, 2014, in was approved by the European Parliament. The Regulation was then formally adopted by the European Council of Ministers and signed into law. All that remains is for the Regulation to be published in the Official Journal of the European Union Publication, which is expected to occur by the end of June. The Regulation will ultimately be binding in its entirety and automatically incorporated into the national laws for all EU Member States and much has been made of a new streamlined application and review process for research sponsors. However, what will be the practical changes that we will see? Many of the current issues stem from the rules being laid out in a Directive, so are implemented nationally, which has lead to: yy Lack of consistency and ultimately divergent applications yy Increased administration yy Ignoring the global nature of trials A key component of the new Regulation is a central, web-based, Portal through which all applications for trials in the EU must be submitted, electronically. However, the Portal is currently being developed, with no deadline set. As transition will not be complete until six months after the EU Portal is fully functional, full enforcement of the Regulation is not expected until mid2016 at the very earliest. 12 Rather than submit separate applications to each Member State (MS) for individual approval, a single submission to the Portal can be made, regardless of the number of MS in which the Sponsor intends to conduct the trial. Upon submission, the Sponsor must identify one of the Member States as their preferred Reporting Member State (RMS), responsible for coordinating and facilitating the review of the application. Should the Sponsor’s proposed RMS not wish to take on the role, there is a period during which, the MS can discuss which is willing to assume the position. Should there be no agreement, the Sponsor’s proposed RMS stands. Six days after the submission of the application, the RMS will be confirmed. The RMS must also validate the application for completeness and compliance within ten days and report back to the Sponsor, either validating the application; providing the Sponsor with ten days to provide the additional documentation; decline to validate the application. There will then be two parts to the application assessment. Part I Part I is a harmonised, scientific assessment of the application and is expected to take up to 76 days (with extended timelines for advanced therapies). The initial assessment of the application will occur within 26 days of validation and will be coordinated by the RMS. A Draft Assessment The new Regulation aims to cut red-tape and bring patientorientated research back to Europe. Click here to go to Contents page Report is circulated to the other MS, who have 12 days to respond to the RMS on the application and Draft Assessment Report. Within seven days of the coordinated review, the RMS will produce a final Assessment Report which the Sponsor and other MS will receive through the Portal. This report will either accept the application; accept the application subject to conditions; reject the application. During the period of coordinated review and production of the final Assessment Report, the RMS can request additional information from the Sponsor, extending the overall deadline by up to 31 days. Part II Part II is a national review by each MS, ensuring that the trial complies with domestic laws and regulation. Application for this stage of review can be done in parallel with Part I, but must occur within two years of Part I. This also includes review by an Independent Ethics Committee (IEC), for which the process of coordination with must be determined by individual MS. As with Part I, there is a 45 day timeline for national assessment, with up to an additional 31 days to clarify any questions. It is not clear whether the IEC review will be via the Portal, or whether a separate submission will still be required. Potential Problems yy Big IT projects are always risky. Nearly all communication around the conduct of a trail will be conducted via the portal (e.g. notification that the trial has started recruiting, suspended, DSUR submission etc.), so must be fit for purpose. As previously mentioned, no deadline has been set for the portal to Go-Live and is expected to take at least two years. yy Coordinating IEC approvals within MS. It is down to individual countries to determine how the Part II assessments are carried out. Within the UK, the MHRA and Health Research Authority are working closely to implement a system of single approval. This should lead result in an efficient review process, but is not guaranteed to be the norm across Europe. yy Workloads for some MS if frequently chosen to the RMS. Even if an MS does not wish to be the RMS, they cannot refuse is no other MS accepts the position. This could lead to unsustainable workloads for certain authorities. Hopefully, applicants will be smart with their submissions and ensure a healthy spread. A final decision as to whether the trial has been authorised, authorised with certain conditions attached, or not authorised will be communicated to applicants via the portal. This notification will take place within five days of the Part II reporting date, or the last day of the Part II assessment, whichever is later. Click here to go to Contents page 13 The Role of Clinical Pharmacists in Pharmacovigilance: Some Thoughts from India Subashchander Krishnamurthy – Pharmacovigilance Associate, Oviya MedSafe Ganesan Ramakrishnan – Senior Drug Safety Manager, Oviya MedSafe Subashchander Krishnamurthy Dr J Vijay Venkatraman – Managing Director & CEO, Oviya MedSafe Summary: The fraternity of Pharmacy has encountered noteworthy advancements in the recent years, across the globe. It has seen a movement from product focus to patient centredness. With India being the fourth largest manufacturer of pharmaceuticals in the world and with newer medications getting introduced through clinical trials continually, the dire need to strengthen and structure the drug safety system in the country is evident. Pharmacists, especially the ones working in developing countries like India, are gradually coming to terms with the fact that their jobs are no more mere assignments to be completed behind the curtains. This article acquaints aspiring pharmacists with one such calling where they can widen their horizons as key service providers as opposed to just being handlers of prescription, in the value chain. The Past and The Present The discipline of Pharmacy is concerned with but not limited to the planning or administering of medications. The professional practice has always shown its commitment to serving community. Ever since the inception of the concept “pharmaceutical care” coined by C. D. Hepler and L. M. Strand, the profession has seen itself extending far beyond dispensing. It emphasises and recognises the role of pharmacist as an integral part of the healthcare system. WHO explains pharmaceutical care as “a philosophy of practice in which the patient is the primary beneficiary of the pharmacist’s actions. Pharmaceutical care focuses on the attitudes, behaviours, commitments, concerns, ethics, functions, knowledge, responsibilities and skills of the pharmacist on the provision of drug therapy with the goal of achieving definite therapeutic outcomes toward patient health and quality of life”. Speaking globally, the role of pharmacists has changed or grown: perhaps now, it has become more 14 patient-focused. Pharmacists have an important responsibility in monitoring the ongoing safety of medicines as part of their professional practice. The agreed common aspect known about pharmacists is counselling patients on potential unfavourable impacts of medications. This one-on-one, two-way interaction can be utilised further by asking them to report any undesirable occurrence which can serve as an effective feedback system. Adding to this, pharmacists’ expertise can play a vital role in early detection of unintended adverse effects. But does that indicate drugs are unsafe? – Definitely not. The intricacies of drugs need to be considered as a concept, with benefit-risk assessment applied to treat patients through proven or commonly agreed measures where benefits outweigh risks. Unless otherwise proven, mere existence of negative data can never be conclusive or deemed immaterial for further analysis. Numbers speak volumes about ubiquity and are never actual. A drug’s benefit-risk profile is better derived from clinical judgement. Ganesan Ramakrishnan Dr J Vijay Venkatraman Click here to go to Contents page It is here that the role of pharmacists assumes more significance, as it could result in gradual accumulation of drug safety information. Drug Safety and The Pharmacist The history of drug usage goes way back to 5000 BC, with suggested use of opium. But not until 1960s after the thalidomide tragedy that it was deemed necessary to closely control the quality, efficacy, and safety of medicines. This ultimately led to the discipline called pharmacovigilance. WHO defines pharmacovigilance as the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug-related problem. It actively contributes to the protection of patients’ and public health. It would be prudent to say pharmacovigilance deals with the collection of adverse drug reaction (ADR) reports from various stakeholders responsible for monitoring the safety profile of drugs. An adverse drug reaction is a noxious and unintended response to the administration of a medicinal product. It may arise from the use of a product within or outside the terms of its marketing authorisation, and a pharmacist knows better on why any drug is non-inclusive of its safety profile in entirety before it enters the market. Pharmacists have an important contribution to make with regard to post-marketing surveillance. Whether it is a non-prescription drug or a prescribed therapy, most drugs accessible to the patients are made available through the pharmacists. India still has traditional pharmacy practice where it sees more floating prescriptions and over-the-counters (OTCs), which remain a challenge. A Step Beyond So, how can pharmacists be proactively involved? Well, the answer is simple. The whole purpose of pharmacovigilance is to minimise the potential for harm that is associated with the drug. Pharmacists form the core of the interdisciplinary healthcare system where safety monitoring is the integral part of clinical practice. It would be a myth to consider a drug entirely safe and a misperception to consider it entirely harmful. From a pharmacovigilance perspective, healthcare professionals need both good clinical judgement of the adverse drug reaction and sound insights on the Click here to go to Contents page effectiveness of a drug to arrive upon the metacentre creating a relationship between benefit and risk. Safety as a Concept Safety when defined can be “relative absence of harm”. But that does not mean safety is never doing anything and hoping nothing has happened. In pharmacovigilance, safety means collection of reports of adverse effects of drugs. Safety can mean generating data and arriving upon a solution to decide on further usage of drugs. Apart from routinely reporting ADRs, a pharmacist must also be involved in the collection of data that might be useful in longitudinal pharmacoepidemiological studies. There is a trendy rule where manufacturers deem their drugs as safe until proven harmful, and the regulatory authorities consider every drug might be harmful until proven safe. As a matter of fact, the pharmacists should consider both. Their roles are not just confined to reporting adverse events but there has to be a proactive approach in preventing the drug-related adverse events. Staying Vigilant One of the safety concerns is timely identification of the early warnings of adverse effects. The process is usually dependent on doctors suspecting something and trying to find an association between the drug and the disease. This gives an opportunity for the pharmacists to step in and make a difference by identifying the initial users of new drugs through prescriptions and to monitor them systematically rather than waiting for someone to recognise a possible adverse effect. This concept is better known as prescription event monitoring. In addition to the pharmacist’s responsibilities relating to the reporting of adverse events, they can also involve themselves in areas such as record keeping, education and monitoring the over-the-counter drugs. A major step in empowering the pharmacists is by providing access to the medication records of the patient, thereby maximising the benefit and minimising the risks of medication use. As a result, assessments of potential drug interactions and any adverse event are possible. In case of changes in the labelling or when a drug has been withdrawn from the market, it becomes absolutely necessary for the pharmacist to ensure that the patients get a change in therapy and continue to take medications for chronic conditions. In Clinical Practice Pharmacists are one of the major stakeholders along with the physicians, patients and other healthcare professionals. Pharmacists play a key role in the management and prevention of the adverse events associated with the drug. Safety concerns are often considered to be implied when drugs are approved or authorized, but the scope has widened now. It includes all safety-related activity right from the moment humans are first exposed to the new drug. It should be noted that the pharmacist has an added advantage of getting in direct contact with the patients who may not be involved in the clinical trials for ethical reasons. Safety when defined can be “relative absence of harm”. But that does not mean safety is never doing anything and hoping nothing has happened. 15 Making two factual considerations, hospital pharmacists can play a major role in drug safety. One, most serious adverse drug events occur in hospitals; two, adverse events account for a significant percentage of all hospital admissions. Pharmacists can actually help in substantially reducing the incidence of the adverse events by recording as well as by ensuring timely communication of adverse events occurring in hospitals to further control the harmful effects of these events. This can be achieved through direct involvement in patient care and having a proper reporting system in place. The adverse event information obtained from hospitals can be quite advantageous because of their highquality documentation. In fact, hospital pharmacists have access to sophisticated computer systems and databases which augurs well for effective retrieval of information. Indian Framework The Central Drugs Standard Control Organisation (CDSCO), Directorate General of Health Services under the aegis of the Ministry of Health and Family Welfare, Government of India in collaboration with Indian Pharmacopeia Commission (IPC), Ghaziabad, has initiated a nation-wide Pharmacovigilance Programme of India (PvPI). The programme is being coordinated by IPC as a National Coordinating Centre (NCC). The centre operates under the supervision of a Steering Committee which has the Drug Controller General of India (DCGI) as its ex-officio chairman and the Officer-inCharge (New Drugs), CDSCO, New Delhi, as its ex-officio Member Secretary. PvPI currently has more than 150 functional ADR monitoring centres (AMCs) across the country. With increased nutritive interest shown by government, regulators, and industry, the numbers are expected to soon increase involving both private and government medical colleges. Accessibility It is observed that underreporting can be significantly reduced by actively involving pharmacists in the surveillance of drug safety. It is highly recommended to increase the participation of pharmacists especially in a country like India, which cites 16 unawareness as primary reason for underreporting. Negligence of reporting is also considered to be a factor for the steady dearth in reporting and seen as a major setback amongst Indian HCPs. The challenge now remains in creating awareness. Back in the olden days, information about possible adverse effects of drugs was spread through medical literature which was then the available effective way. Accessibility is far more accessible now and the needed information just a swipe away. Information technology can be used constructively to improve communication. Pharmacists have quick access to a pool of information on medication safety. India now has a dedicated drug safety website with a PvPI toolkit for the stakeholders. PvPI recently launched a toll-free helpline (1800-180-3024) to facilitate reporting of adverse reactions. There are various national level workshops held providing training to the stakeholders and generating awareness. Moreover, it has become almost imperative for pharmacists to use the Suspected Adverse Drug Reaction Reporting (SADRR) Forms to report suspected adverse drug events. The interaction of much of the stakeholders with the AMCs is primarily through the SADRR forms. This helps AMCs to maintain a database of all the adverse events associate with a drug. Suggestions are being proposed at various levels to consider incorporation of pharmacovigilance concepts in education curriculum. Take a Leap Recently drug safety has earned concerns by regulatory authorities worldwide. Stringent laws are being adopted. Global pharmaceutical companies are looking at India for their pharmacovigilance activities, which is an added benefit to the existing outsourcing hub. India now homes leading PV service providers, clinical research organisations, and IT majors. India is one of the largest pharmaceutical stations with many global players. Pharmaceutical companies are now setting up in-house PV units. With more AMCs being added, medical colleges and hospitals are in ardent need for trained professionals.The industry is all set to see much growth both in private as well as in government sector. For aspirants who wish to take up pharmacovigilance as their career, this might be the right time as future can see a demand in qualified pharmacovigilance specialists. Observation A lifesaving drug today may receive a ban tomorrow. It is very unlikely that harm of medicines can always be predicted and prevented but limiting the numbers is surely attainable. A pharmacist can perhaps be entrusted as an effective messenger in collection and reporting of ADRs as they are present at all levels of medical care; right from community pharmacy to primary healthcare centres, government hospitals to corporate hospitals. Most importantly, pharmacies work as a department at affordable and approachable locations. This serves as best takeaway point as pharmacists are actively involved in the final stages of patient care. Pharmacists can create a trusted environment by counselling patients to reduce medication errors, improve safety, and quality of care; above all, it gets them all the attention a pharmacist deserves. Epilogue Getting pharmacists recognised as healthcare service providers would help overcome the barriers in positioning their role in a healthcare team. At the same time, pharmacists should step out; challenge themselves to take up the responsibilities while working in complex healthcare settings. They should stay tuned, re-invent themselves and be prepared. A budding pharmacist should realise that clinical pharmacy is not just drug-drug, drug-food interaction, but a process involving tracking adverse drug effects, reducing medication errors, monitoring patients’ compliance and counselling patients. Healthcare is all about collective accomplishment and pharmacists do deserve a little credit. A bit of us lies in contributing back to society and if that giving involves our profession too, what better a service can it be? For further information, please contact Dr J Vijay Venkatraman at [email protected] Click here to go to Contents page Meeting Customer Expectations – Is There An App For That? Steve Hutson Innovative technologies such as mobile websites and smartphone applications have impacted many of our traditional businesses including high street banking, music, film and retail and in some instances companies have failed. But for a new technology to bring about a business failure it would need to be supported by other factors e.g. a change in customer requirements and values. A good example of this occurred in the mid 2000s when mobile users were beginning to demand more from their phones, in particular quick and easy access to email and the Internet whilst on the move. Although existing smartphones provided these capabilities they were not easy to use. The launch of Apple’s first iPhone in 2007 not only satisfied the growing requirement for email and Internet access, but also delivered this capability through a multitouch interface and easy to use apps. This innovative approach transformed the iPhone into an intuitive hand held computer that was easy to use whilst on the move. Giving your customers what they want when they need it allows you to stay ahead of the competition and potentially expand your market at their expense, a maxim that applies to the pharmaceutical industry as much as any other. Today a high percentage of pharma customers e.g. healthcare professionals (HCPs) and patients, own smartphones and use apps in their day-to-day activities and want to use this technology when dealing with the industry. But is pharma addressing this fundamental change in its customer base? In those areas where HCPs and patients work closely with the industry, Click here to go to Contents page such as drug development and marketing, we find the adoption of mobile technology patchy. Marketing are very active in this field and across the industry there are over 2000 smartphone apps for customers, a marked contrast to drug development, where only a few apps are available. There are a number of reasons why drug development might want to address this lack of mobile adoption. Firstly, as already mentioned, many HCPs and patients are mobile savvy and now expect to use this technology at every opportunity including reporting clinical trial data for instance. Secondly, industry regulators have picked up on this change in customer expectations and have begun to engage with them through mobile technologies. For example, since 2012 the FDAs MedWatcher app has been available for HCPs and the general public to report adverse events directly to the regulator. More recently the FDA has made their adverse event reporting system (FAERS) accessible to app developers through the OpenFDA project. These two projects demonstrate a growing desire amongst HCPs and patients to get involved in drug safety and the FDA recognizes the potential these initiatives have in helping anticipate safety issues before they become a problem. yy A UK initiative, Care.data, has similar potential. This project plans to release anonymous patient information from GP records and when combined with similar data from hospital records will create a huge resource for medical research. Such a dataset could, like the FAERS data, be accessed using mobile apps. yy Leading technology companies also recognize the potential for healthcare data. Apple, Google, Samsung and IBM have already developed tools that allow health and fitness apps to work together and pool results into big data sets suitable for medical research. I’m sure it will not be long before these companies have mobile apps for medical researchers to interrogate these data. For the pharmaceutical industry to catch up with what the regulators, customer groups and IT companies are already doing with mobile technologies will require considerable effort. However, the recent announcement by Novartis to explore the potential of Google’s “smart lens” to address ocular conditions suggests that the industry is waking up to the potential of mobile technologies. Steve Hutson Co-founder, VirtualPV Ltd. 17 How Will The Change in European Device Regulations Affect Vigilance Practises? - A Future without Scandal Matt Tutton, Senior Pharmacovigilance Scientist, Quanticate Limited. Matt Tutton In October 2013 the European Parliament agreed to new proposals for regulation on medical devices and in vitro diagnostic medical devices. Outdated legislation and high profile scandals have made changes a necessity. This is despite disagreements between legislators and industry over the content of the new legislation. But how will this impact vigilance procedure and will it stop medical device scandals from ever happening again? Before we look forward it is worth looking back to the early 1990s. This was a time before the internet had taken over our lives. Politically and geographically the fall of the Berlin Wall symbolised the dawn of a new Europe at this time. What has this got to do with Medical Devices I dare you ask? It was around the same time that the medical device legislation was first drafted. These are the same directives we work with today (if you exclude numerous amendments). This backdrop is significant because the legislation was initially drafted at a time when there were fewer EU Member States and fewer advances in medical device technology. The three areas of current device legislation are: yy Medical Devices Directive (93/42/ EEC) yy Active Implantable Medical Devices Directive (90/385/EEC) 18 complete overhaul. Recent scandals within the medical device industry, most notably the Poly Implant Prothese (PIP) breast implant scandal, have raised public concerns and accelerated the urgency for change. The new legislation aims to toughen up the rules of the industry and ensure mistakes and misconduct are not likely to occur Who will be affected by the changes? The answer is really everyone who is exposed to a medical device both directly and indirectly, such as the patients/consumers, healthcare professionals and manufacturers. It is important to note there is an acceptance that change is necessary for reasons already mentioned; however there are objections from the industry to the proposed changes in their entirety due to factors such as cost and feasibility. The proposed regulations are: yy Medical Devices Regulation yy In Vitro Diagnostic Medical Devices Directive (98/79/EC) yy In Vitro Diagnostic Medical Devices Regulation Fast forward a couple of decades and proposals for the new legislation on medical devices and in vitro diagnostic medical devices were approved in October 2013. Even more recently was the passing of the first reading of the proposals in April 2014. This is the first time since the early 1990s that Medical Device legislation has been given a So what of the proposed changes? The first change to note is that the updated legislation documents have been drafted as regulations rather than directives. This leaves each member state without the ability to interpret legislation and then write them into their own national law. The new regulations are legally binding across all …there is an acceptance that change is necessary for reasons already mentioned; however there are objections from the industry to the proposed changes… Click here to go to Contents page EU member states in a uniform manner. From a device vigilance perspective this will ensure that there are fewer ambiguities between member states. The main areas that will impact device vigilance are: yy Medical Devices Qualified Person (QP) yy Improved Market Surveillance and Vigilance yy Timeframe for reporting Adverse Incidents yy Centralised EU Device Reporting Portal Does the role “Qualified Person” sound familiar? This is a role that is very familiar to everyone associated with authorised medicinal products in the EU. This regulatory and compliance role has now been adopted within the proposed medical device regulations. The manufacturer will be required to employ an expert on Medical Devices. Amongst a range of responsibilities that the QP will have is the oversight of vigilance reporting obligations including the reporting of adverse incidents. The QP will overlook the manufacturer’s adherence to a new 15 day reporting timeframe signalling a change to the current reporting timeframes listed in MEDDEV 2.12/1 guidance. This timeframe applies for reporting of incidents where there is a causal relationship to the device is a reasonably possibility. The “Qualified Person” will be responsible for oversight of an improved system of vigilance on medical devices. One such improvement will be the introduction of an EU portal, or electronic system, to quote the regulations. The manufacturer will be required to submit serious incidents and corrective actions via the portal. This may not be an altogether new idea as those of us who have submitted incidents via the MHRA device reporting portal; the Manufacturer’s On-Line Environment (MORE), can justify. The EU portal aims to go further with its capabilities to include automatic forwarding of reports to the appropriate national authorities. This will alleviate the administrative burden of creating multiple submissions to numerous authorities. In addition it is hoped that authorities will be Click here to go to Contents page encouraged to have a greater level of communication themselves. So will these changes help to stave off future scandals? That is certainly the objective of legislators and politicians. Tighter regulations will certainly make it less likely. Notified bodies will be under greater scrutiny to fulfil their position. Manufacturers will potentially have inspectors knocking at their door unscheduled. This looks like good news for the safety of patients who are exposed to medical devices. Although critics argue that prices may hike up the costs of being able to implement these regulations into everyday work. This should make life easier from a device vigilance perspective. Teething problems are almost inevitable leading up to the implementation of the new regulations due in the next few years. There were big changes in 2012 for post-authorisation medicinal products in Europe and the new device regulations will have a comparable impact on the medical device community in the next few years. Emphasis on improved and transparent vigilance procedures will enable us to ensure patient safety is paramount. After all, change is long overdue as the landscape of 2014; with technological advances and evolving map of Europe, is a very different one to that of 1990. 19 Risk Management: How Well Are We Doing? A report of a Datapharm Information Meeting held on 18th June 2014 at the Royal Society, London. By Lawrence Berry, CEO Datapharm Lawrence Berry Representatives from 43 pharmaceutical companies attended Datapharm’s latest Information Mee ting designed to provide pharmaceutical company members with essential information on medicines information and risk management. Capturing off-label use – a presentation on the challenges for industry The challenges that companies face in capturing off-label use of medicines were addressed by Jayne Packham from Jayne Packham Consultancy, a company specialising in medical information for the pharmaceutical industry. Companies are obliged by law to capture reports of off-label use. Jayne therefore emphasised the importance of a clear definition of “off-label”. The legislation states that it refers to medicines being “intentionally used”, and “not in accordance with the authorised product information”. Different companies interpreted the legislation in different ways, she said, with some probing deeply to ascertain off-label use in customer interactions, and others capturing information only if the customer discusses actual off-label use. One challenge for companies, said Jayne, was training staff to recognise and capture off-label use. Clear reporting processes are needed for Sales staff to ensure they don’t breach the ABPI Code of Practice. Working together on risk – perspectives from industry Speakers from major pharmaceutical companies provided examples of how their medical information, pharmacovigilance and regulatory 20 departments address issues surrounding risk. Lorna Bailey, coordinator of risk management plans at Roche Products Ltd, explained that risk management plans as specified in legislation cover both routine and additional pharmacovigilance and risk minimization activities. Additional risk management measures (RMMs) included activities such as targeted education and training programmes. At Roche, EMAapproved RMM materials go through local adaptation and implementation planning, and the adapted materials are then submitted to the Medicines and Healthcare Products Regulatory Agency (MHRA) for approval. Once approved, the materials are distributed to external health care professionals. Lorna said that one of the challenges was to distribute materials effectively to the right professionals. “And once they’re out there, what are they doing with them? Providing guidance through face-to-face training is better than mass mailings, but it is not always possible.” Another question was whether sales people should talk about risk management materials: there was a risk that it could be seen as promotion. “Defining promotional versus nonpromotional purpose is all very well on paper, but in practice clear definition can sometimes be problematic” she explained. Another challenging area which Lorna highlighted was around …one of the challenges was to distribute materials effectively to the right professionals. “And once they’re out there, what are they doing with them? Click here to go to Contents page measures of effectiveness. “We need to measure the effectiveness of RMMs. Some outcomes are easier to measure than others.” she said. Jacquie Warner, Operations Manager at the Medical Collaboration Centre of Novartis Pharmaceuticals UK, explained the processes used by Medical Information at her company for reporting adverse events to their Pharmacovigilance department. Novartis is introducing electronic reporting systems, she said, which will link Medical Information systems directly with the Pharmacovigilance systems. There were many benefits of doing this, including real time reporting, a simpler, quicker process, reduced paperwork and filing, but such systems took time to test and develop. “As yet, at Novartis, it’s early days in terms of electronic reporting, but we’re hoping to go live globally by the end of this year.” Demand for and supply of medicines information is soaring – the Datapharm experience Demand for medicines information is growing exponentially, with nearly 20 million visits to the eMC website during the past year, compared with just over five million five years ago. Click here to go to Contents page Lawrence Berry, Chief Executive of Datapharm which provides the eMC, explained that the growth was not just down to increased internet use, but to the growing amount of information now available on the eMC. There are currently more than 10,000 PILs and SPCs available on the website, compared with around half that number in 2006. This was good news for pharmaceutical companies that are members of Datapharm – their subscription costs had gone down 30% in real terms over a period when the cost per document had gone down by almost 50%. “With your help, we spend a lot of time keeping the eMC up to date,” Lawrence told the 80 pharmaceutical company representatives at the meeting. “Without that, the quality of the eMC would disappear.” He reported that 33% of all visits to the eMC today are by mobile devices. Datapharm’s iComply service, providing tailored medicines information to companies’ sales force, is now being used by 800 company representatives in the field – a figure expected to double by the end of the year. “This compliance tool is fast becoming the industry standard,” he said. Dr Peter Carroll, Datapharm’s Director of Business Development, went on to explain that 11 companies are now using the eMC to host their risk management materials (RMMs). “We think it makes sense for companies to centralise their assets on the eMC, and we’d like to further develop this free service.” Lawrence Berry also announced that Datapharm will continue to be the official provider of consumer medicines information for the NHS Choices website. This service will be funded by the NHS rather than Datapharm’s members. If you would like to contact Datapharm to discuss your company’s requirements or to enquire about eMC membership, please contact Rick Hindle on 01372 371407 21 DISCUSSION FORUMS PIPA Discussion Forum AE follow-up. Has anyone done a study on how to increase the effectivness of AE follow up? Do certain methods produce better response rates than others? From discussions with others in PV I get the impression that the response rate to requests for follow up information to AE reports is very low. I was wondering if anyone had any experience of looking into this and maybe making changes to improve it. Notification of SmPC & product updates Which publications and relevant bodies do people notify when they have SmPC updates / product changes such as supply issues and discontinuations? Does anyone notify UKMI via the email mailing list you can purchase for £100, how often do you obtain an updated mailing list? How do you maintain your PV system master files? Is this done by PV or QA? PSMF maintenance My experience has been that while it is a PV document and is likely to be written by someone with more of a PV background, many sections, particularly some of the annexes (e.g system performance and that around audit/inspection findings) are better handled by QA, or at least the person responsible for PV compliance activities (e.g CAPA management). Day zero - AIFA portal In Italy, companies download regulatory authority ICSRs from a portal. I’d like to know what people consider to be day zero for cases downloaded from this portal - is it the day the MAH is aware of the valid case – i.e., the day they search the database OR the day the case was available (e.g the day it was uploaded to the portal by the authority). Performance indicators I would like to get some thoughts regarding Performance indicators mentioned within the PSMF. At the moment we are considering: timeliness of ICSR reporting and PSUR Submission - timeliness of variation implementation - adherence to RMP and safety commitments What other indicators do you use? Training MSLs and Sales to report off label Company staff are trained about reporting adverse events, but do companies now routinely train MSLs and Sales and all staff for that matter, and get them to report off label use (without an adverse event). I know that an MHRA speaker suggested this at the last PIPA Conference. Are companies doing this now? I assume that in VII.B.5.5.2, section 3 ‘other sources available to the MA holder’ could include all company staff. Data Protection responsibilities for PV Service Providers Is anyone able to provide advice/guidance regarding the data protection responsibilities specifically for PV Service Providers? In particular, would PV Service Providers be considered Data Controllers or Data Processors? Would it be acceptable to assume that the client (MAH) is the data controller as they are the organisation who has the legal basis for collection personal data and that the PV Service Provider is the data processor? Obtaining written or verbal consent from HCPs for Reps to refer their enquiry to Medical Information Would anyone be willing to share whether their Reps obtain written consent from a HCP (either by signing a form or using an app for example) for referring their unsolicited enquiry to Medical Information to respond? Medical Information requirements in Europe I would be interested to know the requirements for Medical Information in Europe especially for France, Germany, Italy and Spain. Do you happen to know the answers to these questions or experience of dealing with these from within your own company and would li ke to share it with our readership? Please email the editors at [email protected] and we will pass on your knowledge in our next edition of PIPELINE. 22 Click here to go to Contents page FORUMS AND DIARY Forums and Online Learning PIPA has a long tradition of providing opportunities for members to discuss hot topics, share best practice and learn from each others experiences. Each year we run a series of forums which combine formal presentations with discussion and debate. PIPA also holds an annual conference in order to allow members and non-members to network and exchange ideas. The scientific programme runs over two days and includes presentations, exhibitions and workshops. Diary PIPA Conference 2014 Delegates Thursday 6th & Friday 7th November 2014 Getting the Best Out of Searching: Optimise your Search Skills Wednesday 8th October 2014 Chartridge Lodge, Chartridge Lane, Chesham, Buckinghamshire, HP5 2TU If you’re looking to get the best out of searching and to optimise your search skills, this course is for you. With the increasing amount of resources available on the internet, this course will enable you to choose the most valuable resources and will also provide you with the skills and tips to be able to effectively search within these resources. The course will also provide tips on the best ways to search for publications within PubMed. This 1 day course is aimed at individuals who are new to Medical Information or a similar area, or would like up-to-date tips on how to search for information. This course has been recently updated in light of feedback from previous delegates and advancements in resources and searching technology. Attending this course will enable delegates to: • • • • • • • Search the internet with confidence Improve search accuracy Identify resources and search tools Identify reliable information Assess the quality of internet sites Learn more about literature searching (e.g. PubMed) Have an increased awareness of servies such as electronic Medicines Compendium (eMC) and dictionary of medicines and devices browser (dm+d) • Keep abreast of social media channels and searching within these channels Interactive workshops within a small group provide you with a good opportunity and forum for sharing experiences, best practice and networking. Click here to go to Contents page 23 Online Learning Introduction to Pharmacovigilance for non-Pharmacovigilance Personnel Pharmaceutical companies are required, by law, to provide adequate training to their employees so that they are able to identify and report adverse events. While many companies will have a programme in place to facilitate this training, it may not always be possible to make this immediately available to every new starter. Equally, the existing programme may be labour intensive to maintain or deliver. PIPA has therefore developed a cost-effective generic online training course that provides the legally required level of training for all employees working for a Pharmaceutical Company so that they are able to recognise and report adverse events. It has been developed by senior Pharmacovigilance experts working within the Pharmaceutical Industry so you can be confident of the quality and relevance of the training. It is up-to-date with current regulations and, once you have passed the test at the end of the training course, a formal certificate is provided to confirm course completion and understanding. As a not-for-profit association, PIPA can only make this course available to members. However non-members may easily access the course by becoming a ‘member for a month’* for a £15.00 supplement (£18.00 for overseas members) This fee provides you with temporary PIPA membership for the month that you are completing the course 24 Click here to go to Contents page NEW MEMBERS Welcome to … Mr William Ray Pharmacovigilance Associate Aptiv Solutions 2014153 Mr Mallesh Anemoni 2014156 Mrs Margaret Walters Director, European Pharmacovigilance and Deputy EU QPPV Merck Sharp & Dohme Ltd. 2014157 Dr Samer Taslaq PV officer Fontus Health Ltd 2014159 Miss Holly Withers Mr Venkatesh Sapavat Miss Kru Doshi Dr Christine Blanc Wilkinson Dr Praful Bhooma OBG Pharmaceuticals LTD 2014214 Central Medical Affairs Executive Norgine Ltd. 2014183 UK Drug Safety & Quality Assurance Assistant Roche Products UK 2014188 Retinal Grader/Screener Oxford University Hospitals NHS 2014189 Pharmacy consultant Unik pharma services ltd. 2014209 Chief Medical Officer PsiOxus Therapeutics Ltd 2014210 Dr John Paul Kinsey Miss Kate Bowerman Mr David Olatunya Drug Safety Scientist Janssen Cilag Ltd. 2014216 Mr David Olatunya Medical information Officer Genzyme Therapeutics 2014217 Medical Affairs Manager Octapharma 2014167 Mr David Olatunya Medical information Officer Genzyme Therapeutics 2014217 Miss Sandra Wallik Mr Sameer Wagle Medicines Information Scientist Merck Serono Limited 2014219 Ms Stephanie Duncan Medical Information Officer Janssen Cilag Ltd. 2014222 Miss Liesbeth Meulenberg Pharmacovigilance/Regulatory Affairs Officer (EU-QPPV) Disphar International B.V. 2014164 Dr Paul Riley 2014170 Dr Alexandr Meszaros Senior PV Manager Acino 2014173 Mr Kurt Strittmatter Associate Director Pharmacovigilance & Risk Management Shire Pharmaceuticals 2014176 Mr Florence Denance Habek Marketing Manager PrimeVigilance 2014177 Mr Daniel Houde Director, R&D Quality Assurance Ariad Pharmaceuticals, Inc. 2014178 Mr Stephen Ormesher Reglatory Associate Ayrton Saunders Limited 2014180 Click here to go to Contents page Medinfo scientist Gsk 2014190 Medical Information Assistant Martindale Pharma 2014190 GlaxoSmithKline UK Ltd. 2014190 Senior Medical Information Officer Teva UK Ltd. 2014193 Senior Medical Affairs Associate United Therapeutics Europe Ltd. 2014194 Mr Deepak Vyas 2014197 Ms Amy Walker Medical Information Officer Boehringer Ingelheim Ltd. 2014203 Miss Lauren Davis Miss Lauren Davis Miss Poonam Dhokia Ms Jenny Lau Miss Sophie Keddie PV Consultant Adamas Consulting 2014224 Miss Ella Jobson Senior Consultant Hobson Prior International Limited 2014229 Mr Om Prakash Maurya asst. manager in swift ltd 2014205 Mrs Sai Maneesha Chundi Senior Associate Global Safety Amgen Ltd 2014208 25 NOTICEBOARD Contribute an Article to PIPELINE Do you enjoy reading the articles and meeting reports in PIPELINE? Then have you ever considered writing one yourself? We are committed to providing you with an excellent range of topical articles from changes in the regulatory environment, promotion of new initiatives to sharing best practices as just a few examples and always welcome any contribution. There is also a reward for your hard work as well as getting to see you words in print. PIPA will give you a voucher for a well-known high street store; the value depends on whether you have submitted an article or meeting review. Also in recognition for not only attending our meetings and forums we can also provide a subsidised or free place if you offer to write-up the session for us. Just tell Sharon (PIPA administrator) when you book. Interested? Then please contact us at [email protected] We look forward to hearing from you. 26 Click here to go to Contents page COMMITTEE MEMBERS 2014 President Vice President Treasurer Baxter Healthcare Tel: 01635 206861 Email: [email protected] Novarits Pharmaceuticals UK Ltd Tel: 01276 698594 Email: [email protected] Eusa Pharma Tel: 07500 849613 Email: [email protected] Vicki Page-Clark (HonMPIPA) PIPA Administrative and Treasurer Support PIPELINE Co-editor Sarah Dunnett (FPIPA) Chair of Training and Development Working Party Sarah Hall (FPIPA) Takeda UK Ltd Tel: 01628 537966 Email: [email protected] PIPA Membership and Events Co-ordinator Christine Needham (FPIPA) PIPA Operations Manager Anne Turnbull Tel: 07531 899537 PO Box 254, Haslemere, Surrey GU27 9AF Email: [email protected] Archimedes Tel: 0118 931 5092 Email: [email protected] or [email protected] Sanjay Motivaras (MPIPA) Sarah Anthony PIPELINE Co-editor Committee Member Committee Member Baxter Tel: Email: [email protected] Cancer Research UK Tel: +44(0)20 3469 6905 Email: [email protected] Amdipharm Mercury Company Ltd Tel: +44 (0) 208 588 9048 Email: [email protected] Tel: 07722 411409 Email: [email protected] Tom Nichols Committee Member Committee Member Committee Member Napp Medical Information Manager Tel: Email: Head of Pharmacovigilance for Boehringer Ingelheim Tel: 01344 741479 Email: [email protected] Associate Safety Risk Lead for Pfizer Limited Tel: 01737 331386 Email: [email protected] Phil Ball Meetings Co-ordinator (PV) Chair of Signal Detection Working Party Tel: 07512 317656 Email: [email protected] or [email protected] Giulia Brichetto Graglia Sharon Braithwaite Emma Boulton (HonFPIPA) Blessing Gombedza Members of the PIPA Training Working Party Sarah Hall Avile Rodriguez Elizabeth Gwynne Manju Bhandari Patricia Walcott Jacquie Warner Shirley-Ann van der Spuy Alpa Shah Jennifer Lean Lisa Hughes Click here to go to Contents page Tel: 07773 818912 Email: [email protected] Betty Mwesigwa Ester Strachan Members of the Benefit Risk Management Working Party Vicki Page-Clark, ProStrakan Group (Chair) John Barber, Dr Reddy’s Laboratories Jeff Guillon, Alliance Pharmaceuticals Katerina Chinenyeze, Eisai Bharat Amlani, Norgine Sally Francis, Panacea Pharma Projects Anne Lloyd, Sanofi-Aventis Emily Barrett, Eli Lilly Claire Longman, Pope Woodhead & Associates 27 www.pipaonline.org










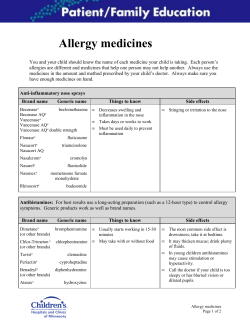

© Copyright 2026