
Nasal vaccination using novel mucosal adjuvants - with main
Linköping University Medical Dissertations No. 1460 Nasal vaccination using novel mucosal adjuvants - with main focus on influenza A virus Tina Falkeborn Department of Clinical and Experimental Medicine Division of Molecular Virology Faculty of Health Sciences, Linköping University, SE-581 85 Linköping, Sweden Linköping 2015 © Tina Falkeborn, 2015 Cover illustration made by Rada Ellegård. The front page illustrates IgG and sIgA antibodies moving towards an influenza virus particle. Published articles have been reprinted with permission from respective copyright holder. ISBN: 978-91-7519-060-0 ISSN 0345-0082 Printed in Sweden by LiU-Tryck, Linköping, Sweden, 2015 To Rikard and the little one Supervisor: Jorma Hinkula, Professor Division of Molecular Virology Department of Clinical and Experimental Medicine Linköping University, Linköping, Sweden Co-supervisors: Marie Larsson, Professor Division of Molecular Virology Department of Clinical and Experimental Medicine Linköping University, Linköping, Sweden Britt Åkerlind, Med Dr, Smittskyddsläkare Department of Clinical and Experimental Medicine Linköping University, Linköping, Sweden Opponent: Anders Wallensten, Docent The Public Health Agency (Folkhälsomyndigheten) Stockholm, Sweden Table of Contents List of papers .................................................................................................................... 1 Publications not included in this thesis ............................................................................................... 1 Abstract ............................................................................................................................ 2 Populärvetenskaplig sammanfattning ............................................................................... 3 Abbreviations ................................................................................................................... 5 Introduction ..................................................................................................................... 7 History ................................................................................................................................................. 7 Classification and structure ................................................................................................................. 7 Influenza A virus .............................................................................................................................. 8 Replication ......................................................................................................................................... 10 Immune response towards influenza A virus ................................................................... 11 Innate immune response .................................................................................................................. 11 Professional antigen presenting cell - the dendritic cell ............................................................... 12 Adaptive immune response .............................................................................................................. 12 Cell-mediated immune response .................................................................................................. 13 Humoral immune response ........................................................................................................... 13 Mucosal immunity ............................................................................................................................. 14 Mucosal IgA/Secretory IgA ............................................................................................................ 15 Immune escape mechanisms of the influenza virus ......................................................................... 15 Transmission and symptoms ............................................................................................................. 16 Treatment ...................................................................................................................................... 17 Influenza vaccines ........................................................................................................... 18 Parenteral vaccination....................................................................................................................... 19 Immune response after inactivated influenza vaccination ........................................................... 19 Advantages and disadvantages with inactivated vaccines............................................................ 19 Mucosal vaccination .......................................................................................................................... 20 Immune response stimulated after mucosal vaccination ............................................................. 20 Advantages and disadvantages with live attenuated vaccines administered intra nasally .......... 21 Influenza vaccination of risk groups .................................................................................................. 21 Children ......................................................................................................................................... 21 Elderly ............................................................................................................................................ 22 Correlation of protection .................................................................................................................. 22 DNA-vaccination ............................................................................................................. 24 Adjuvants ....................................................................................................................... 26 Adjuvants used in influenza vaccines ................................................................................................ 26 Aluminum salt, the first adjuvant .................................................................................................. 26 MF59.............................................................................................................................................. 27 Virosomes ...................................................................................................................................... 27 ASO3 .............................................................................................................................................. 27 Very potent but toxic mucosal adjuvants- CT and LT ........................................................................ 28 Adjuvants studied in this thesis ......................................................................................................... 28 Endocine™ ..................................................................................................................................... 28 N3, N3OA and N3OASq.................................................................................................................. 31 Flagellin (FliC) ................................................................................................................................ 31 Severe adverse events observed after influenza vaccination............................................ 32 Guillain-Barré syndrome ................................................................................................................... 32 Bell´s palsy ......................................................................................................................................... 32 Narcolepsy ......................................................................................................................................... 32 Aim of the thesis ............................................................................................................. 34 Methods ......................................................................................................................... 35 Enzyme-linked immunosorbent assay (ELISA)................................................................................... 35 Cell culturing and virus propagation ................................................................................................. 36 Hemagglutination assay and Hemagglutination inhibition assay (HAI) ............................................ 36 Tissue culture infectious dose 50 (TCID50) and Neutralization assay/Virus neutralizing assay ......... 36 ELISpot ............................................................................................................................................... 37 Flow cytometry of stimulated DCs .................................................................................................... 37 Results and discussion .................................................................................................... 38 Paper I. Endocine™, N3OA and N3OASq; Three Mucosal Adjuvants That Enhance the Immune Response to Nasal Influenza Vaccination.......................................................................................... 38 Paper II. DNA-Encoded Flagellin Activates Toll-Like Receptor 5 (TLR5), Nod-like Receptor Family CARD Domain-Containing Protein 4 (NLRC4), and Acts as an Epidermal, Systemic, and MucosalAdjuvant ............................................................................................................................................ 38 Paper III. Comparison of the mucosal adjuvant Endocine™ with two well-known adjuvants: cholera toxin and alum ................................................................................................................................... 39 Paper IV. The mucosal adjuvant Endocine™ increases immune responses to influenza antigen in aged mice .......................................................................................................................................... 40 Concluding remarks ........................................................................................................ 42 Acknowledgements ........................................................................................................ 44 References ...................................................................................................................... 46 |1 List of papers This thesis is based on the following publications, which will be referred to in the text by their roman numerals: I. Endocine™, N3OA and N3OASq; Three Mucosal Adjuvants That Enhance the Immune Response to Nasal Influenza Vaccination. Falkeborn T, Bråve A, Larsson M, Åkerlind B, Schröder U, Hinkula J. PLoS One, 2013. 8(8): p. e70527. II. DNA-Encoded Flagellin Activates Toll-Like Receptor 5 (TLR5), NOD-like Receptor Family CARD Domain-Containing Protein 4 (NLRC4), and Acts as an Epidermal, Systemic, and Mucosal-Adjuvant. Nyström S, Bråve A, Falkeborn T, Devito C, Rissiek B, Johansson JX, Schröder U, Uematsu S, Akira S, Hinkula J, Applequist SE. Vaccines 2013, 1(4), 415-443 III. Comparison of the mucosal adjuvant Endocine™ with two well-known adjuvants: cholera toxin and alum. Falkeborn T, Asahara N, Hayashi M, Arai M, Hinkula J and Maltais AK Submitted IV. The mucosal adjuvant Endocine™ increases immune responses to influenza antigen in aged mice. Falkeborn T, Hinkula J, Lindberg A and Maltais AK Manuscript Publications not included in this thesis Real-time PCR detection of human herpesvirus 1-5 in patients lacking clinical signs of a viral CNS infection. Sunden B, Larsson M, Falkeborn T, Paues J, Forsum U, Lindh M, Ydrenius L, Åkerlind B, Serrander L. BMC Infect Dis, 2011. 11: p. 220. Limited effect on NS3-NS4A protein cleavage after alanine substitutions within the immunodominant HLA-A2-restricted epitope of the hepatitis C virus genotype 3a non-structural 3/4A protease. Ahlén G, Chen A, Roe B, Falkeborn T, Frelin L, Hall WW, Sällberg M, Söderholm J. J Gen Virol. 2012 Aug;93(Pt 8):1680-6. A novel class of anti-HIV agents with multiple copies of enfuvirtide enhances inhibition of viral replication and cellular transmission in vitro. Chang CH, Hinkula J, Loo M, Falkeborn T, Li R, Cardillo TM, Rossi EA, Goldenberg DM, Wahren B. PLoS One. 2012;7(7):e41235 2| Abstract Influenza viruses have sporadically caused pandemics during the last century, with the most severe occurring in 1918 when the “Spanish flu”, an A/H1N1 influenza virus, passed around the globe killing about 20-100 million people. Today 250 000-500 000 deaths occur annually due to influenza virus or secondary infection after influenza, e.g. pneumonia. Influenza viruses cause severe infections in susceptible age groups like children and elderly and in individuals with impaired immune response due to other medical conditions. The best way to prevent an influenza epidemic is by vaccination. Since the 1950´s we have vaccines against seasonal flu, but vaccine efficacy is not 100 % and there is a need to develop better and more effective vaccines, especially for the risk groups. Since the virus enters the host through the nasal cavity, nasal vaccination is a good approach. By stimulating a mucosal immune response already in the nasal cavity, the goal with nasal vaccination is to stop the virus before it enters the host. Nasal vaccination also reduces the risk of transmission of blood-borne diseases, and is less painful and easier to administer, compared to injectable vaccines. In order to be able to use less immunogenic antigens, like split and subunit antigens, as nasal vaccine components, an adjuvant is needed to enhance the immune response. At the moment there is no licensed mucosal adjuvant for human use. Several studies are ongoing, but it is a complicated and long way to reach the market. In this thesis nasal vaccination with influenza antigen together with the mucosal adjuvant Endocine™ and other mucosal adjuvants has been evaluated. The Endocine™ adjuvant has been shown to be safe and well tolerated in clinical trials. Depending on the pathogen of interest, different approaches are necessary. For HIV, DNA-vaccination has been evaluated together with a plasmid encoding Salmonella typhimurium flagellin C and the mucosal adjuvant N3. The results found in paper I-IV show that by adding adjuvant to the antigen enhances the protective immune response towards the antigen. Enhanced systemic, mucosal and cell-mediated immunity were observed. Hopefully in the future these adjuvants evaluated in this thesis, will be used in vaccines for humans. |3 Populärvetenskaplig sammanfattning Varje år dör 250 000-500 000 människor runt om i världen av influensa eller av en sekundär infektion efter att ha haft influensa. I Sverige slår influensan till under hösten och når sin kulmen runt sportlovet. De typiska symptomen vid en influensasjukdom är hög feber, ont i kroppen, hosta och halsont. Influensaviruset tillhör familjen Orthomyxoviridae och kan delas in i fem olika stammar; influensa A, influensa B, influensa C, thogoto- och isavirus. De tre influensastammarna som kan orsaka sjukdom hos människor är A, B och C, men Influensa C är ovanligare och orsakar i regel bara vanlig förkylning. Influensa A däremot kan infektera många olika arter och den naturliga bäraren är vattenlevande fåglar. Influensa A viruset kan delas upp i flera olika stammar baserade på ytproteinerna hemagglutinin (HA) och neuraminidas (NA). Det finns idag 18 kända HA och nio kända NA typer. Det bästa sättet att minska spridningen av influensa är genom att vaccinera befolkningen. Världshälsoorganisationen (WHO) rekommenderar bland annat vaccination av riskgrupperna unga barn, äldre och människor med andra underliggande sjukdomar vilka löper en högre risk att drabbas av influensa och dess bieffekter. Redan på 1940-talet kom det första injicerbara vaccinet mot influensa och det bestod av levande försvagade eller avdödade viruspartiklar. På grund av biverkningar utvecklade man istället s.k. split-vaccin på 60-talet. Det är ett vaccin där man har behandlat viruset med en detergent och på så vis sönderdelat viruspartikeln, vilket gör att vaccinet fortfarande innehåller alla virusproteiner. Detta är ett inaktiverat influensavaccin (IIV), men det finns även ett levandeförsvagat influensa vaccin (LAIV) som ges med hjälp av nässpray (nasal vaccination). I Sverige är LAIV endast tillåtet för barn mellan 2-18 år, medan IIV kan ges från 6 månaders ålder och uppåt. Både IIV och LAIV ges som säsongsinfluensavaccin och innehåller tre eller fyra olika stammar av influensa; en A/H1N1, en H3N2 och en eller två B influensor. Tyvärr ger inte dagens influensavaccin ett hundraprocentigt skydd, vilket gör att det finns ett behov av att utveckla nya mer effektiva vacciner och eventuellt nya vaccinationsvägar. För att kunna använda sig av vaccinantigener som är immunologiskt svaga, som split- och DNA-vaccin, för nasal vaccination, behöver man tillsätta ett immunologiskt förstärkande hjälpämne, ett s.k. adjuvant, till vaccinet för att öka responsen av immunförsvaret mot antigenet. Fördelen med att ge ett vaccin nasalt är att det stimulerar ett försvar i form av lokala IgA antikroppar i slemhinnorna som kan hindra viruset från att ta sig in i värdcellerna, d.v.s. förhindra smitta. Detta klarar ej vaccin som ges med nål under huden eller i muskelvävnaden. Utmaningen i att hitta bra adjuvant ligger i att hitta ett ämne som är ofarligt men samtidigt immunstimulerande för den som vaccineras. Denna balans mellan risk/nytta är mycket viktig. I denna avhandling har framförallt det nasala adjuvantet Endocine™ studerats. Det är ett fettbaserat adjuvant och som i kliniska studier har visat sig vara säkert och 4| tolererbart hos människor. Endocine™ ges tillsammans med influensaantigen nasalt i form av näsdroppar. Även två andra nasala adjuvant, N3OA och N3OASq, har utvärderats tillsammans med influensaantigen. Influensavaccin med Endocine™, N3OA och N3OASq har visat sig kunna öka både antikropps- och cellmedierat immunsvar i möss, jämfört med icke adjuvanterat antigen som administrerats nasalt. Endocine™ i kombination med influensavaccin visade sig även kunna stimulera liknande mängd skyddande serumantikroppar som det effektiva, men giftiga, slemhinnestimulerande adjuvantet koleratoxin och högre serum och slemhinne-IgA antikroppar jämfört med det äldsta förekommande adjuvantet, aluminium. Studier i äldre möss visade även att Endocine™-adjuvanterat influensavaccin kan bidra med ökat immunsvar hos äldre. Beroende på vaccinkomponenten och målet med vaccinationen, kan olika adjuvant behövas. För humant immunbristvirus (HIV), är troligen inte ett antikroppssvar tillräckligt utan även ett cellmedierat immunsvar är nödvändigt. Genom att använda sig av olika vektorer, bärarsystem, för DNA-vaccination, kan man lyckas stimulera båda delarna av immunförsvaret. I denna avhandling studerades två adjuvant; DNA-plasmiden som kodar för bakteriell Salmonella typhimurium flagellin C (FliC) samt N3, tillsammans med plasmider kodande för HIV-proteiner. Studien visar att en kombination av dessa två adjuvant och DNA-plasmiden som kodar för HIV-proteinerna stimulerar både delarna av immunförsvaret. I den här avhandlingen har fem nya nasala adjuvant studerats och lovande resultat har visats. Det finns ett behov av att utveckla nya effektivare vaccin, men även att utveckla vaccin för de patogener (sjukdomsframkallande bakterier och virus) som fortfarande inte har ett vaccin. I framtiden kan förhoppningsvis dessa nya nasala adjuvant komma till god nytta i dessa sammanhang. |5 Abbreviations Ad5 Adenovirus 5 vector AE Adverse event APC Antibody presenting cell ASC Antibody secreting cell CT Cholera toxin CTL Cytotoxic T cell DAMP Danger-associated molecular pattern DC Dendritic cell DLN Draining lymph node dsRNA double-stranded RNA EMA/CHMP European Medicines Agency/Committee for Medical Products HA Hemagglutinin HAI Hemagglutinin inhibition HBV Hepatitis B virus HPV Human papilloma virus iDC Immature dendritic cell IFN Interferon Ig Immunoglobulin IIV Inactivated influenza vaccine ILI Influenza like illness i.n. Intra nasal LAIV Live-attenuated influenza vaccine LN Lymph node LP Lamina propria LRT Lower respiratory tract LT Escherichia coli heat label toxin MALT Mucosal-associated lymphoid tissue MHC Major histocompatibility complex NA Neuraminidase NALT Nasopharyngeal-associated lymphoid tissue NOD nucleotide binding oligomerization domain NS non-structural NF-κB Nuclear factor-κB 6| OPD O-phenylenediaminedihydrochloride PAMP Pathogen-associated molecular pattern pDC Plasma dendritic cell pIgR Polymeric immunoglobulin receptor pNPP p-nitrophenyl phosphate RdRP RNA-dependent RNA polymerase RIG-I Retinoic acid-inducible gene I RT Respiratory tract s.c. Subcutan sIgA Secretory IgA ssRNA Single-stranded RNA TFH Follicular helper T TH Helper T Treg Regulatory T TCID50 Tissue culture infectious dose 50 TIV Trivalent inactivated vaccine TLR Toll-like receptor URT Upper respiratory tract vRNA Viral RNA |7 Introduction History As early as 412 BC Hippocrates described the acute respiratory disease influenza [1], but it was not until 1933 the first human influenza virus was isolated [2]. Today we know that influenza virus can be divided into several groups based on the two glycoproteins, hemagglutinin (HA) and neuraminidase (NA) [3]. Influenza virus is constantly present around the world and cause diseases, and occasionally pandemics and epidemics occur. The first well documented influenza virus outbreak was during the 1890´s and it may have been caused by an H3 influenza A virus [3]. In 1918 the extremely virulent H1N1 influenza A virus swept around the world and infected approximately 30 % of the world’s population and caused 20-100 million deaths [1-3]. The pandemic was called the “Spanish flu”. The mortality rate was highest in healthy young adults and the virus showed high replication rate and spread in the lungs. It is believed that the virus enhanced the cytokine production and caused a “cytokine storm” that led to great damage of organs and other tissues [4]. In 1957 the next major outbreak of influenza occurred, The “Asian flu” caused by an H2N2 influenza A virus. The new subtype was a reassortment of human and avian genes and caused 1-2 million deaths globally [3]. The “Hong Kong flu” in 1968 was caused by an H3N2 influenza A virus, and was also a reassortant virus with avian genes. This pandemic was however milder. In 1977 the H1N1 type virus returned and was detected in Siberia, this outbreak was named the “Russian flu” [1]. Since this influenza A virus was similar to the one circulating before 1957, only small and mostly mild outbreaks occurred among the younger age group. Until 1997 only H1, H2 and H3 influenza A viruses were known to infect and cause disease in humans. However, today H5 and H7 viruses have also been found and shown to cause disease in humans. The highly pathogenic influenza H5N1, the “Bird flu”, caused an outbreak in Hong Kong 1997. Six out of 18 infected people died, however no human to human transmission was observed [1,2]. The “Bird flu” returned in 2003 in Asia, with a mortality rate of 80 %. The latest pandemic that occurred, the ”Swine flu”, was again caused by an H1N1 virus in year 2009 and it was antigenically similar to the “Spanish flu” [1]. Although the virus was not as virulent as the one in 1918, most deaths occurred in the young population. Classification and structure Influenza belongs to the family of Orthomyxoviridae and can be divided into five genera; influenza A, influenza B, influenza C, thogoto- and isavirus [3]. The viruses in this family are enveloped, segmented negative-polarized single stranded RNA-viruses. They are classified based on their antigenic structure, genetic, and epidemic differences. All three influenza virus strains can cause disease in humans, but Influenza C is rare and only cause common cold in humans [5]. Influenza A and B cause influenza like illness (ILI) in humans. Influenza B mainly infects humans [2] and consist of two different lineages; 8| Victoria and Yamagata [6]. Influenza A virus are able to infect many different species [7] and this thesis will focus on influenza A. Influenza A virus The natural reservoir for influenza A virus is the aquatic birds [3,8]. However poultry, aquatic birds and porcines can all transfer the virus to humans (Fig 1). Influenza A can further be divided based on the proteins on the surface of the virus particle, i.e. HA and NA. At the moment there are 18 known HA and nine known NA types, where the two latest HA types have only been found in bats [9,10]. Figure 1. Natural hosts of influenza A. Modified from Wahlgren J, 2011 [7] The influenza A virus has a lipid bilayer envelope and the genome is segmented in eight fragments and encodes for 11 proteins (Fig 2) [2,3,11]. These eight fragments contains the genetic information that is necessary for the virus to be able to infect and multiply itself. Nine of the 11 proteins are structural (HA, NA, nucleoprotein (NP), matrix 1 (M1), matrix 2 (M2), polymerase basic 1 (PB1), polymerase basicF2 (PB1-F2), polymerase basic 2 (PB2) and polymerase acidic (PA)) and two are non-structural (NS1 and NS2) (Table 1). The PB2, PB1 and PA are encoded by the three largest RNA segments and form a heterotrimeric RNA-dependent RNA polymerase (RdRP). PB1-F2 protein is also encoded by the PB1 segment and have an apoptotic function [12]. M1 is a matrix protein and M2 form an ion-channel. NP is the nucleoprotein that binds to the viral RNA (vRNA) fragments and encapsulates them. The most important structural and virulent parts of the virus, are the HA and NA proteins. |9 Figure 2. Schematic picture of the influenza A virus [13]. Printed with permission from Nature publishing Group, April 2015 The HA protein consist of a trimer of three identical units that interact together and form a binding pocket. Each unit contains two subunits: HA1 and HA2. These take form after cleavage of the precursor protein HA0. The HA1 unit contains the viral binding site and binds to sialic acid on the surface of the host cell. In humans the receptor is α-Gal(2,6) and is expressed on respiratory epithelial cells, while avian flu utilize the receptor α-Gal(2,3). The HA2 subunit contains the fusion domain, which is used when the virus envelope and the endosome fuses. The NA protein consists of four identical subunits and is an enzyme. The NA enzyme cleaves the sialic acid to provide virus release in active form and this seems necessary for the virus to bud off from the infected host cell. NA also protects the infected cell from becoming infected by daughter viruses. The ratio between HA and NA is approximately 5:1. [2,3,11,14] Table 1. Influenza A virus proteins and their functions. Gene segment 1 2 3 4 Protein Polymerase basic 2 (PB2) Polymerase basic 1 (PB1) Polymerase basic 1-F2 (PB1-F2) Polymerase acidic (PA) Hemagglutinin (HA) 5 6 Nucleoprotein (NP) Neuraminidase (NA) 7 Matrix 1 (M1) Matrix 2 (M2) Nonstructural 1 (NS1) 8 Nonstructural 2 (NS2) Function/s RNA transcription and replication Induce apoptosis RNA transcription and replication Major surface glycoprotein, used for cellreceptor binding and fusion Nucleocapsid protein, associates with RNA Major surface glycoprotein, used for virus release Matrix protein, protects RNP-core Ion channel Interact with host mRNA, inhibits interferon production Nuclear export protein 10 | Replication When the virus binds to the receptor on the host cell, the virus particle is endocytosed. Acidification of the endosome with the help of the viral M2 channel induces conformational changes in HA1 and HA2, which move them away from each other. The HA2 fusion peptide acts as an anchor in the endosome and the virus envelope and the endosome membrane are moved towards each other. Fusion occurs and the eight RNA fragments are released into the cytosol. The viral replication occurs in the nucleus where the virus steals the 5’ cap from the cellular host mRNA with the help of NS1 and PB2. The 5’ cap works as a primer. Since influenza A is a negative strand RNA virus, it must carry RdRp or PA to be able to produce mRNA. RdRp binds to the 5’ cap and starts the transcription of the vRNA. The mRNA is translated into proteins in the cytosol and the HA, NA and M2 proteins continue to the endoplasmic reticulum and golgi apparatus to become glycosylated before they are attached to the cell surface. The NS-proteins are transported back into the nucleus and support the production of new vRNA copies. The NP, PA, PB1 and PB2 proteins and the eight RNA fragments form a ribonucleoprotein (RNP)-core. The M1-protein builds a shell around the RNP-core which moves towards the HA, NA and M2 proteins attached to the cell membrane. A new immature virus particle is produced and NA facilitates the virus budding off from the host cell surface by cleavage of sialic acid. [11,15] | 11 Immune response towards influenza A virus The first defense against a pathogen is the innate immune system. This is a nonspecific immune reaction and the innate immune cells react rapidly with a cascade of actions with the aim of destroying the pathogen. This is not a long lasting protection, but it facilitates activation of the adaptive immune response. The adaptive immune response consists of specialized cells that recognize pathogens that have infected the host earlier and this response is required to recover from the infection [16]. The hallmark of vaccination should be to induce a long-lasting adaptive immune response with memory T and B cells, preferably in mucosal and systemic immune tissues. The entry site for influenza viruses is through the respiratory tract (RT) and it can be divided into two parts; the upper (URT) and the lower respiratory tract (LRT). The URT consists of the mouth, nose and pharynx, while the LRT consists of the bronchi, lungs and trachea [17]. The airway lymphoid tissue is called Waldeyer´s ring, and is located in the border between URT and LRT [17]. The RT is covered with mucosa that acts as a physical and biological barrier against invading pathogens [18,19]. The mucosa consists of a layer of epithelial cells with tight junctions, a thick layer of mucins, and antimicrobial peptides (defensins). The innate and adaptive immune cells are located underneath this layer, ready to fight the pathogen. Innate immune response The epithelial cells of the URT are the primary targets for influenza virus. If the pathogen succeeds to pass these cells the next step to overcome is the components of the immune system. The innate immune system initiates an antiviral first line of defense against the detected virus. The antiviral response is initiated through the recognition of vRNA or proteins, i.e. pathogen-associated molecular patterns (PAMPs) [19]. They are only present on, or induced by, pathogens and not by the body´s own cells. PAMPs are recognized by pattern recognition receptors (PRRs), which are located on many cells including macrophages, neutrophils and dendritic cells (DCs) [19]. There are three different PRRs; tolllike receptors (TLRs), retinoic acid-inducible gene I (RIG-I), and the nucleotide binding oligomerization domain (NOD-like) receptor family (NLR) that recognize influenza proteins [20-22]. The PRRs initiate an antiviral signal cascade after recognition of PAMPs. Several TLRs recognize different parts of the influenza virus. TLR7 and TLR8 recognize single stranded RNA (ssRNA) [19,23,24] while TLR3 recognizes double-stranded RNA (dsRNA) [25-27]. TLR3 is expressed by DCs in the RT and is probably activated through phagocytosis of dying influenza-infected cells [28]. TLR2 and TLR4 located on the host cell surface recognize the viral envelope proteins [21,29]. Cytoplasmic ssRNA is detected by RIG-I [30] and cytoplasmic dsRNA by NLRs [31]. NLRs induce caspase-1 activation, while activation of TLRs and RIG-I receptors lead to the activation of nuclear factor-κB (NF-κB) and IRF3, which give rise to the production 12 | and secretion of type 1 interferons (IFNs), pro-inflammatory cytokines, and chemokines [16]. Macrophage and DC production of type 1 IFNs, IFN-α and IFN-β further stimulate the production of more IFNs by neighboring cells, which will limit the viral replication [32]. The pro-inflammatory cytokines and eicosanoids cause fever and anorexia while chemokines attract other immune cells to the infected area [16]. Neutrophils, monocytes, and natural killer cells are recruited to the area, and clear and kill infected cells. Macrophages phagocytose apoptotic cells, while DCs present viral antigens to naïve T cells, which activate the adaptive immune system [19,33]. Cytokines produced during the innate immune response, e.g. IL-1, IL-6 and IL-18, also promote the activation of the adaptive immune system. Professional antigen presenting cell - the dendritic cell Dendritic cells (DCs) connect the innate with the adaptive immune response. Under the epithelial cells, the DCs lie ready to detect viral intruders. The DCs are probably the most efficient antigen presenting cells (APCs) [19,34-37] and they are also important for the continued immune response. Immature DCs (iDCs) can engulf pathogens by receptor-mediated endocytosis or take up pathogens by micropinocytosis and then degrade them intracellularly [19]. The iDCs then migrate to the lymph nodes (LNs) and on their way they become mature and start to express co-stimulatory molecules on their surface, that are needed for T cell activation. Different DC subsets reach the LNs with viral antigens and here, the DCs present the antigen to naïve CD4+ and CD8+ T cells, and activate them with the help of a second signal from the co-stimulatory molecules (CD80 and CD86) [19]. Studies on skin DCs have demonstrated that within 24 hours, DCs will take up the antigen, process it for presentation and then migrate to the draining lymph node (DLN) to present it to naïve T and B cells [38-40]. However studies in lungs show that after 2-4 days the DLN contain the maximum number of CD103+ DCs, while after 57 days the CD11bhi DCs peak [34,41]. These two subsets of DCs are preferentially localized in the airway and submucosa of the RT. Plasmacytoid DCs (pDCs) are major producers of type 1 IFNs, but can also transport antigen to the DLN but are weak activators of naïve T cells [17]. After the DC-T cell interaction the T cells undergo three different steps; activation, proliferation and differentiation to become effector cells [17]. They are then able to migrate to the site of infection and continue the immune response. Adaptive immune response The adaptive immune responses consist of two branches; the cell-mediated and the humoral immune responses. The cell-mediated immune response provides help to activate B cells of the humoral immune response and kill infected cells. The humoral immune response leads to antibody production through activation of B cells. | 13 Cell-mediated immune response The cell-mediated immune response consists of activated CD4+ and CD8+ T cells that help to activate B cells and to kill infected cells. Both CD4+ and CD8+ T cells can secrete IL-2, IFN-γ and TNF-α. IL-2 is important for further CD4+ and CD8+ T cell proliferation, while IFN-γ and TNF-α have antiviral and inflammatory effects [19,42]. After T cell activation the main role for CD4+ T cells are to activate B cells and support their differentiation [19,43] and this will lead to antibody production. However CD4+ cells are also needed for activation of CD8+ T cells. Activated CD4+ T cells produce IL-2 and express CD40 that will bind to CD40 ligands on the APC, which will help the APC to enhance the surface markers that are needed for CD8+ T cell activation [19]. The CD4+ T cells can differentiate to different types of effector cells; Thelper cells (TH1, TH2, TH17), follicular helper T cells (TFH cells) and regulatory T cells (Treg) [19]. The TH1 cells activate macrophages that will help to kill infected cells. The TFH cells are believed to be the ones that helps to activate B cells and promote antibody production, however the TH2 cells are also of importance [19]. The main role for activated CD8+ T cells, cytotoxic T cells (CTLs), is to promote cell lysis and apoptosis of infected cells or produce pro-inflammatory cytokines at the site of infection [44-47]. The CTLs contain large cytoplasmic granules with serine proteases, granzymes (grz), and pore forming enzyme (perforin) that can be released and induce cell death of infected cells [48]. Perforin seems to be of importance for influenza clearance [44], however mice lacking grz A and B can still clear the viral infection. In grz AB-/- mice cytotoxicity was still observed, and in CTLs grz K can be expressed and is suggested to contribute to cytolysis [49]. Memory T and B cells will be developed days to weeks after infection, and can offer lifelong protection [19,50] by rapidly differentiating to effector cells when needed [42]. A recent study in humans showed that pre-existing CD4+ memory T cells correlated with less severe influenza disease [51]. Humoral immune response The process of producing antibodies specific for antigens that the immune system has been exposed to was proposed by Macfarlane Burnet in the 1950s [19]. B cells are developed from pluripotent hematopoietic stem cells in the bone marrow, where they undergo different steps and negative selection before they are released into the blood stream. They then continue their development in the spleen and undergo negative selection again before they are mature [52]. They are now naïve B cells that will be located in the spleen, lymph nodes and in the bone marrow until they are activated by antigens. When the naïve B cells or memory B cells meet the antigen, they will become plasma cells. It is the plasma cells that will produce antibodies. In the germinal center of the lymph node, follicular B 14 | cells will be activated by the antigen and TH cells. This may also occur in the mucosa of the URT [53]. The B cells will move to the border of the T-B cell zone and receive help from CD4+ T cells [54]. The activated B cells will undergo affinity maturation and class-switch recombination of immunoglobulins (Igs). The B cells will also undergo clonal expansion, which means that they will divide into many identical short-lived plasma cells [19,54]. It will take 10-14 days for the response to peak in the germinal center and the plasma and memory B cells will then leave the germinal center [52]. The memory B cell can then re-enter to the circulation or remain in secondary lymphoid tissue like the spleen or the mucosa epithelium of the tonsils [55,56]. Compared to naïve B cells, the memory B cells will react fast the next time they recognize the antigen [57]. The half-life of serum antibodies is short [58] and a continuous presence of antibody secreting cells (ASCs) is necessary. A study with cytomegalovirus infection showed that the APC are gone from the spleen within two weeks after an infection, but they remain in the bone marrow for more than a year [59] and maybe for life. During the 2009 H1N1 pandemic in the US, about 33 % of the people above 60 years of age had cross-reactive antibodies towards the virus [54]. Antibodies in the RT have better correlation with protection from re-infection as compared to serum antibodies [60]. In the URT the dominating antibody is IgA, while in the LRT IgM develops first and the IgG is then slightly more common [61]. B cells produce antibodies mainly towards HA and NA. These antibodies, when directed against the neutralizing epitopes, will inhibit the virus attachment to host cells and limit the spread of the virus. The production of IgG antibodies towards influenza envelope proteins are correlated with long-lasting protection [62], while secretory IgA (sIgA) produced in the mucosa protect the airways from infection [63,64]. Mucosal immunity In humans the nasal cavity, adenoids and the tonsils, represent one part of the mucosa-associated lymphoid tissue (MALT) and this is where the antigen-specific immune response is initiated in the RT. The adenoids and tonsils are functionally related to nasopharyngeal-associated lymphoid tissue (NALT) in rodents [65] and are the inductive sites for humoral and cellular immune response [66]. In MALT specialized antigen-sampling cells are located, i.e. M cells [19] and they are not covered with glycoproteins and do not secrete mucus or enzymes. Recently it was shown that this type of antigensampling cells, M cells, is also located in the URT [67]. The M cells take up the antigen through endocytosis or phagocytosis and the antigen is transported across the cell to the basal surface where the antigen is taken up by DCs [18]. The DCs process the antigen and present it through major histocompatibility complex (MHC) I or II to naïve T cells in the T cell zones in the LNs, and antigen- | 15 specific T cells are generated. In the B cell zone and germinal center, the antigen-specific TH-cells stimulate IgA class switching and somatic hypermutation of B cells with the help of cytokines that promote IgA production [18]. After maturation, the IgA committed B cells migrate to the effector site, the lamina propria (LP), with the help of the mucosal homing integrin α4β7 and the chemokine receptors CCR9 and CCR10. Final differentiation into plasma cells occurs under the influence of TH2 cytokines (IL-5 and IL-6) in the LP. The B cells synthesize dimeric or polymeric IgA that is transported across the epithelium with the help of polymeric immunoglobulin receptors (pIgRs). A part of the receptor (secretory component) will be attached to the IgA dimer after release and the antibody is then termed secretory IgA (sIgA) [18,19]. Mucosal IgA/Secretory IgA In the URT the mucosal/secretory antibody IgA (sIgA) is the dominating antibody subtype and has been shown to have many important properties [68]. Mucosal sIgA is produced by plasma cells in the mucosa wall, while serum IgA is produced in the bone marrow [19]. In the mucosa, sIgA is produced as a polymer, usually a dimer antibody linked by a J chain [19,65]. The polymeric form is able to protect against influenza virus infection [64]. Secretory antibodies are able to neutralize pathogens at the mucosal site before they enter the host cell [19,64]. sIgA can also neutralize virus inside cells, without destroying the host cell [19,69,70]. IgA deficient mice have been shown to be highly susceptible to influenza infection [71,72]. Another important property of sIgA is that it has been shown to have crossprotection properties against both homotypic and heterotypic strains [63,64,72,73]. Immune escape mechanisms of the influenza virus Influenza virus does not have proofreading of the genomic RNA during viral replication which results in the development of viral quasi species. During replication small mutations occur in the genome, but the viruses are still related to each other. Small amino acid changes/mutations occur constantantly in the HA and NA proteins and this is called antigenic drift [3], which results in loss of antibody recognition by the host. The virus may also undergo antigenic shift, major antigenic changes, that can happen if a host is infected with two different influenza strains at the same time [3]. Swine have the receptor for both human and avian influenza, α-Gal(2,6) and α-Gal(2,3), which makes it possible for reassortment, i.e. switching of gene segments between two different viral strains [3,33,74]. This can result in pandemic outbreaks, since the population probably doesn´t have antibodies against the new virus. Recently the quail was also proposed to be able to serve as a mixing vessel for human and avian influenzas [75]. Some of the influenza virus proteins exhibit immune inhibition properties. The multifunctional protein NS1 is very important for the virus and is involved in different steps of the viral life cycle. NS1 is also 16 | able to modulate the innate immune response by inhibiting the RIG-I receptor [30] and other proteins in the RIG-I signal pathway [76-78] to limit the cytokine production. However various influenza strains have different abilities to affect the IFN system and thereby they differ in virulence [3]. Some variants of the PB2 and PB-F2 proteins act downstream of RIG-I, and they may limit the IFN-β production [79,80]. Since PB2, PB1 and PA perform cap-snatching [81-83], this reduces the host gene expression and thereby also limit IFN-β production. The PA-X protein is a rather newly discovered protein and seems to be able to suppress cellular gene expression [84] and thereby control the kinetics of inflammatory response, apoptosis and T cell-signaling. Both NP and M2 are able to bind to human heat shock protein 40, which reduces the IRF3 and IFN-β production [85,86]. Some influenza virus strains are more pathogenic than others and the above described escape mechanisms play an important role in their pathogenesis. Transmission and symptoms Influenza is spread from human to human via aerosol/droplets and direct contact [87,88]. Every droplet contains around 100 000- 1 000 000 viruses. In dense populations, closed or badly ventilated areas, the risk of virus transmission increases. In the northern hemisphere, due to dry air, the autumn and winter is the major influenza season, with the peak usually in February or March (Fig 3). The incubation time is 1-5 days and the virus secretion is highest between 1-2 days after symptoms. Influenza causes acute disease and infects mainly the URT (nose, throat and bronchi). The main symptoms are high fever, headache, muscle pain, cough, nausea and inflammation in the airways. For people with other medical conditions like chronic heart-and lung failure, immunocompromised or people of high age, there is a risk of a more severe disease. Young children have a higher risk of getting otitis and pseudo-croup. It is estimated that 3-5 million people worldwide are infected by influenza each year and 250 000-500 000 people die of influenza or by secondary infection after influenza illness [88]. Figure 3. Influenza cases during the last four influenza seasons in Sweden. Printed with permission from The Public Health Agency (Folkhälsomyndigheten), April 2015. | 17 Treatment There are two drugs available for influenza treatment; M2 and NA inhibitors. The M2 inhibitors; Amantadine and Rimantadine, prevent the fusion between the endosome and virus particle and the assembly of new particles [3,89]. The NA inhibitors, Zanamivir and Oseltamivir, inhibit the release of new virus from the host cell. In addition, Ribavirin, a drug not specific for influenza, can also be used to treat the infection by inhibiting the RNA-polymerase so no viral replication can occur [90]. Influenza vaccines are also available on the market and are usually distributed from September or October in Sweden. 18 | Influenza vaccines It was during the 1940s in the US the first influenza vaccine was developed [91] and in 1945 the vaccine was licensed for civilian use [92]. The vaccine contained whole inactivated virus. Due to reactogenicity and side-effects towards the vaccine, especially in children and infants, split vaccine was developed during the 1960s [93]. In a split vaccine the whole virus is treated with a detergent to deconstruct the virus particle into viral subunits [94]. In 1970, quantification of the vaccine was possible and the vaccine was standardized to contain 15 µg HA/strain per dose [92]. Since the development of inactivated influenza vaccines (IIV) several billion people have been vaccinated worldwide. The influenza vaccines available today are usually trivalent inactivated vaccines (TIV), which means that three different viral strains are added to the vaccine; one A/H1N1, one A/H3N2 and one influenza B strain. However, since the season 2013/2014 quadrivalent vaccines are available that contain both B linages (Victoria and Yamagata) [92]. These two B lineages have been co-circulating since 2004. There is also one live-attenuated influenza vaccine (LAIV) available called Flumist®. The vaccines have to be reformulated each year depending on which strains that are circulating. It is the World health organization’s (WHO´s) Global Influenza Surveillance and Response System (GISRS) that recommends which strains that should be included in the vaccine. The traditional way of growing virus is through embryonated hen´s egg where the allantoic fluid is harvested and processed. Cell-based vaccines are available on the market, but they are not as common as egg-grown. WHO recommend vaccination of people with high risk of getting severe complications after an influenza infection, people in contact with these people, elderly, and people with chronically medical conditions, pregnant women, health care workers and children age 6-24 months [88]. Studies show that vaccination of adults results in reduced absence from work and school and less use of antibiotics, while vaccination of children results in decreased need of medical care [92]. Today, whole virus, split, subunit, recombinant, virosome, and whole live attenuated vaccines are available on the market (Fig 4). However they are distributed in different ways. There are two different vaccination strategies available for influenza vaccines; parenteral and mucosal delivery. Figure 4. Different influenza vaccines; whole live, split, subunit, recombinant, live attenuated virus and virosome vaccine. Modified from www.ifpma.org | 19 Parenteral vaccination The traditional way of vaccination is parenteral injection, either intramuscular or intradermal. Today split and subunit vaccines are most common, the whole inactivated vaccines are being replaced [95]. In subunit vaccines the HA and NA proteins have been further purified to remove the other viral proteins [94]. Parenteral vaccines can also be given together with an adjuvant. The virosome-based vaccine Inflexal®V contains the influenza virus outer membrane proteins HA and NA that are purified and incorporated into a lipid membrane to form a virus without any genetical material [96]. The only age restrictions for parenterally given vaccines is that infants less than 6 months are not allowed to be vaccinated [95]. Naïve children, children that do not previously have antibodies towards influenza virus, are given two doses, with one month apart [95]. There is also an MF59 adjuvanted subunit vaccine (Fluad®) licensed for elderly ≥ 65 years of age. HA is the main immunogen in IIV and it is used to standardize the vaccine dose. The amount of NA is not quantified and can vary between manufacturers. The antibodies generated after parenteral vaccination are mainly targeting the HA protein. The protective efficacy of vaccination in different age groups is varying, depending on studies and the best efficacy are seen in adults, while elderly and young children respond with less antibody titers [92,97]. Adverse events (AEs) seen after vaccination are usually pain at injection site, swelling, malaise, arm tenderness, fever and redness [98,99]. Immune response after inactivated influenza vaccination Inactivated influenza vaccines stimulate primarily a systemic immune response directed towards HA. The main antibody after parenteral vaccination is serum IgG and the serum antibody response peak 24 weeks after vaccination and decline by 50% over 6-12 months [92]. Antibodies against NA and cellmediated immunity is also of importance, but it is usually the HA specific antibody response that is measured. Sasaki et al showed that, 7-12 days after immunizations, influenza specific IgG and IgA ASCs are detected in the blood of children and adults [100] and that IgG ASCs were more common than IgA ASCs. The number of circulating influenza specific memory B-cells were significantly increased by TIV. Krosor Krnic et al showed that the number of CTLs increases 7 days after vaccination and peaks around day 28 and then returns to baseline within a year [101], however the number of CTLs was rather low. About 7 days after a booster vaccination, plasma cells are circulating in the peripheral blood again and memory B cells peak 1-2 weeks later [102]. Advantages and disadvantages with inactivated vaccines The advantages with IIV are that they have been used during a long time, with billions of doses distributed worldwide. The AEs are mild and the vaccine can be used from 6 months of age and in adults, the IIV have shown good efficacy [97]. The disadvantages with IIVs are that they have poor 20 | cross-reactivity to other influenza strains and are poor stimulators of mucosal IgA responses [98,103106] and cell-mediated immune responses [106-108]. Injectable vaccines stimulate high titers of hemagglutination inhibiting antibodies (HAI), but studies have also shown that NA-directed antibodies [109-111], mucosal IgA [63,64] and cell-mediated immune response [112] are of importance. Mucosal vaccination To use the nasal route for vaccination is rather new, even if scientists have studied it for a long time. Since the natural entry site for influenza is through the nasal cavity, nasal vaccination stimulates the first line of defense at the site activated by the virus infection. In 2003 the mucosal vaccine FluMist® was licensed in US in the form of a nasal spray, and it is a LAIV vaccine. LAIV has a backbone from the cold adapted virus strain A/Ann Arbor/6/60 (H2N2) or B/Ann Arbor/1/66, where the current HA and NA are incorporated by genetic reassortment into the backbone strain [3,92]. LAIV is produced as a quadrivalent vaccine and contains both B linages. In the US, FluMist® is restricted to people between 2-49 years of age and is not licensed for elderly, infants or people with underlying medical conditions [97]. In 2011, FluMist® was approved to be used in children 2-18 years of age in Europe and since 2012 it is provided in Sweden [113]. Immune response stimulated after mucosal vaccination The attenuated strain is able to replicate in the mucosal tissue in the nasal cavity and throat and thereby stimulate an immune response similar to a natural influenza infection and provide protection. Both a humoral and cellular immune responses are stimulated. At day 7-12, a peak in influenza specific IgA and IgG ASCs is seen as for TIV, however less memory B cells are produced after LAIV immunization [100]. Since LAIV is a whole live attenuated vaccine it does not generate as high systemic immune response as IIVs, but stimulate a local response with mucosal IgA and a cell-mediated immune response [98,114]. The advantage of getting a cell-mediated immune response is that the immune cells often target the conserved internal proteins, which may give a broader response [115]. LAIV have been shown to be more effective in children compared to TIV [97,103,114,116]. Children respond best to the vaccine and 85 % of the young children develop a mucosal response [92]. The efficacy in 15-72 months old children have shown to be as high as 91-95 %, while it decreases with age and in elderly people it was shown to be only 42 % [92]. In a systematic review of influenza efficacy DiazGranados et al showed that LAIV gave 80 % protection while TIV only gave 48 % protection in children [117]. In a Cochrane report from 2012, they found that the efficacy of LAIV in children >2 years of age was better than TIV [116]. However the relative effectiveness was similar between LAIV and TIV (33 and 36 %). A reason why LAIV is effective in children may be that they are more likely to induce a mucosal immune response with the support of a cell-mediated response rather than mainly a systemic | 21 response, and children also have less preexisting immunity towards many influenza strains than adults [118]. Few studies have been conducted in children less than 2 years of age. Advantages and disadvantages with live attenuated vaccines administered intra nasally The advantage of LAIV is that it stimulates a first line of defense at the entry site for influenza virus and is thereby able to stop the virus from entering the host. Since the vaccine is administered by nasal spray, no needles are used and the risk of blood borne transmission of other diseases is eliminated. LAIV is also easier to administer and may be more accepted by vaccinees. There is, however a relatively small risk of reversion to virulence and reactogenicity by the vaccine strain, but some severe AEs have been reported. Belshe et al reported a higher risk of wheezing in infants 6 to 11 months of age after LAIV [103]. The risk of bronchospasm is also a reason why LAIV is not allowed in infants [118]. Common AEs seen after vaccination are bad taste, runny nose, nasal congestion, headache, sore throat, malaise, decreased appetite and cough [98,119]. Influenza vaccination of risk groups Children Influenza naïve children and infants have shown to be difficult to vaccinate as two doses of vaccine are needed to create a robust immune response to the vaccine [113]. A study by Bodewes et al in the Netherlands showed that before 6 months of age, the influenza antibodies in the children are maternal, but after this time the children start to produce their own antibodies [120]. During these first months of life, the children start to develop the nasopharyngeal tonsils (adenoids) that are a part of the lymphoid tissue of Waldeyer´s ring. The adenoids and tonsils have an important role in host defense against pathogens invading the URT [121]. At age one the children have detectable antibodies against influenza and this increases gradually to the age of 6, when all children have antibody response towards at least one influenza A strain [120]. The highest influenza virus infection rates were seen at age 2-3 years. The very young children were also the group that had the highest risk of being hospitalized with more severe LRT symptoms from the influenza infection [122]. Six European countries, Austria, Estonia, Finland, Latvia, Slovakia, and Slovenia, have included the influenza vaccine into the pediatric vaccination schedule [123]. However, there are doubts about the ability of the influenza vaccine to induce a protective immune response in children in their first years of life and this is one reason why more countries have not included the vaccine into their pediatric programs. By administering two larger TIV vaccine doses, 0.5 ml instead of 0.25 ml, Skowronski et al proved that it was possible to significantly increase the antibody response in 6-11 month old infants and thereby increase the chance of a protective immune response [124]. A virosome adjuvanted vaccine study showed similar results in children less than 35 months of age [125]. Studies with the MF59 adjuvanted TIV showed higher efficacy than TIV in children 6-35 months old [126], however there are some 22 | concerns about the safety and tolerability in children [118]. By using a higher dose or adjuvant it may be possible to induce a strong and protective immune response also in young children. Elderly The aging population worldwide is increasing and in 2050 it is estimated that 21 % of the population will be over 60 years of age [127]. The elderly population has the highest risk of dying of influenza or secondary infections after influenza. In the US, 90 % of the influenza associated deaths occurred in people over 65 years of age [128]. During the season 2011/2012, 1000 deaths were reported in Sweden and 75 % of these deaths occurred in people above 85 years of age [129]. Elderly people and young children’s hospitalization rates are similar during influenza illness, however the mortality rate is almost 35 times higher in the people over 70 years of age [130]. Vaccination of school children in Japan reduced the mortality in elderly ≥ 65 years of age [131]. The main goal with vaccination of elderly is to reduce the risk of severe influenza related complications. Vaccination of elderly may not reduce the risk of influenza illness, but may reduce severity and prevent deaths [88]. However the aged population is a difficult group to vaccinate and to achieve a protective immune response in and this is due to immunosenescence, aging of the immune system. It is more difficult to initiate an immune response towards novel antigens [132], but also against previously known pathogens with elevated age [133]. In elderly the bone marrow site for B cells is decreasing which leads to decreased naïve B-cell production [133,134] and to a decreased B cell repertoire [135,136]. The size and number of germinal centers is also decreasing with age [133,137] and this will lead to a loss of Ig diversity, B-cell class switching [138] and affinity [134]. The vaccine efficacy in elderly after parenteral vaccination is only 17-53 % [139] while in the younger population vaccine efficacy can be 70-90 % [97]. A quantitative review done by Goodwin et al showed that younger had about 2-4 times higher antibody response compared to elderly towards the seasonal influenza vaccine [139]. However by adding adjuvant to the vaccine for elderly, an increased immune response can be detected compared to TIV alone [140-143]. Correlation of protection The HA of influenza is the major target for neutralizing antibodies. Vaccine efficacy and preexisting antibodies are measured using the hemagglutination inhibition test (HAI/HI) and this is the golden standard method to evaluate the immunogenicity of an influenza vaccine. A HAI titer of 1:40 is considered protective and this reduces the risk of getting sick in influenza with 50 % [54]. Traditionally it is only antibodies against HA that are measured, but antibodies towards NA are also of importance. People with titers higher than 1:160 against both HA and NA had a very little risk of getting influenza [144]. Antibodies are the best correlation of protection against many infectious diseases [145], this | 23 was already noticed in 1949 by Salk and Suriano [146]. However the HAI test does not evaluate functional antibodies, instead a virus neutralization test (VN/NT) may be more correct. This assay studies the titers of antibodies that actually have the ability to neutralize the virus and stop the viral internalization. An important question is if the serum antibody levels best correlate with protection towards influenza? Since the virus is entering the host through the RT, this is the place where mucosal antibodies first fight the virus. However, in elderly the antibody response is declining and the cellular immune response may be more correct to use for measuring protection in this group [147]. A study by McElhaney et al in 2006 showed that in elderly ex vivo stimulation of PBMCs and measurement of the ratio of IFN-γ:IL2 correlated better with protection against influenza [147]. In children, the LAIV influenza vaccines are more effective. This could be due to that both humoral and cell-mediated immunity is stimulated but with lower titers [107]. Thereby the HAI/HI test may not be the most correct way to measure protection in this age group. 24 | DNA-vaccination In the 1990´s DNA-plasmid immunization was found to be able to induce immune response after injection [148,149]. The first human clinical trials with DNA-plasmid were performed in 1997-98 and the plasmid was shown to be safe and well tolerated [150,151]. In 1997 the plasmid contained the genes for HIV rev and env and the antibody levels increased in the groups that received the highest dose, however no changes in cell-mediated immune response was seen. In the Swedish therapeutic HIV-1 DNA plasmid phase I trial the genes for HIV-1 were nef, tat and rev and predominately good safety and cell-mediated immunity was monitored, but no or only very modest clinical effect was shown. It might have been a poor uptake and delivery of the plasmids, which generated modest or low responses [152]. DNA-vaccines have now been tested towards many different pathogens and at the moment almost 800 clinical trials with DNA vaccines are registered on clinicaltrials.gov. DNA-vaccines against HIV and different forms of cancer are popular fields. The DNA-vaccines today are able to induce a broader immune response with both humoral as well as cell-mediated immune response [152]. Much work has been focused on the codon-optimization and DNA delivery, complemented with addition of adjuvant and antigen or immunization design. One way to deliver the DNA-plasmid is by using gene-gun. It is a needle free system, were the DNA is coated onto gold particles and then delivered with high pressure to the skin [153]. A Hepatitis B virus (HBV) study in HBV antibody-naïve volunteers showed increased protective antibody and cell-mediated immune response after using gene-gun vaccination [154]. Dermal patches [155] and electrical pulses [156] have also been evaluated as delivery ways. By delivering the DNA-vaccine together with adjuvant or by adding other inserts to the same plasmid encoding for example a cytokine or chemokine, the immune response can be increased [152]. To some degree the DNA-plasmids, originating from bacterial DNA, carry their own PAMP sequences, such as the CpG-repeats that function as PRR immune triggers via TLR9 [20,157]. Different vectors can be used. One important and potent vector is the modified vaccinia virus Ankara and another is the adenovirus 5 (Ad5) vector [158]. The mechanism behind the immune response achieved by DNA-vaccination, is believed to be that the DNA-plasmid is entering the nucleus of host cells (APCs or keratinocytes for example) and the cells start producing pathogen proteins. The DNA-transfected APC will migrate to the DLN where peptides from the proteins will be presented in the context of both MHC I and II molecules to naïve T cells. Since both MHC I and II are activated, humoral as well as cell-mediated immunity are stimulated. In addition, the DNA-transfected cell can secrete antigens, which are subsequently endocytosed by APC and then presented on MHC II molecules. [158] | 25 There are many advantages with DNA-vaccines [158]. The vaccine is safe, there is no risk of reversion to virulence and no detergents are needed. The vaccine is easy to design and the production is fast and can be made in large scale. DNA vaccines seem to be more temperature stable and have long shelf time. So far of the DNA vaccine candidates tested, hardly any AEs have been experienced. However, there are some safety concerns regarding DNA-vaccination, such as integration of DNA into the host DNA, autoimmunity, and antibiotic resistance development. At the moment there is no DNA-vaccine licensed for humans, but in the veterinary field several gene-based vaccines are available [152,159161]. 26 | Adjuvants Adjuvant comes from the Latin word adjuvare, which means “to help” [162]. By using adjuvants it is possible to use less immunogenic antigens as vaccine components, i.e. split antigens, subunit antigens and DNA-plasmids. The purpose of adding adjuvant may be to enhance the immune response, sustain and direct the immunity to the antigen for a specific response, modulate appropriate immune response, reduce the amount of antigen needed, reduce the number of doses or improve the vaccination in children, elderly and immune compromised individuals [162]. Adjuvants can consist of in principal anything that can help to deliver and/or stimulate the immune response. Of course it has to be tolerated and non-toxic for the host. The different properties of the adjuvant depend on which kind of adjuvant and substance it is (Fig 5). Figure 5. Different kinds of adjuvant and properties [162]. Printed with permission from Nature publishing Group, April 2015. The first and still the most commonly used adjuvant, aluminum salt (alum), was discovered by Alexander Glenny and colleagues in the early 1920s and since 1926 it has been used as an adjuvant [163]. There are only five adjuvants approved for human use; aluminum, ASO3, MF59, virosome and ASO4. All except ASO4 are used in influenza vaccines. ASO4 is instead used in HBV and human papilloma virus (HPV) vaccines [164]. All these are used parenterally and at the moment there are no mucosal adjuvants licensed for human use. There are many mucosal adjuvants under development, for example: Endocine™ [165-168], CAF01 [169,170], nanoemulsion W805EC [171,172], GPI-0100 [173], CCS [174,175], cholera toxin (CT) and Escherichia coli heat-label toxin (LT) mutants [176-180]. Adjuvants used in influenza vaccines Aluminum salt, the first adjuvant Since Alexander Glenny and colleagues discovered that alum could enhance the antibody production it has been approved in the US as an adjuvant in many different kinds of vaccines like hepatitis A and B, HPV, Haemophilus influenza and pneumococcal vaccines [163,181]. Alum is not used in seasonal flu vaccines but is used in H5N1 vaccines [182]. Even though alum has been used for almost 90 years, the mechanism and mode of action is still not totally clear. Alum has been shown to absorb antigens to its surface, and thereby stabilizes the vaccines and prevents precipitation. A depot effect has been seen, | 27 which allows the antigen to be slowly released after injection. Studies have also shown that alum induces a strong innate immune response, and might directly bind to DCs [183]. Macrophages are stimulated by alum and release cytokines and chemokines that attract neutrophils, eosinophils, NK cells, monocytes, and DCs. Although, alum has also been shown to have cytotoxic effects, which lead to the release of uric acid that acts as danger associated molecular patterns (DAMPs). Studies have further shown that alum activates the inflammasome and thereby caspase-1 and IL-1β secretion which will induce a TH2, antibody dependent immune response. However all these mechanisms are widely debated [163,181,184,185], and more studies have to be performed. MF59 The adjuvant MF59 was developed during the 1990s and has been used in influenza vaccines since 1997 [186]. MF59 is an oil-in-water emulsion and consists of two non-ionic surfactants, Tween 80 and Span 85, with a squalene core and is about 160 nm in diameter [186,187]. MF59 activates macrophages, monocytes, and granulocytes at the injection site. These cells secrete chemokines, which attracts more immune cells to the injection site, and the monocytes increase their endocytic activity. The adjuvant also increases the uptake of the antigen by differentiating monocytes into DCs, which migrate to the LN where they activate both T and B cells [185]. MF59 is used in the seasonal flu vaccine Fluad® for elderly. Virosomes Virosomes are virus-like particles and have been used since year 2000. They are delivery particles consisting of a phospholipid bilayer where the influenza virus surface proteins are incorporated or integrated into. It is an empty particle with a diameter of 100-200 nm with the ability to stimulate both humoral and cell-mediated immunity [188]. Since the virosome contains HA and NA, natural cellreceptor-binding and viral fusion with the host cells occurs. Since fusion occurs, antigen presentation through MHC I occurs and stimulates CTL response, a MHC II response could also be observed [189]. The mechanism of action of virosomes is suggested to be the direct contact with APCs. Studies reveal that virosomes are able to induce maturation of DCs [188]. Virosomes have also been shown to be safe and highly immunogenic [188]. The injectable virosome based influenza vaccine Inflexal®V is used in Sweden. ASO3 ASO3 is relatively new adjuvant and has been used since 2009 when it was used in Pandemrix. ASO3 is an oil-in-water emulsion containing squalene, α-tocopherol, and polysorbate 80 [190]. The adjuvant has shown to increase the antibody production, stimulate the innate immune system, and enhance 28 | the antigen uptake and presentation in the DLN [190]. More studies are needed regarding the mode of action. Very potent but toxic mucosal adjuvants- CT and LT The cholera toxin (CT) and Escherichia coli heat-label toxin (LT) are two very potent mucosal adjuvants. They share 80 % sequence homology [191] and have shown to induce a strong mucosal immune response when administered intra nasally. Unfortunately they bind to ganglioside GM1 [192,193] and have shown to be the cause of Bell´s palsy. Mutant versions of CT and LT have been made and some show promising results. The CT mutant S61F and E112K [177,194,195] and the LT mutant LTK63 [196198] lack the ADP-ribosyltransferase activity and cAMP formation, so they are considered to be nontoxic. However the LT mutant was recently associated with transient peripheral facial nerve paralysis [199]. Adjuvants studied in this thesis Endocine™ In this thesis the adjuvant Endocine™ has been evaluated as a mucosal adjuvant together with influenza antigens. Endocine™ is a lipid-based dispersion with particles of less than 100 nm (Fig 6). Endocine™ consists of the endogenous lipids mono-olein and oleic acid. The adjuvant has been shown to be safe and well tolerated in both clinical and pre-clinical studies [165-168,200]. Endocine™adjuvanted vaccine induces both serum antibodies and IgA in nasal wash [165,166]. Some TH1 activity has been observed with elevated levels of IFN-y and IL-2 [165]. Further studies to evaluate the balance between TH1/TH2 responses induced by Endocine™ are of interest. Does Endocine™ induce mainly a TH1 or a TH2 response, or is it a balanced TH1/TH2 response? It would also be interesting to evaluate if Endocine™ induces a TH17 response. Figure 6. 2 % Endocine™ consists of lipid particles less than 100 nm in diameter. Printed with permission from Eurocine Vaccines AB, April 2015. Our preliminary data suggest that Endocine™ may stimulate the maturation of DCs and enhance the expression of the surface markers CD86 and MHC II (data not shown). Stability of Endocine™adjuvanted vaccine during a year was also studied. The ampoules contained 100 µg/mL HA from each of the three different strains (H1N1, H3N2 and one influenza B) included and 20 mg/mL Endocine™. The influenza-specific IgG response to the vaccine stored for one year at +5°C was evaluated in mice and not found different from freshly made vaccine (Fig 7). | 29 Figure 7. Storage of Endocine™-adjuvanted vaccine for 12 1000000 Serum IgG (GMT, log 10) months at +5°C does not affect the immunogenicity. 100 µg/mL 100000 HA from each of the three strains (H1N1, H3N2 and influenza B) and 20 µg/mL of Endocine™ were mixed and stored at +5°C for a 10000 year. ELISA IgG end titers in BALB/c mice after immunization with Endocine™-adjuvanted vaccine stored for 0 and 12 months. Data °C at 5 shown represent geometric mean titers with 95 % CI. 12 -m on th s 0m on th s 1000 A clinical phase I/II study in humans with Endocine™-adjuvanted vaccine During my PhD studies I was also part of a human clinical phase I/II study. It was a double blind, multi center, randomized, parallel group study on safety and tolerability of a nasal whole virus influenza vaccine in healthy volunteers and was performed during year 2009-2010. A formal clinical trial report was written but the results have not been published. A total of 229 men in the age 18-50 years were screened at four centers in Sweden. The main exclusion criteria’s were a laboratory-confirmed HAI titer against A/Brisbane/59/2007 (H1N1) of ≥ 30 or hypersensitivity against egg or mercury. 154 of these men were included in the trial and received at least one dose, in total 143 men completed the study. The subjects were divided into 9 vaccination groups (Table 2). Table 2. The nine different study groups in the nasal whole influenza vaccine clinical phase I/II study. Groups Intra nasal vaccine with Endocine™ Intra nasal vaccine, no Endocine™ Intra nasal Endocine™ alone Parenteral vaccine, no Endocine™ Specification of the 9 study groups H1N1, 5 µg HA, 1 % Endocine™ H1N1, 5 µg HA, 2 % Endocine™ H1N1, 15 µg HA, 0.5 % Endocine™ H1N1, 15 µg HA, 1 % Endocine™ H1N1, 15 µg HA, 2 % Endocine™ H1N1, 30 µg HA, 1 % Endocine™ H1N1, 15 µg HA, 0 % Endocine™ H1N1, 0 µg HA, 2 % Endocine™ Fluarix® season 2009/2010 Defined as 5/1 5/2 15/0.5 15/1 15/2 30/1 15/0 0/2 Fluarix® i.m. H1N1: Monovalent inactivated whole virus A/Brisbane/59/2007 (H1N1) Fluarix: Trivalent vaccine A/Brisbane/59/2007 (H1N1), A/Brisbane/10/2007 (H3N2) and B/Brisbane/60/2008 The intra nasal (i.n.) group received 3 doses (150 µL/nostril) with 3 weeks apart, while the parenteral group only received one injection (according to the prescribing information for Fluarix®). The antigen dose in the i.n. vaccination groups ranged between 5-30 µg HA with or without 0.5-2 % Endocine™. Adverse events were seen in 53 subjects and in total 110 AEs were reported (Table 3). Of these 36 were reported as probably and 30 as possibly related to the vaccination. They were mild or of moderate intensity, and no one dropped off because of the AEs. The most common AE was nasopharyngitis, followed by throat irritation and oropharyngeal pain. Nasopharyngitis and 30 | oropharyngeal pain were also reported in the i.m. group. Local tolerability was also examined and 420 % reported pain, redness and swelling, while 20-40 % reported pruritus 15 minutes after vaccination. 98 % of the symptoms were classified as mild. Table 3. Number of adverse events in the different vaccine groups, and corresponding classifications regarding relationship to the vaccine and intensity (severity). AE classifications 5/1 5/2 15/0.5 Vaccine groups* (HA [μg]/EndocineTM [%]) 15/1 15/2 30/1 15/0 0/2 Fluarix® i.m.* Tot. Causal relationship Unlikely Possible Probable Intensity Mild Moderate Severe 3 7 1 10 9 8 6 3 5 5 1 0 6 2 10 4 2 1 4 2 1 0 2 10 6 2 0 110 AE 11 0 0 12 15 0 11 3 0 4 2 0 13 5 0 6 1 0 4 3 0 3 9 0 8 0 0 110 AE No. of subjects 5 10 8 4 6 3 5 7 5 53 * Note that the i.m. vaccine was given once and the intranasal vaccines were given 3 times in each subject. The incidence of AE and the number of subjects reporting them with the two administration forms are therefore a sum of one or three administration occasions, respectively. Serological assays were performed such as HAI and ELISA on serum and nasal washes. All vaccine groups had significantly higher HAI titers compared to the placebo group (0/2). Furthermore, all vaccine groups fulfilled at least one of the European Medicines Agency/Committee for Medical products for human use (EMA/CHMP) criteria [201] after two doses (data not shown). HAI criteria after the last dose is shown in Table 4. Table 4. Fulfillment of European Medicines Agency/Committee for Medical Products for human use (EMA/CHMP) criteria by the intra nasal vaccine groups and comparator groups Intra nasal vaccine groups HAI Criteria* Seroconversion % 47 YES 56 YES 39 44 YES 82 YES 73 YES GMT (fold increase) % 3.1 YES 4.2 YES 3.7 YES 3.3 YES 8,5 YES 6.9 YES Seroprotection % 5/1 47 5/2 44 15/0.5 44 15/1 50 15/2 77 YES 30/1 80 YES Comparator groups 15/0 65 YES 5 YES 53 0/2 0 1 6 Fluarix® 94 YES 31.3 YES 94 YES * All fold increases are calculated against HAI titers at visit 2 (pre-vaccination). Seroconversion: Proportion of subjects achieving a ≥4 fold increase in HAI titer between pre- and postvaccination, if pre-HAI ≥40, or alternatively, a post-HAI ≥40, if pre-HAI <10 (requirement >40% of subjects); GMT (fold increase): Geometric mean of individual pre- to post-vaccination HAI titer fold increases (requirement >2.5 times); Seroprotection: Proportion of subjects achieving HAI titers ≥40 (requirement >70% of subjects); YES: Indicates fulfillment of EMA/CHMP’s HAI criteria. | 31 Regarding the immune response, the 15/2 and 30/1 group had a 4-fold increase in nasal influenzaspecific IgA GMT against A/Brisbane/59/2007 (H1N1). More than 60 % of the subjects in these two groups had a ≥ 4-fold increase. These two groups and the 5/2 and the Fluarix® group induced significantly higher influenza-specific IgG titers than the placebo group (0/2). This study showed that Endocine™ is well tolerated in humans and doesn´t cause any severe AEs and enhanced the immune response. However, after this study, a decision has been made by Eurocine Vaccines AB to switch from whole virus to split virus antigen instead. This decision was partly based on the result from an influenza challenge study in ferrets where split based vaccine performed better than whole virus based vaccine. Split virus antigen is also the dominating form of antigen on the market today. N3, N3OA and N3OASq The N3OA and N3 adjuvants have been studied together with influenza antigen respectively HIV DNAplasmids. Both adjuvants are cationic lipids and N3OA consists of oleylamine, while N3 contains monoolein and oleylamine. They have shown to be able to enhance both humoral as well as cell-mediated immunity [165,202-204]. The mode of action is unknown. The N3OASq has been evaluated together with influenza antigen. The adjuvant is a cationic adjuvant that consist of oleylamine and squalene and it stimulates more of a cell-mediated immune response compared to N3OA. The mode of action is unknown, but data suggests that squalene is shifting the immune response from a mixed response towards cell-mediated immunity. [165] Flagellin (FliC) Flagellin is secreted by many enteric bacteria’s, but the flagellin that was studied in paper II came from Salmonella typhimurium and is known to bind to TLR5 and probably the NLR family receptor [205]. Flagellin, both as protein and expressed with plasmid, has been tested as a mucosal and injectable adjuvant and it has been shown to mainly stimulate cell-mediated immunity, but also with good efficacy a humoral immune response [204,206-208]. The adjuvant has been evaluated together with many different pathogens such as influenza, malaria, HIV and plaque [204,209-211]. 32 | Severe adverse events observed after influenza vaccination When working with vaccines and adjuvant it is important that the components are non-toxic and tolerated. It is also important to consider the closeness to the olfactory nerve when working with nasal vaccination. Sometimes severe AEs have occurred, such as Guillain-Barré syndrome, Bell´s palsy and narcolepsy after vaccination [212-216]. Guillain-Barré syndrome Guillain-Barre syndrome (GBS) is an autoimmune disease that affect the peripheral nervous system and degenerate nerves [217]. In 1976 during the H1N1 swine flu vaccine campaign, the Guillain-Barré syndrome was observed for the first time and the campaign was stopped [212]. However since then there is no evidence of a correlation between GBS and influenza vaccination or other vaccinations [218]. A study by Romio et al concluded that there was no elevated risk of GBS after the latest A(H1N1)pdm09 vaccination [219]. During the season 2012/2013 both IIV and LAIV were evaluated and no increased risk could be observed [220]. Bell´s palsy In year 2000 the first large-scale i.n. influenza vaccination project with inactivated influenza vaccine was introduced in Switzerland. The inactivated virosomal-subunit vaccine contained LT as a mucosal adjuvant. The vaccine was called NasalFlu, but was withdrawn from the market in 2001 due to severe side effects. NasalFlu increased the risk of a facial paralysis, Bell´s palsy after vaccination [213]. LT have shown to bind to ganglioside GM1 [193], which may be the cause of Bell´s palsy. The adjuvant consists of an A and a B subunit and it is believed that it is the A subunit, that contributes to the toxicity of LT [221]. LT is a very potent mucosal adjuvant, and non-toxic mutants has been developed and is under investigation [179,221]. However, the mutant LTK63 recently also showed an association to Bell´s palsy. The conclusion drawn from Lewis et al study is “nasal administration of neuronal-binding LTderived molecules is inadvisable” [199]. Narcolepsy During the late nineteenth century narcolepsy was described for the first time. Narcolepsy is an autoimmune sleeping disorder with daytime sleepiness and cataplex (muscle weakness). Patients with narcolepsy lack the neurotransmitter orexin produced by hypocretin cells. These cells are believed to be destroyed in an autoimmune manner. Narcolepsy have also been associated with human leukocyte antigen (HLA) DQB1*0602 [222]. Before 2010 no associations had been done between influenza vaccination and narcolepsy. However, after the 2009 pandemic when many people were vaccinated against influenza, several cases of narcolepsy was developed in Scandinavian children receiving Pandemrix (ASO3 adjuvanted vaccine). About 67 % of the Swedish children were vaccinated against | 33 pH1N1 with Pandemrix [214]. A study in western Sweden found a 25-fold higher risk of developing narcolepsy after Pandemrix vaccination, and all narcolepsy patients in this study had HLA DQB1*0602 [223]. The report from the Swedish Medical Products Agency (MPA) reported a 6.6 higher risk in children <19 years of age for developing narcolepsy with cataplexy after vaccination [214]. The vaccination coverage of children in Finland was even higher than in Sweden, and they reported a 17fold higher risk of developing narcolepsy post-vaccination [216]. All children with narcolepsy that were HLA typed were positive for HLA DQB1*0602. An increased risk of narcolepsy was also reported in England and Norway after ASO3 adjuvanted pandemic vaccination in young people [224,225]. However in China a 3-fold increased risk of narcolepsy was seen after the 2009 pandemic H1N1 influenza which could not be explained by vaccination, since only 5.6 % were vaccinated [226]. The MF59 adjuvanted and non-adjuvanted vaccine were used in South Korea during the pandemic period and no increased risk of narcolepsy could be detected here [227]. Streptococcal infection have also been associated with the onset of narcolepsy [228] and observed in China, the virus itself can cause narcolepsy [226]. It is hypothesized that a cross-reaction is occurring between H1N1-specific CD4+ T cells and an epitope presented on hypocretin-producing cells [229]. The T cells recognize the epitopes that lead to cytokine and chemokine release that will attract other immune cells that will damage the cells. More studies need to be performed to understand the association between influenza/influenza vaccines and narcolepsy. 34 | Aim of the thesis The general aim during my PhD studies has been to evaluate the potency of the novel mucosal adjuvants Endocine™, N3OA, N3OASq, N3 and pFlic. Paper I. Investigate the humoral and cell-mediated immune response after i.n. immunization with split influenza antigen together with one and each of the three novel mucosal adjuvants, Endocine™, N3OA and N3OASq. Paper II. Study if plasmid-encoded flagellin (pFlic) could act as an adjuvant for non-living/replicating DNA immunizations. Paper III. Compare the mucosal adjuvant Endocine™ with two potent well-known adjuvants. Paper IV. Investigate if Endocine™ could enhance the influenza-specific immune response in elderly mice after i.n. immunization. | 35 Methods During my PhD studies different kinds of immunological assays have been used to detect and analyze the immune response achieved after i.n. vaccination with vaccine and adjuvant. The studies in my papers have been performed mainly in BALB/c mice, which is a good animal model for immunological influenza vaccine related studies [230]. However in paper II, C57BL6/J mice were also used, which is another good model for especially cell-mediated immunological studies [231]. Enzyme-linked immunosorbent assay (ELISA) This assay is a standard method to detect antibodies and was used to determine the prevalence of influenza specific antibodies in sample from vaccinated mice. The main antibodies are IgG and IgA, but subclass IgG has also been evaluated. In mice the subclass antibody response can show if a TH1 or TH2 response is stimulated. Higher IgG1 titers shows a trend towards TH2 [232], while higher IgG2a titers in BALB/c mice [233] and IgG2c in C57BL/6 mice [234,235], indicates a trend towards a TH1 response. For influenza antibody detection, recombinant HA or TIV vaccine was diluted in coating buffer (0.05 M Sodiumcarbonate, pH 9.5) to a concentration of 1-1.5 µg HA/mL and coated onto 96-well plates. In paper II plates were coated with anti-OVA and anti-gp160. The plates were either incubated overnight in room temperature or stored in +4°C. At the day of analysis, the plates were washed three times with washing buffer (0.9 % saline, 0.05 % Tween®-20 in water) and then blocked with 5 % blocking solution (5 % dry milk in PBS) for 1 hour. If subclass IgG was analyzed the blocking step was not needed. Sera was then diluted ten-fold from a starting dilution of 1:100 in ELISA-buffer (2.5 % dry milk and 0.05 % Tween®-20 in PBS)). For IgG measurement, goat-anti-mouse IgG (H+L)-HRP conjugate diluted 1:3 000 was used. IgA and IgG subclasses were measured with a mouse monoclonal antibody isotyping reagent according to the manufacturer’s protocol in conjunction with peroxidase-conjugated anti-Goat IgG diluted 1:20 000. For developing the reaction, O-phenylenediaminedihydrochloride (OPD) was used according to the manufacturer’s protocol. Based on earlier studies, an OD of 0.2 was set as the cut-off value for positive samples. Nasal lavage were analyzed for mucosal IgA against recombinant HA or Inflexal®V. The samples were incubated overnight in +4°C on the plate and then analyzed as above. In paper IV lung homogenates were analyzed for antibodies. The lungs were flushed with PBS and the solution were collected and centrifuged to remove tissue and cell debris. To analyze total IgA, plates were coated with 1 µg/mL of Goat-anti mouse IgA and to analyze influenza specific IgA and IgG, plates were coated with Inflexal®V as above. The samples were then incubated on the plate overnight in +4°C. The same procedure as stated above was performed to detect total IgA, except that Mouse Immunoglobulins AP diluted 1:3 000 and p-nitrophenyl phosphate (pNPP) was used instead. 36 | In paper III Block ACE was used instead of blocking solution and 0.4 % Block Ace was used for dilution of the samples. TMB was used for detection of the antibodies according to manufacturer’s protocol. Cell culturing and virus propagation To be able to perform HAI and NT assay, influenza A/H1N1 virus was grown in MDCK (Madin-Darby canine kidney) cells. The cells were grown in a +37°C humidified incubator with 5 % CO2 in RPMI with 1 % L-glutamin, 4 mM Na-pyruvate, 50 µM 2-mercaptoethanol, 1 % PEST and 8 % BCS. A confluent layer of MDCK cells was incubated with influenza virus and 1 % trypsin. After 1-2 h the virus was discharged and serum free RPMI media with trypsin was added. Four days after incubation the virus was harvested and frozen at -80°C. Hemagglutination assay and Hemagglutination inhibition assay (HAI) To determine the HA titer of the virus stock, the HA assay was used. Depending on strain, A/Brisbane (H1N1) or A/California (H1N1), chicken or guinea pig red blood cells (RBCs) are used. The assay was performed in V-shape plates. The RBCs were washed in PBS and centrifuged on 1800 rpm for 7 minutes without break. The virus was serial diluted in the plate and 50 µl of 0.5-0.75 % of RBCs was added to each well. Mixing was performed carefully by tapping the plate and incubated for 2-4 h in room temperature. The HA titer is the last dilution of the virus that can cause hemagglutination of the RBCs. The HAI was used to determine if samples contains antibodies against the HA on influenza virus surface coated onto the surface of HA-binding erytrochytes and was performed according to standard procedure. The sera was first treated with receptor destroying enzymes (RDE-treated) to inactivate non-specific inhibitors. 25 µl of sera was then serial diluted in V-shape plates and 25 µl of influenza virus with 4 HA units was added. 50 µl RBC was then added as stated above. The HAI titer is the highest dilution of the sera that fully inhibit hemagglutination. Tissue culture infectious dose 50 (TCID50) and Neutralization assay/Virus neutralizing assay First the tissue culture infectious dose 50 (TCID50) of the virus stock was determined. MDCK cells were cultured in 96-well microtiter plates to 80-90 % confluence. The virus was serial diluted and added to PBS washed MDCK cells. The virus was incubated with trypsin for 2 h in +37°C and 5 % CO2. After adsorption the virus was poured away and serum free media with trypsin was added. The plate was cultured for 4 days. At the day of analysis, the plate was centrifuged at 1500 rpm for 10 minutes. The contents were poured away and 100 µl 90 % ice cold acetone was added to each well for 10 minutes. The plates were then dried and an ELISA was performed to detect influenza-positive wells. A human | 37 anti-influenza IgG antibody sample diluted 1:5 000 was used together with a polyclonal Rabbit antihuman IgG/HRP diluted 1:10 000. The plate was developed with OPD as for the ELISA test. In paper I NT assay was used and in paper III VN assay was used. In the NT assay the serum samples were inactivated for 30 minutes in +56°C before use. The sera were serial diluted in two-fold steps and influenza virus of A/Brisbane (H1N1) at 10 TCID50/well was added. Firstly the sera and virus were incubated together for 1 h then the mix was added to the cells and analyzed as above. In paper III, 100 TCID50/mL of A/California (H1N1) was used. After 4 days of incubation the plates were fixed with Midform and then stained with Naphtol Blue Black Solution. VN titers of each serum sample were determined by the maximum dilution ratio that showed higher absorbance than average of positive and negative controls. ELISpot In paper I IFN-γ and IL-2 ELISpot was used to measure the cell-mediated immune response. Splenocytes from vaccinated mice were analyzed for influenza-specific cytokine secretion. 96-well ELISpot plates were activated with ethanol and then coated with the capture antibody for IFN-γ or IL-2 and placed at +4°C overnight. Next day 250 000 splenocytes were added to each well either with or without stimulatory agents. Stimulatory agents were whole A/Brisbane (H1N1) and A/California (H1N1) virus, nucleoprotein peptide mix (SNLNDATYQRTRALV141-155, TYQRTRALV147-155 and TRALVRTGMDPRMCS151165) and the positive control provided in the commercial kit. As negative control, plain RPMI media was used. The plates were incubated in +37°C and 5 % CO2 overnight. Biotinylated detection antibodies were added the next day and followed by streptavidin-ALP. The plates were developed with the substrate solution provided in the kit and the color reaction was stopped with tap water. The plates were air dried and then read in an ELISpot reader. Flow cytometry of stimulated DCs In the end of my PhD studies, we started to evaluate the working mechanism of Endocine™. We cultured human PBMCs and stimulated these with adjuvant and influenza antigen to see if the DCs were affected. The preliminary results are discussed in the text. PBMCs were derived from buffy coats or healthy volunteer by Ficoll-Paque PLUS gradient centrifugation. Monocyte-derived iDCs were propagated as previously described [236]. The iDC cultures were assessed for surface marker expression (CD11c+, CD1a+ and CD14-) with fluorescentlabeled antibodies before use. iDCS were exposed to various concentrations of Endocine™ (ranging from 0.0004-0.04 %) and Vaxigrip® (0.09-9 µg HA/mL) for 1-4 days. After 24 h of stimulation, DCs were stained for anti-CD86, anti-MHC class II and anti-CCR7 and analyzed by flow cytometry (FACS Canto II). 38 | Results and discussion Paper I. Endocine™, N3OA and N3OASq; Three Mucosal Adjuvants That Enhance the Immune Response to Nasal Influenza Vaccination In paper I, the humoral and cell-mediated immune responses after i.n. immunization with three different mucosal adjuvants; Endocine™, N3OA and N3OASq, were evaluated. These three adjuvants are all based on lipids. The mild anionic Endocine™ (formerly known as L3B) consists of mono-olein and oleic acid, while the cationic adjuvants N3OA and N3OASq consist of oleylamine with or without squalene. The study was conducted in BALB/c mice that were i.n. vaccinated with split influenza vaccine (Vaxigrip) with or without the adjuvants. The Endocine™-adjuvanted vaccine was shown to significantly enhance the systemic immune response (IgG, HAI and NT) compared to non-adjuvanted vaccine. Serum IgG subclasses were also evaluated and Endocine™ stimulated a significantly higher production of all four classes. Nasal wash and serum IgA were also significantly enhanced towards the A/Brisbane (H1N1) strain included in the vaccine. Regarding the cell-mediated immune response a significantly enhanced production of IFN-γ and IL-2 were observed against whole A/Brisbane (H1N1) virus and NP from Brisbane, and enhanced titers were also seen against the homologous A/California (H1N1) strain. The N3OA adjuvant responded in a similar manner as Endocine™, but with lower serum IgG response. Instead the cell-mediated immune response was slightly higher. By adding squalene to the N3OA adjuvant, a decrease to lower systemic immune response than the non-adjuvanted vaccine was seen. Instead the squalene increased the cell-mediated immune response compared to the other two adjuvants. After depletion of CD4+ T cells, the squalene group still produced low amount of IFN-γ and IL-2, which shows that CD8+ T cells were present and stimulated. The Endocine™ and N3OA groups lost most of their cytokine production after CD4+ T cell depletion. By adding squalene to N3OA, the production of IFN-γ and IL-2 increased. To conclude: Endocine™ and N3OA significantly induced both humoral and cell-mediated immunity, while N3OASq mainly induced cell-mediated immunity. However all three adjuvants induced mucosal antibody response. Paper II. DNA-Encoded Flagellin Activates Toll-Like Receptor 5 (TLR5), Nod-like Receptor Family CARD Domain-Containing Protein 4 (NLRC4), and Acts as an Epidermal, Systemic, and Mucosal-Adjuvant In paper II a plasmid encoding flagellin (pFliC(-gly)) was studied. Flagellin from Salmonella typhimurium is known to be capable of activating two innate immune receptors, TLR5 and NRLC4 [205]. In this study we investigated if pFliC(-gly) could act as an adjuvant and three different vaccinations routes were | 39 studied; dermal, systemic and mucosal. Additional adjuvants, N3 and L3B (now called Endocine™), were also tested together with pFlic(-gly). First studies with the model antigen ovalbumin were performed and all three routes increased the serum response and MHC I cellular immune response. Using i.n. vaccination with both pFliC(-gly) and N3, both mucosal antibodies and MHC II cellular immune response were induced. Studies with an HIV DNA-plasmid encoding gp160/p24gag were also evaluated using different combinations of pFliC(-gly), N3 and L3B adjuvant. The combination of pFliC(-gly) and N3 showed to be able to stimulate systemic, mucosal and cell-mediated immune response. Priming with N3 and boosted with L3B also showed similar, but slightly lower cell-mediated immune response. Gene-gun and i.n. vaccination worked more efficiently with plasmid-DNA vaccination, but to stimulate mucosal immune response, the i.n. route was superior. The DNA-plasmids have a slightly negative charge and the N3 adjuvants consists of cationic lipids. It might be that N3 encapsulate the plasmid DNA and protect it from being degraded in the mucosal environment and it is thereby able to stimulate APCs. DNA-vaccination has been studied for different pathogenic species during a long time. Since DNAplasmids are non-living vaccines they have several advantages compared to living delivery vectors; production costs are low, the stability is increased, the safety profile is high and they have been shown to induce protection against viral infections [158]. Adenovirus-vector Ad5 has been tried for HIVvaccinations, but led to an increased risk of being infected with the virus instead [237]. This is another preferable reason to continue working with non-living vectors. For HIV, DNA-vaccination has been studied since the beginning of the 1990s, in general, in combination with other vaccine vectors as booster immunizations [238]. To fight HIV, antibody response will not be enough, and cell-mediated immunity is definitively also needed. By obtaining this goal with non-live vaccine candidates, the DNAplasmids by their functional nature have and are showing a great immune priming property and promise. The main finding in paper II is that a combination of the adjuvant pFliC(-gly) and N3 is able to stimulate both a TH1 and a TH2-cell mediated immune response when delivered together with plasmid-DNA encoding gp160 and p24gag. Paper III. Comparison of the mucosal adjuvant Endocine™ with two well-known adjuvants: cholera toxin and alum In paper III we continued to evaluate the immune response achieved by using Endocine™ as an adjuvant together with three different split influenza antigens: A/California/7/2009 (H1N1), A/Victoria/210/2009 (H3N2) and B/Brisbane/60/2008. We compared the immune response induced 40 | by Endocine™ with two other well-known adjuvants, CT and aluminum salt (alum) in mice. Endocine™ was also evaluated with different amounts of antigen given i.n. CT is a very potent mucosal adjuvant, but can cause severe AEs when used in its native form, and is not licensed for nasal use. CT binds to the GM1 receptor like LT, on nerves and may cause Bell´s palsy [192,193,213]. Alum is not used in any seasonal influenza vaccine on the market, but it is used in licensed avian influenza vaccines [182]. Alum has been used for a long time as an adjuvant and billions of doses have been distributed worldwide. Alum is known to mainly stimulate a TH2 response after parenteral vaccination [163]. Both these two adjuvants are strong immune response inducers, but in different ways, and thereby very suitable for comparison of the potency of Endocine™. As expected the CT-adjuvanted mice responded with higher IgA titers, both in nasal wash and in serum compared to Endocine™. Otherwise the serum IgG and VN titers in serum were similar between CTand Endocine™-adjuvanted mice. Already after 1 dose, serum IgG titers could be detected in mice receiving 0.1 as well as 1 µg HA with adjuvant. Regarding the alum adjuvant, the mice responded with high serum IgG and VN titers. Already after one dose, the alum-vaccinated mice responded with serum IgG titers significantly higher than Endocine™adjuvanted mice. However when the California strain was used in the anti-viral functional VN test, similar titers were seen in mice vaccinated with alum and Endocine™. Furthermore, Endocine™ significantly enhanced the mucosal and serum IgA response compared to alum-vaccinated mice. We also found that Endocine™ had a dose-sparing effect in i.n. vaccinated mice. The non-adjuvanted group received 1 µg HA i.n. while Endocine™-adjuvanted groups received 0.01-1 µg HA i.n. The lowest amount (0.01 µg) of antigen given together with Endocine™ showed similar result as the nonadjuvanted group that received 1 µg antigen. While giving 10 times less antigen with Endocine™ overall the influenza-specific titers were higher (serum IgG, IgA, nasal wash IgA and VN) compared to the nonadjuvanted group. The main finding in paper III is that Endocine™-adjuvanted vaccine is able to induce similar serum IgG and VN titers as CT and significantly higher serum and mucosal influenza specific-IgA titers compared to alum. A dose-sparing effect could be observed when Endocine™ was added to the antigen. Paper IV. The mucosal adjuvant Endocine™ increases immune responses to influenza antigen in aged mice The number of elderly in the human population is increasing and in 2050 it is estimated that 21 % of the population in the world will be above 60 years of age [127]. The influenza vaccines available on the market today for elderly are given parenterally and an estimated vaccine efficacy in elderly is 17-53 % | 41 [139]. The elderly have in general 2-4 times lower antibody response compared to adults [139]. This knowledge support the fact that there is a need to develop new and more effective vaccines for the elderly. In paper I and III the immune response after i.n. immunization with and without Endocine™ and influenza antigen in BALB/c mice, 8-10 weeks old was evaluated. In paper IV similar studies were performed but in aging mice, from 15-25 months old. The mice were vaccinated with split influenza A/California/07/2009 (H1N1) antigen i.n. or subcutaneously (s.c.) with or without Endocine™. The 1824 months old mice corresponds immunological to 56-69-years old humans [239]. The results seen in paper IV showed that already at 15-months of age there is a decrease in immune responses compared to 2-months old mice given i.n. vaccination with Endocine™. At 20-months of age, the mice are still able to reach HI titers above 40, but compared to young mice, the antibody levels are decreasing sooner. However compared to s.c. immunization in 20-month old mice, Endocine™adjuvanted vaccine stimulates significantly higher HI, serum IgG and influenza specific IgA titers in lung homogenates. When the mice reach 25-months of age, they start to die due to old age and the antibody levels in these mice are low or absent. Both the humoral and cell-mediated immune responses are affected by age. Elderly people becoming ill of influenza have a switch in their cytokine production, which results in lower levels of IFN-γ and higher IL-10 levels and thereby less CTL activity [147]. In addition, there is an impairment in the CD8+ T cell expansion in influenza infected elderly and due to this impaired CTL activity the elderly get prolonged duration of infection and shedding [240,241]. In infected mice, the elderly had a significantly higher expansion of Tregs compared to young mice and Tregs suppress CD8+ T cells [242]. Lanzer et al showed that there was a delay of CD4+ T cells in the lungs of mice, however the cytokine profile was not changed and the cells were not as impaired as the CD8+ T cells [243]. However, the elderly mice produce less IFN-y, IL-2, IL-6 and IL-10 but more IL-4. Studies in elderly also show that they have a reduced expansion of antigenic B cells and germinal center expansion [137]. Elderly also have a reduced ability to respond to new antigens and this may be due to reduced B cell clonal diversity and defect B cell class switching and this lead to decreased antibody production [135,136,138,244]. The main finding in paper IV was that in 20-month old mice receiving Endocine™-adjuvanted vaccine significantly enhanced serum IgG, HI and mucosal IgA response compared to mice vaccinated parenterally with the same antigen. This suggests that an Endocine™ formulated vaccine can be more efficient than non-adjuvanted parenteral vaccines in elderly. 42 | Concluding remarks At the moment there is no mucosal adjuvant on the market for human use and by using adjuvant in vaccines an enhancement of the desired immune response can be achieved. By using adjuvants less immunogenic antigens like split antigen and DNA-plasmids can be used as vaccine components when administered together with adjuvant. During my PhD studies I have been evaluating different novel adjuvants mainly for influenza split antigen and mucosal use. One study, Paper II, was done using HIV DNA-plasmids and ovalbumin as model antigens. The first paper investigating the properties of the predecessor adjuvants of Endocine™ was published by the inventor Ulf Schröder already in 1999 [245]. However there is a long way from idea to product. Except for studies with influenza antigen, Endocine™ has also been evaluated with diphtheria, HIV and tuberculosis (BCG antigens) [245-247]. These studies have shown that Endocine™ is capable of enhancing the immune response after i.n. vaccination. The N3 adjuvant was first published and evaluated in 2006 together with HIV DNA [202]. It was an i.n. vaccination study and the mice were boosted with a peptide and Endocine™ adjuvant mix. The study showed that primary N3 vaccination with a booster of Endocine™ induced broadly neutralizing antibodies towards HIV-1 in serum and mucosa. An immunization strategy termed heterologous DNA-prime and peptide booster vaccination was used. The results from paper I, III and IV, show that Endocine™ has the property to enhance the immune response towards the influenza vaccine antigen. Humoral binding and neutralizing responses as well as cell-mediated immunity were obtained after i.n. vaccination with adjuvant, a procedure that furthermore stimulate the production of mucosal IgA. When Endocine™ was compared to CT and alum, Endocine™ was shown to share a similar magnitude of immune activating capacity as these two wellknown adjuvants. In the last paper Endocine™ was shown to enhance the immune response in aged mice compared to s.c. immunized mice. This shows that the mucosal adjuvant Endocine™ has a potential to be used in young as well as elderly. Further clinical studies are needed to show efficacy in humans. Previous studies have also shown that mice vaccinated with influenza antigen and Endocine™ have less influenza RNA in the lungs after i.n. virus challenge [166]. A challenge study in Endocine™ vaccinated ferrets showed that the animals were completely protected against influenza and high levels of HAI and VN titers were seen [167]. In paper II using DNA-vaccination, a combination of TLR5-agonist of S. typhimurium flagellin C and the N3-adjuvant showed promising results. Also here humoral as well as cell-mediated immunity, and mucosal immune response was achieved. Depending on the vaccine component and the goal with | 43 vaccination, different adjuvants will be needed. For HIV, antibody response may not be enough, and a cell-mediated immune response will most likely be necessary, and by using different vectors it is possible to stimulate both branches of the immune system. In this thesis five novel mucosal adjuvants have been investigated and promising results have been obtained. Hopefully in the future these adjuvants will be available on the market for human use. There is a need to develop more effective vaccines, but also to develop vaccines for pathogens that do not yet have a vaccine. These novel adjuvants can hopefully be helpful in this process. 44 | Acknowledgements I have really enjoyed my time as a PhD student at floor 13, it is a very a friendly and encouraging environment for doing science. I wish you all the best! I would like to send a special thanks to my supervisor Jorma Hinkula who remembered me two years after our first meeting and took me in as a PhD student. Even if you are very busy you always have the time for questions and discussions, and it feels like no question is too stupid to be asked. Thanks for your guidance during these years! Marie Larsson, my co-supervisor, who loves to discuss science and have a good eye for scientific writing. Thanks for all the help and collaboration! Britt Åkerlind, my second co-supervisor and former boss on Clinical Microbiology. You have a burning interest in the virology field and is a source of encouragement. Even though I have been the only PhD student in my group, I have never felt lonely. I have always had the support from Lennart’s and Marie’s group members and I am thankful for that. Johan, my statistic advisor, thanks for all help and good luck in finding a house. Sumit, who knows a lot and is good source to get help from. Marie H, you are always so helpful and happy. Sonja, my office mate, thanks for all nice chats. Bea, nice to have someone to talk renovation with, good look with your bathroom! Eli, thanks for the help at lab and I love your Italian pastry! Sofia and Mohammad thanks for the help in lab and nice coffee room chats. A special thanks to Rada, what would I have done without your illustrator knowledge? Thanks for all the help with the pictures and for being such a good friend. I hope we will have many more bridge evenings. Thanks also to my lovely friends and colleagues/former colleagues on floor 13, Amanda, Camilla, Emmy, Josefine and Johanna for all enjoyable fikas and social activities. I hope that we one day can have “Kräftskiva” again with all children as well. =) Pia, Lotta and Carolin, the ones to ask when you don´t know where to order stuff from or need lab advice. We have had many nice lunch and fika-chats, thanks. Thanks to Eurocine Vaccines AB and a special thanks to Anki, for the support with projects and scientific writing. I would also like to thank the rest of the people at floor 13 for all lovely discussions and fika-times. I wish you all the best of luck with your own projects and future work. | 45 I would also like to thank my former colleagues Maria, Marie and Susanne, “spelgänget”, for wonderful after works and discussion. It is not so much playing games anymore, it’s more of wine drinking, chatting and popcorn eating! To my family, Disa and Hans, I would like to thank them for their support during all these years. Sofi and Jesper, good luck with your house and thanks for all light advices. Björn, I love your homemade bread and I´m looking forward to get the chili plants. Thanks for our nice family-gatherings. A special thanks to my lovely husband Rikard, who has always supported me and believed in me. We have discovered the wonderful underwater world together and this has giving me energy to continue focus on the research. There is still more to discover, but first we have to think of the little one growing inside me, that will change our lives. I´m really looking forward to what the future has to offer us! I love you! 46 | References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. Monto AS, Webster RG (2013) Influenza pandemics: History and lessons learned. Textbook of influenza (2nd Edition): John Wiley & Sons. pp. 20-33. Strauss JH, Strauss EG Viruses and Human disease. Elsevier Country, 2008; pp. 162-175. Lamb RA (2008) Influenza. In: Mahy BWJ, Regenmortel MHVv, editors. Encyclopedia of Virology. Oxford: Academic Press. pp. 95-104. Kobasa D, Takada A, Shinya K, Hatta M, Halfmann P, et al. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 2004, 431: 703-707. Hayden FG, de Jong MD (2013) Human influenza: Pathogenesis, clinical features, and management. Textbook of Influenza (2nd Edition): John Wiley & Sons. pp. 373-391. Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, et al. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175: 59-68. Wahlgren J Influenza A viruses: an ecology review. Infect Ecol Epidemiol 2011, 1. Wallensten A, Munster VJ, Karlsson M, Lundkvist A, Brytting M, et al. High prevalence of influenza A virus in ducks caught during spring migration through Sweden. Vaccine 2006, 24: 6734-6735. Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, et al. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci U S A 2012, 109: 4269-4274. Tong S, Zhu X, Li Y, Shi M, Zhang J, et al. New world bats harbor diverse influenza A viruses. PLoS Pathog 2013, 9: e1003657. Das K, Aramini JM, Ma LC, Krug RM, Arnold E Structures of influenza A proteins and insights into antiviral drug targets. Nat Struct Mol Biol 2010, 17: 530-538. Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 2001, 7: 1306-1312. Horimoto T, Kawaoka Y Influenza: lessons from past pandemics, warnings from current incidents. Nat Rev Micro 2005, 3: 591-600. Klenk HD, Garten W, Matrosovich M (2014) Pathogenesis. Textbook of influenza (2nd Edition): John Wiley & Sons. pp. 157-171. Wagner EK, Hewlett MJ Basic Virology. Blackwell Publishing Country, 2004; pp. Iwasaki A, Pillai PS Innate immunity to influenza virus infection. Nat Rev Immunol 2014, 14: 315-328. Braciale TJ, Sun J, Kim TS Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol 2012, 12: 295-305. Lamichhane A, Azegamia T, Kiyonoa H The mucosal immune system for vaccine development. Vaccine 2014, 32: 6711-6723. Murphy K, Travers P, Walport M, Janeway C Janeway's immunobiology. Garland Science New York Country, 2012; pp. Blasius AL, Beutler B Intracellular toll-like receptors. Immunity 2010, 32: 305-315. Takeuchi O, Akira S Innate immunity to virus infection. Immunol Rev 2009, 227: 75-86. Pang IK, Iwasaki A Inflammasomes as mediators of immunity against influenza virus. Trends Immunol 2011, 32: 34-41. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 2004, 101: 5598-5603. Iwasaki A, Peiris M (2013) Innate Immunity. Textbook of Influenza (2nd Edition): John Wiley & Sons. pp. 269-281. Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, et al. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 2005, 280: 5571-5580. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413: 732-738. | 47 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, et al. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol 2007, 178: 3368-3372. Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433: 887-892. Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol 2007, 178: 2448-2457. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science 2006, 314: 997-1001. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 2006, 281: 36560-36568. Chelbi-Alix MK, Wietzerbin J Interferon, a growing cytokine family: 50 years of interferon research. Biochimie 2007, 89: 713-718. Van de Sandt CE, Kreujtz JHCM, Rimmelzwaan GF Evasion of Influenza A viruses from innate and adaptive immune responses. Viruses 2012: 1438-1476. Kim TS, Braciale TJ Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One 2009, 4: e4204. Lawrence CW, Braciale TJ Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J Immunol 2004, 173: 1209-1218. Stumbles PA, Upham JW, Holt PG Airway dendritic cells: co-ordinators of immunological homeostasis and immunity in the respiratory tract. Apmis 2003, 111: 741-755. Steinman RM The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 1991, 9: 271-296. Macatonia SE, Edwards AJ, Knight SC Dendritic cells and the initiation of contact sensitivity to fluorescein isothiocyanate. Immunology 1986, 59: 509-514. Hill S, Edwards AJ, Kimber I, Knight SC Systemic migration of dendritic cells during contact sensitization. Immunology 1990, 71: 277-281. Cumberbatch M, Illingworth I, Kimber I Antigen-bearing dendritic cells in the draining lymph nodes of contact sensitized mice: cluster formation with lymphocytes. Immunology 1991, 74: 139-145. Moltedo B, Li W, Yount JS, Moran TM Unique type I interferon responses determine the functional fate of migratory lung dendritic cells during influenza virus infection. PLoS Pathog 2011, 7: e1002345. Turner SJ, Doherty PC, Kelso A (2013) Cell-mediated Immunity. Textbook of Influenza (2nd Edition): John Wiley & Sons. pp. 298-309. Topham DJ, Doherty PC Clearance of an influenza A virus by CD4+ T cells is inefficient in the absence of B cells. J Virol 1998, 72: 882-885. Topham DJ, Tripp RA, Doherty PC CD8+ T cells clear influenza virus by perforin or Fasdependent processes. J Immunol 1997, 159: 5197-5200. Brincks EL, Katewa A, Kucaba TA, Griffith TS, Legge KL CD8 T cells utilize TRAIL to control influenza virus infection. J Immunol 2008, 181: 4918-4925. Hufford MM, Kim TS, Sun J, Braciale TJ Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J Exp Med 2011, 208: 167-180. La Gruta NL, Kedzierska K, Stambas J, Doherty PC A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 2007, 85: 85-92. Peters PJ, Borst J, Oorschot V, Fukuda M, Krahenbuhl O, et al. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med 1991, 173: 1099-1109. 48 | 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. Jenkins MR, Trapani JA, Doherty PC, Turner SJ Granzyme K expressing cytotoxic T lymphocytes protects against influenza virus in granzyme AB-/- mice. Viral Immunol 2008, 21: 341-346. Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med 2005, 11: 748756. Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 2012, 18: 274-280. Shapiro-Shelef M, Calame K Regulation of plasma-cell development. Nat Rev Immunol 2005, 5: 230-242. Randall TD Bronchus-associated lymphoid tissue (BALT) structure and function. Adv Immunol 2010, 107: 187-241. Baumgarth N, Carroll MC, Gonzalez S (2013) Antbody-mediated Immunity. Textbook of Influenza (2nd Edition): John Wiley & Sons. pp. 283-297. Liu YJ, Barthelemy C, de Bouteiller O, Arpin C, Durand I, et al. Memory B cells from human tonsils colonize mucosal epithelium and directly present antigen to T cells by rapid upregulation of B7-1 and B7-2. Immunity 1995, 2: 239-248. Dunn-Walters DK, Isaacson PG, Spencer J Analysis of mutations in immunoglobulin heavy chain variable region genes of microdissected marginal zone (MGZ) B cells suggests that the MGZ of human spleen is a reservoir of memory B cells. J Exp Med 1995, 182: 559-566. Tangye SG, Avery DT, Deenick EK, Hodgkin PD Intrinsic differences in the proliferation of naive and memory human B cells as a mechanism for enhanced secondary immune responses. J Immunol 2003, 170: 686-694. Fahey JL, Sell S THE IMMUNOGLOBULINS OF MICE. V. THE METABOLIC (CATABOLIC) PROPERTIES OF FIVE IMMUNOGLOBULIN CLASSES. J Exp Med 1965, 122: 41-58. Slifka MK, Matloubian M, Ahmed R Bone marrow is a major site of long-term antibody production after acute viral infection. J Virol 1995, 69: 1895-1902. Couch RB, Kasel JA Immunity to influenza in man. Annu Rev Microbiol 1983, 37: 529-549. Tamura S, Kurata T Defense mechanisms against influenza virus infection in the respiratory tract mucosa. Jpn J Infect Dis 2004, 57: 236-247. Jones PD, Ada GL Persistence of influenza virus-specific antibody-secreting cells and B-cell memory after primary murine influenza virus infection. Cell Immunol 1987, 109: 53-64. Tamura S, Funato H, Hirabayashi Y, Suzuki Y, Nagamine T, et al. Cross-protection against influenza A virus infection by passively transferred respiratory tract IgA antibodies to different hemagglutinin molecules. Eur J Immunol 1991, 21: 1337-1344. Tamura S, Funato H, Hirabayashi Y, Kikuta K, Suzuki Y, et al. Functional role of respiratory tract haemagglutinin-specific IgA antibodies in protection against influenza. Vaccine 1990, 8: 479-485. Brandtzaeg P Potential of nasopharynx-associated lymphoid tissue for vaccine responses in the airways. Am J Respir Crit Care Med 2011, 183: 1595-1604. Zuercher AW, Coffin SE, Thurnheer MC, Fundova P, Cebra JJ Nasal-associated lymphoid tissue is a mucosal inductive site for virus-specific humoral and cellular immune responses. J Immunol 2002, 168: 1796-1803. Kim DY, Sato A, Fukuyama S, Sagara H, Nagatake T, et al. The airway antigen sampling system: respiratory M cells as an alternative gateway for inhaled antigens. J Immunol 2011, 186: 4253-4262. Renegar KB, Small PA, Jr., Boykins LG, Wright PF Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J Immunol 2004, 173: 1978-1986. Ogra P L MJ, Lamm M E, Strober W, Bienenstock J, McGhee J R Mucosal Immunology. Academic Press San Diego, US Country, 1999; pp. 485-490. | 49 70. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. 87. 88. Armstrong SJ, Dimmock NJ Neutralization of influenza virus by low concentrations of hemagglutinin-specific polymeric immunoglobulin A inhibits viral fusion activity, but activation of the ribonucleoprotein is also inhibited. J Virol 1992, 66: 3823-3832. Asanuma H, Zamri NB, Sekine S, Fukuyama Y, Tokuhara D, et al. A novel combined adjuvant for nasal delivery elicits mucosal immunity to influenza in aging. Vaccine 2012, 30: 803-812. Asahi Y, Yoshikawa T, Watanabe I, Iwasaki T, Hasegawa H, et al. Protection against influenza virus infection in polymeric Ig receptor knockout mice immunized intranasally with adjuvantcombined vaccines. J Immunol 2002, 168: 2930-2938. Asahi-Ozaki Y, Yoshikawa T, Iwakura Y, Suzuki Y, Tamura S, et al. Secretory IgA antibodies provide cross-protection against infection with different strains of influenza B virus. J Med Virol 2004, 74: 328-335. Doherty PC, Turner SJ, Webby RG, Thomas PG Influenza and the challenge for immunology. Nat Immunol 2006, 7: 449-455. Thontiravong A, Kitikoon P, Wannaratana S, Tantilertcharoen R, Tuanudom R, et al. Quail as a potential mixing vessel for the generation of new reassortant influenza A viruses. Vet Microbiol 2012, 160: 305-313. Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5: 439-449. Talon J, Horvath CM, Polley R, Basler CF, Muster T, et al. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 2000, 74: 7989-7996. Wang X, Li M, Zheng H, Muster T, Palese P, et al. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol 2000, 74: 1156611573. Varga ZT, Ramos I, Hai R, Schmolke M, Garcia-Sastre A, et al. The influenza virus protein PB1F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog 2011, 7: e1002067. Graef KM, Vreede FT, Lau YF, McCall AW, Carr SM, et al. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol 2010, 84: 8433-8445. Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol 2008, 15: 500506. Sugiyama K, Obayashi E, Kawaguchi A, Suzuki Y, Tame JR, et al. Structural insight into the essential PB1-PB2 subunit contact of the influenza virus RNA polymerase. Embo j 2009, 28: 1803-1811. Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, et al. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458: 914-918. Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337: 199204. Sharma K, Tripathi S, Ranjan P, Kumar P, Garten R, et al. Influenza A virus nucleoprotein exploits Hsp40 to inhibit PKR activation. PLoS One 2011, 6: e20215. Guan Z, Liu D, Mi S, Zhang J, Ye Q, et al. Interaction of Hsp40 with influenza virus M2 protein: implications for PKR signaling pathway. Protein Cell 2010, 1: 944-955. Folkhälsomyndigheten. Folkhälsomyndigheten. (2013) Available from: http://www.folkhalsomyndigheten.se/amnesomraden/smittskydd-ochsjukdomar/smittsamma-sjukdomar/influensa-/. (Accessed on March 18 2015). WHO. Influenza (Seasonal). Available from: http://www.who.int/mediacentre/factsheets/fs211/en/index.html. (Accessed on 18 March 2015). 50 | 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 100. 101. 102. 103. 104. 105. 106. 107. Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH The molecular basis of the specific antiinfluenza action of amantadine. Embo j 1985, 4: 3021-3024. von Itzstein M The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov 2007, 6: 967-974. Stokes J, Chenoweth AD, Waltz AD, Gladen RG, Shaw D RESULTS OF IMMUNIZATION BY MEANS OF ACTIVE VIRUS OF HUMAN INFLUENZA. J Clin Invest 1937, 16: 237-243. Keitel WA, Neuzil KM, Treanor J (2013) Immunogenicity, efficacy of inactivated/live virus seasonal and pandemic vaccines. Textbook of influenza (2nd Edition): John Wiley & Sons. pp. 313-326. Ellebedy AH, Webby RJ Influenza vaccines. Vaccine 2009, 27 Suppl 4: D65-68. WHO. Influenza. (2015) Available from: http://www.who.int/biologicals/vaccines/influenza/en/. (Accessed on. WHO. Seasonal influenza - Vaccine. (2015) Available from: http://www.who.int/ith/vaccines/seasonal_influenza/en/. (Accessed on 26th of March 2015). Mischler R, Metcalfe IC Inflexal V a trivalent virosome subunit influenza vaccine: production. Vaccine 2002, 20 Suppl 5: B17-23. CDC. Prevention and Control of Influenza. Recommendations of the Advisory Committe on Immunization preacties (ACIP). MMWR Morb Mortal Wkly Rep (2008) 57(RR07);160:[Available from: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5707a1.htm. (Accessed on 29 of december 2014). Atmar RL, Keitel WA, Cate TR, Munoz FM, Ruben F, et al. A dose-response evaluation of inactivated influenza vaccine given intranasally and intramuscularly to healthy young adults. Vaccine 2007, 25: 5367-5373. Forrest BD, Steele AD, Hiemstra L, Rappaport R, Ambrose CS, et al. A prospective, randomized, open-label trial comparing the safety and efficacy of trivalent live attenuated and inactivated influenza vaccines in adults 60 years of age and older. Vaccine 2011, 29: 3633-3639. Sasaki S, Jaimes MC, Holmes TH, Dekker CL, Mahmood K, et al. Comparison of the influenza virus-specific effector and memory B-cell responses to immunization of children and adults with live attenuated or inactivated influenza virus vaccines. J Virol 2007, 81: 215-228. Kosor Krnic E, Gagro A, Drazenovic V, Kuzman I, Jeren T, et al. Enumeration of haemagglutinin-specific CD8+ T cells after influenza vaccination using MHC class I peptide tetramers. Scand J Immunol 2008, 67: 86-94. Wrammert J, Smith K, Miller J, Langley WA, Kokko K, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453: 667-671. Belshe RB, Edwards KM, Vesikari T, Black SV, Walker RE, et al. Live attenuated versus inactivated influenza vaccine in infants and young children. N Engl J Med 2007, 356: 685-696. Johnson PR, Feldman S, Thompson JM, Mahoney JD, Wright PF Immunity to influenza A virus infection in young children: a comparison of natural infection, live cold-adapted vaccine, and inactivated vaccine. J Infect Dis 1986, 154: 121-127. Johnson PR, Jr., Feldman S, Thompson JM, Mahoney JD, Wright PF Comparison of long-term systemic and secretory antibody responses in children given live, attenuated, or inactivated influenza A vaccine. J Med Virol 1985, 17: 325-335. Cheng X, Zengel JR, Suguitan AL, Jr., Xu Q, Wang W, et al. Evaluation of the humoral and cellular immune responses elicited by the live attenuated and inactivated influenza vaccines and their roles in heterologous protection in ferrets. J Infect Dis 2013, 208: 594-602. Hoft DF, Babusis E, Worku S, Spencer CT, Lottenbach K, et al. Live and inactivated influenza vaccines induce similar humoral responses, but only live vaccines induce diverse T-cell responses in young children. J Infect Dis 2011, 204: 845-853. | 51 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. 118. 119. 120. 121. 122. 123. 124. 125. 126. Basha S, Hazenfeld S, Brady RC, Subbramanian RA Comparison of antibody and T-cell responses elicited by licensed inactivated- and live-attenuated influenza vaccines against H3N2 hemagglutinin. Hum Immunol 2011, 72: 463-469. Schulman JL, Khakpour M, Kilbourne ED Protective effects of specific immunity to viral neuraminidase on influenza virus infection of mice. J Virol 1968, 2: 778-786. Johansson BE, Bucher DJ, Kilbourne ED Purified influenza virus hemagglutinin and neuraminidase are equivalent in stimulation of antibody response but induce contrasting types of immunity to infection. J Virol 1989, 63: 1239-1246. Marcelin G, Sandbulte MR, Webby RJ Contribution of antibody production against neuraminidase to the protection afforded by influenza vaccines. Rev Med Virol 2012, 22: 267279. Murasko DM, Bernstein ED, Gardner EM, Gross P, Munk G, et al. Role of humoral and cellmediated immunity in protection from influenza disease after immunization of healthy elderly. Exp Gerontol 2002, 37: 427-439. Folkhälsomyndigheten. Principer för influensavaccination och tillgängliga vaccintyper (2015) Available from: http://www.folkhalsomyndigheten.se/amnesomraden/smittskydd-ochsjukdomar/vaccinationer/vacciner-a-o/influensa/principer-for-influensavaccination-ochtillgangliga-vaccintyper/. (Accessed on 28th of March 2015). Carter N, Curran M Live Attenuated Influenza Vaccine (FluMist®; Fluenz™). Drugs 2011, 71: 1591-1622. Thomas PG, Keating R, Hulse-Post DJ, Doherty PC Cell-mediated protection in influenza infection. Emerg Infect Dis 2006, 12: 48-54. Jefferson T, Rivetti A, Di Pietrantonj C, Demicheli V, Ferroni E Vaccines for preventing influenza in healthy children. Cochrane Database Syst Rev 2012, 8: CD004879. DiazGranados CA, Denis M, Plotkin S Seasonal influenza vaccine efficacy and its determinants in children and non-elderly adults: a systematic review with meta-analyses of controlled trials. Vaccine 2012, 31: 49-57. Esposito S, Tagliabue C, Tagliaferri L, Semino M, Longo MR, et al. Preventing influenza in younger children. Clin Microbiol Infect 2012, 18 Suppl 5: 42-49. Block SL, Reisinger KS, Hultquist M, Walker RE Comparative immunogenicities of frozen and refrigerated formulations of live attenuated influenza vaccine in healthy subjects. Antimicrob Agents Chemother 2007, 51: 4001-4008. Bodewes R, de Mutsert G, van der Klis FR, Ventresca M, Wilks S, et al. Prevalence of antibodies against seasonal influenza A and B viruses in children in Netherlands. Clin Vaccine Immunol 2011, 18: 469-476. Ogasawara N, Kojima T, Go M, Takano K, Kamekura R, et al. Epithelial barrier and antigen uptake in lymphoepithelium of human adenoids. Acta Otolaryngol 2011, 131: 116-123. Oshansky CM, Gartland AJ, Wong SS, Jeevan T, Wang D, et al. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am J Respir Crit Care Med 2014, 189: 449-462. Blank PR, Schwenkglenks M, Szucs TD Vaccination coverage rates in eleven European countries during two consecutive influenza seasons. J Infect 2009, 58: 446-458. Skowronski DM, Hottes TS, Chong M, De Serres G, Scheifele DW, et al. Randomized controlled trial of dose response to influenza vaccine in children aged 6 to 23 months. Pediatrics 2011, 128: e276-289. Esposito S, Marchisio P, Ansaldi F, Bianchini S, Pacei M, et al. A randomized clinical trial assessing immunogenicity and safety of a double dose of virosomal-adjuvanted influenza vaccine administered to unprimed children aged 6–35 months. Vaccine 2010, 28: 6137-6144. Vesikari T, Knuf M, Wutzler P, Karvonen A, Kieninger-Baum D, et al. Oil-in-water emulsion adjuvant with influenza vaccine in young children. N Engl J Med 2011, 365: 1406-1416. 52 | 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 139. 140. 141. 142. 143. 144. 145. 146. 147. United Nations, ESA. World population ageing 1950-2050. (2002) Available from: http://www.un.org/esa/population/publications/worldageing19502050/. (Accessed on 1st of July 2014). Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003, 289: 179-186. Smittskyddsintitutet (2013) Influenza in Sweden, Season 2011-2012 Smittskyddsinstitutet: Smittskyddsinstitutet. 2013-101-4 2013-101-4. 42 p. Couch RB, Kasel JA, Glezen WP, Cate TR, Six HR, et al. Influenza: its control in persons and populations. J Infect Dis 1986, 153: 431-440. Charu V, Viboud C, Simonsen L, Sturm-Ramirez K, Shinjoh M, et al. Influenza-related mortality trends in Japanese and American seniors: evidence for the indirect mortality benefits of vaccinating schoolchildren. PLoS One 2011, 6: e26282. Hernandez-Vargas EA, Wilk E, Canini L, Toapanta FR, Binder SC, et al. Effects of aging on influenza virus infection dynamics. J Virol 2014, 88: 4123-4131. Siegrist C-A, Aspinall R B-cell responses to vaccination at the extremes of age. Nat Rev Immunol 2009, 9: 185-194. Aw D, Silva AB, Palmer DB Immunosenescence: emerging challenges for an ageing population. Immunology 2007, 120: 435-446. Colonna-Romano G, Aquino A, Bulati M, Di Lorenzo G, Listi F, et al. Memory B cell subpopulations in the aged. Rejuvenation Res 2006, 9: 149-152. Gibson KL, Wu YC, Barnett Y, Duggan O, Vaughan R, et al. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 2009, 8: 18-25. Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL CD4 T cell memory derived from young naive cells functions well into old age, but memory generated from aged naive cells functions poorly. Proc Natl Acad Sci U S A 2003, 100: 15053-15058. Frasca D, Riley RL, Blomberg BB Humoral immune response and B-cell functions including immunoglobulin class switch are downregulated in aged mice and humans. Seminars in Immunology 2005, 17: 378-384. Goodwin K, Viboud C, Simonsen L Antibody response to influenza vaccination in the elderly: a quantitative review. Vaccine 2006, 24: 1159-1169. Minutello M, Senatore F, Cecchinelli G, Bianchi M, Andreani T, et al. Safety and immunogenicity of an inactivated subunit influenza virus vaccine combined with MF59 adjuvant emulsion in elderly subjects, immunized for three consecutive influenza seasons. Vaccine 1999, 17: 99-104. Iob A, Brianti G, Zamparo E, Gallo T Evidence of increased clinical protection of an MF59adjuvant influenza vaccine compared to a non-adjuvant vaccine among elderly residents of long-term care facilities in Italy. Epidemiol Infect 2005, 133: 687-693. Mannino S, Villa M, Apolone G, Weiss NS, Groth N, et al. Effectiveness of adjuvanted influenza vaccination in elderly subjects in northern Italy. Am J Epidemiol 2012, 176: 527-533. Puig-Barbera J, Diez-Domingo J, Perez Hoyos S, Belenguer Varea A, Gonzalez Vidal D Effectiveness of the MF59-adjuvanted influenza vaccine in preventing emergency admissions for pneumonia in the elderly over 64 years of age. Vaccine 2004, 23: 283-289. Dowdle WR, Coleman MT, Mostow SR, Kaye HS, Schoenbaum SC Inactivated vaccines. 2. Laboratory indices of protection. Postgraduate Medical Journal 1973, 49: 159-163. Plotkin SA Vaccines: correlates of vaccine-induced immunity. Clin Infect Dis 2008, 47: 401409. Salk JE, Suriano PC Importance of antigenic composition of influenza virus vaccine in protecting against the natural disease; observations during the winter of 1947-1948. Am J Public Health Nations Health 1949, 39: 345-355. McElhaney JE, Xie D, Hager WD, Barry MB, Wang Y, et al. T cell responses are better correlates of vaccine protection in the elderly. J Immunol 2006, 176: 6333-6339. | 53 148. 149. 150. 151. 152. 153. 154. 155. 156. 157. 158. 159. 160. 161. 162. 163. 164. 165. 166. 167. 168. 169. Tang DC, DeVit M, Johnston SA Genetic immunization is a simple method for eliciting an immune response. Nature 1992, 356: 152-154. Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science 1993, 259: 1745-1749. MacGregor RR, Boyer JD, Ugen KE, Lacy KE, Gluckman SJ, et al. First human trial of a DNAbased vaccine for treatment of human immunodeficiency virus type 1 infection: safety and host response. J Infect Dis 1998, 178: 92-100. Calarota S, Bratt G, Nordlund S, Hinkula J, Leandersson AC, et al. Cellular cytotoxic response induced by DNA vaccination in HIV-1-infected patients. Lancet 1998, 351: 1320-1325. Ferraro B, Morrow MP, Hutnick NA, Shin TH, Lucke CE, et al. Clinical applications of DNA vaccines: current progress. Clin Infect Dis 2011, 53: 296-302. Chen D, Maa YF, Haynes JR Needle-free epidermal powder immunization. Expert Rev Vaccines 2002, 1: 265-276. Roy MJ, Wu MS, Barr LJ, Fuller JT, Tussey LG, et al. Induction of antigen-specific CD8+ T cells, T helper cells, and protective levels of antibody in humans by particle-mediated administration of a hepatitis B virus DNA vaccine. Vaccine 2000, 19: 764-778. Lisziewicz J, Calarota SA, Lori F The potential of topical DNA vaccines adjuvanted by cytokines. Expert Opin Biol Ther 2007, 7: 1563-1574. Otten G, Schaefer M, Doe B, Liu H, Srivastava I, et al. Enhancement of DNA vaccine potency in rhesus macaques by electroporation. Vaccine 2004, 22: 2489-2493. Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci U S A 1996, 93: 2879-2883. Kutzler MA, Weiner DB DNA vaccines: ready for prime time? Nat Rev Genet 2008, 9: 776-788. Ng T, Hathaway D, Jennings N, Champ D, Chiang YW, et al. Equine vaccine for West Nile virus. Dev Biol (Basel) 2003, 114: 221-227. Merial. Oncept, Canine Melanoma Vaccine, DNA (2012) Available from: http://www.petcancervaccine.com/pages/default.aspx. (Accessed on. FASS. Improvac. (2015) Available from: http://www.fass.se/LIF/product?userType=1&nplId=20070918000076. (Accessed on. Guy B The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol 2007, 5: 505517. Marrack P, McKee AS, Munks MW Towards an understanding of the adjuvant action of aluminium. Nat Rev Immunol 2009, 9: 287-293. Tritto E, Mosca F, De Gregorio E Mechanism of action of licensed vaccine adjuvants. Vaccine 2009, 27: 3331-3334. Falkeborn T, Brave A, Larsson M, Akerlind B, Schroder U, et al. Endocine, N3OA and N3OASq; Three Mucosal Adjuvants That Enhance the Immune Response to Nasal Influenza Vaccination. PLoS One 2013, 8: e70527. Petersson P, Hedenskog M, Alves D, Brytting M, Schroder U, et al. The Eurocine L3 adjuvants with subunit influenza antigens induce protective immunity in mice after intranasal vaccination. Vaccine 2010, 28: 6491-6497. Maltais AK, Stittelaar KJ, Kroeze EJ, van Amerongen G, Dijkshoorn ML, et al. Intranasally administered Endocine formulated 2009 pandemic influenza H1N1 vaccine induces broad specific antibody responses and confers protection in ferrets. Vaccine 2014, 32: 3307–3315. Brekke K, Lind A, Holm-Hansen C, Haugen IL, Sorensen B, et al. Intranasal Administration of a Therapeutic HIV Vaccine (Vacc-4x) Induces Dose-Dependent Systemic and Mucosal Immune Responses in a Randomized Controlled Trial. PLoS One 2014, 9: e112556. Martel CJ, Agger EM, Poulsen JJ, Hammer Jensen T, Andresen L, et al. CAF01 Potentiates Immune Responses and Efficacy of an Inactivated Influenza Vaccine in Ferrets. PLoS One 2011, 6: e22891. 54 | 170. 171. 172. 173. 174. 175. 176. 177. 178. 179. 180. 181. 182. 183. 184. 185. 186. 187. 188. Christensen D, Foged C, Rosenkrands I, Lundberg CV, Andersen P, et al. CAF01 liposomes as a mucosal vaccine adjuvant: In vitro and in vivo investigations. Int J Pharm 2010, 390: 19-24. Das SC, Hatta M, Wilker PR, Myc A, Hamouda T, et al. Nanoemulsion W805EC improves immune responses upon intranasal delivery of an inactivated pandemic H1N1 influenza vaccine. Vaccine 2012, 30: 6871-6877. Stanberry LR, Simon JK, Johnson C, Robinson PL, Morry J, et al. Safety and immunogenicity of a novel nanoemulsion mucosal adjuvant W805EC combined with approved seasonal influenza antigens. Vaccine 2012, 30: 307-316. Liu H, Patil HP, de Vries-Idema J, Wilschut J, Huckriede A Evaluation of mucosal and systemic immune responses elicited by GPI-0100- adjuvanted influenza vaccine delivered by different immunization strategies. PLoS One 2013, 8: e69649. Even-Or O, Joseph A, Itskovitz-Cooper N, Samira S, Rochlin E, et al. A new intranasal influenza vaccine based on a novel polycationic lipid-ceramide carbamoyl-spermine (CCS). II. Studies in mice and ferrets and mechanism of adjuvanticity. Vaccine 2011, 29: 2474-2486. Even-Or O, Samira S, Rochlin E, Balasingam S, Mann AJ, et al. Immunogenicity, protective efficacy and mechanism of novel CCS adjuvanted influenza vaccine. Vaccine 2010, 28: 65276541. Watanabe I, Hagiwara Y, Kadowaki SE, Yoshikawa T, Komase K, et al. Characterization of protective immune responses induced by nasal influenza vaccine containing mutant cholera toxin as a safe adjuvant (CT112K). Vaccine 2002, 20: 3443-3455. Ohmura M, Yamamoto M, Kiyono H, Fujihashi K, Takeda Y, et al. Highly purified mutant E112K of cholera toxin elicits protective lung mucosal immunity to diphtheria toxin. Vaccine 2001, 20: 756-762. Isaka M, Zhao Y, Nobusawa E, Nakajima S, Nakajima K, et al. Protective effect of nasal immunization of influenza virus hemagglutinin with recombinant cholera toxin B subunit as a mucosal adjuvant in mice. Microbiol Immunol 2008, 52: 55-63. Pine S, Barackman J, Ott G, O'Hagan D Intranasal immunization with influenza vaccine and a detoxified mutant of heat labile enterotoxin from Escherichia coli (LTK63). J Control Release 2002, 85: 263-270. Peppoloni S, Ruggiero P, Contorni M, Morandi M, Pizza M, et al. Mutants of the Escherichia coli heat-labile enterotoxin as safe and strong adjuvants for intranasal delivery of vaccines. Expert Rev Vaccines 2003, 2: 285-293. Kool M, Fierens K, Lambrecht BN Alum adjuvant: some of the tricks of the oldest adjuvant. J Med Microbiol 2012, 61: 927-934. SAGE. WHO Strategic Advisory Group of Experts (SAGE); Use of licensed H5N1 influenza vaccines in the interpandemic period. (2009) Available from: www.who.int/immunization/sage/SAGE_H5N1_26Mayb.pdf. (Accessed on. Flach TL, Ng G, Hari A, Desrosiers MD, Zhang P, et al. Alum interaction with dendritic cell membrane lipids is essential for its adjuvanticity. Nat Med 2011, 17: 479-487. Lambrecht BN, Kool M, Willart MAM, Hammad H Mechanism of action of clinically approved adjuvants. Current Opinion in Immunology 2009, 21: 23-29. Seubert A, Monaci E, Pizza M, O'Hagan DT, Wack A The adjuvants aluminum hydroxide and MF59 induce monocyte and granulocyte chemoattractants and enhance monocyte differentiation toward dendritic cells. J Immunol 2008, 180: 5402-5412. O'Hagan DT, Ott GS, De Gregorio E, Seubert A The mechanism of action of MF59 - an innately attractive adjuvant formulation. Vaccine 2012, 30: 4341-4348. Calabro S, Tritto E, Pezzotti A, Taccone M, Muzzi A, et al. The adjuvant effect of MF59 is due to the oil-in-water emulsion formulation, none of the individual components induce a comparable adjuvant effect. Vaccine 2013, 31: 3363-3369. Huckriede A, Bungener L, Stegmann T, Daemen T, Medema J, et al. The virosome concept for influenza vaccines. Vaccine 2005, 23 Suppl 1: S26-38. | 55 189. 190. 191. 192. 193. 194. 195. 196. 197. 198. 199. 200. 201. 202. 203. 204. 205. Bungener L, Serre K, Bijl L, Leserman L, Wilschut J, et al. Virosome-mediated delivery of protein antigens to dendritic cells. Vaccine 2002, 20: 2287-2295. Garcon N, Vaughn DW, Didierlaurent AM Development and evaluation of AS03, an Adjuvant System containing alpha-tocopherol and squalene in an oil-in-water emulsion. Expert Rev Vaccines 2012, 11: 349-366. Dallas WS, Falkow S Amino acid sequence homology between cholera toxin and Escherichia coli heat-labile toxin. Nature 1980, 288: 499-501. Holmgren J, Lonnroth I, Svennerholm L Tissue receptor for cholera exotoxin: postulated structure from studies with GM1 ganglioside and related glycolipids. Infect Immun 1973, 8: 208-214. Sugii S, Tsuji T Binding specificities of heat-labile enterotoxins isolated from porcine and human enterotoxigenic Escherichia coli for different gangliosides. Can J Microbiol 1989, 35: 670-673. Yamamoto S, Kiyono H, Yamamoto M, Imaoka K, Fujihashi K, et al. A nontoxic mutant of cholera toxin elicits Th2-type responses for enhanced mucosal immunity. Proc Natl Acad Sci U S A 1997, 94: 5267-5272. Yamamoto S, Takeda Y, Yamamoto M, Kurazono H, Imaoka K, et al. Mutants in the ADPribosyltransferase cleft of cholera toxin lack diarrheagenicity but retain adjuvanticity. J Exp Med 1997, 185: 1203-1210. Douce G, Fontana M, Pizza M, Rappuoli R, Dougan G Intranasal immunogenicity and adjuvanticity of site-directed mutant derivatives of cholera toxin. Infect Immun 1997, 65: 2821-2828. Douce G, Turcotte C, Cropley I, Roberts M, Pizza M, et al. Mutants of Escherichia coli heatlabile toxin lacking ADP-ribosyltransferase activity act as nontoxic, mucosal adjuvants. Proc Natl Acad Sci U S A 1995, 92: 1644-1648. Giannelli V, Fontana MR, Giuliani MM, Guangcai D, Rappuoli R, et al. Protease susceptibility and toxicity of heat-labile enterotoxins with a mutation in the active site or in the proteasesensitive loop. Infect Immun 1997, 65: 331-334. Lewis DJ, Huo Z, Barnett S, Kromann I, Giemza R, et al. Transient facial nerve paralysis (Bell's palsy) following intranasal delivery of a genetically detoxified mutant of Escherichia coli heat labile toxin. PLoS One 2009, 4: e6999. Hinkula J, Falkeborn T, Pauksens K, Maltais A-K, Lindberg A, et al. (2012) A nasal influenza vaccine with unique safety profile and robust immunogenic properties. Abstract for the 4th International conference on: Modern Vaccines Adjuvant and Delivery Systems, Copenhagen, Denmark, 4-6 July. EMA/CHMP. The European Agency for the Evaluation of Medicinal Products. Note for guidance on harmonisation of requirements for influenza vaccines. (1997) Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/W C500003945.pdf. (Accessed on 9th of April 2015). Hinkula J, Devito C, Zuber B, Benthin R, Ferreira D, et al. A novel DNA adjuvant, N3, enhances mucosal and systemic immune responses induced by HIV-1 DNA and peptide immunizations. Vaccine 2006, 24: 4494-4497. Hinkula J, Hagbom M, Wahren B, Schroder U Safety and immunogenicity, after nasal application of HIV-1 DNA gagp37 plasmid vaccine in young mice. Vaccine 2008, 26: 51015106. Nyström S BA, Falkeborn T, Devito C, Rissiek B, Johansson DX, Schröder U, Uematsu S, Akira S, Hinkula J, Appelquist SE DNA-Encoded Flagellin Acitvates Toll-Like Receptor 5 (TLR5), Nodlike Receptor Family CARD Domain-Containing Protein 4 (NRLC4), and Acts as an Epidermal, Systemic, and Mucosal-Adjuvant. Vaccines 2013, 1: 415-443. Keestra-Gounder AM, Tsolis RM, Baumler AJ Now you see me, now you don't: the interaction of Salmonella with innate immune receptors. Nat Rev Micro 2015, 13: 206-216. 56 | 206. 207. 208. 209. 210. 211. 212. 213. 214. 215. 216. 217. 218. 219. 220. 221. 222. 223. 224. 225. McSorley SJ, Ehst BD, Yu Y, Gewirtz AT Bacterial flagellin is an effective adjuvant for CD4+ T cells in vivo. J Immunol 2002, 169: 3914-3919. Van Maele L, Fougeron D, Janot L, Didierlaurent A, Cayet D, et al. Airway structural cells regulate TLR5-mediated mucosal adjuvant activity. Mucosal Immunol 2014, 7: 489-500. Lai CH, Tang N, Jan JT, Huang MH, Lu CY, et al. Use of recombinant flagellin in oil-in-water emulsions enhances hemagglutinin-specific mucosal IgA production and IL-17 secreting T cells against H5N1 avian influenza virus infection. Vaccine 2015. Qian F, Guo A, Li M, Liu W, Pan Z, et al. Salmonella flagellin is a potent carrier-adjuvant for peptide conjugate to induce peptide-specific antibody response in mice. Vaccine 2015, 33: 2038-2044. Honko AN, Sriranganathan N, Lees CJ, Mizel SB Flagellin Is an Effective Adjuvant for Immunization against Lethal Respiratory Challenge with Yersinia pestis. Infection and Immunity 2006, 74: 1113-1120. Chaung HC, Cheng LT, Hung LH, Tsai PC, Skountzou I, et al. Salmonella flagellin enhances mucosal immunity of avian influenza vaccine in chickens. Vet Microbiol 2012, 157: 69-77. Langmuir AD, Bregman DJ, Kurland LT, Nathanson N, Victor M An epidemiologic and clinical evaluation of Guillain-Barre syndrome reported in association with the administration of swine influenza vaccines. Am J Epidemiol 1984, 119: 841-879. Mutsch M, Zhou W, Rhodes P, Bopp M, Chen RT, et al. Use of the inactivated intranasal influenza vaccine and the risk of Bell's palsy in Switzerland. N Engl J Med 2004, 350: 896-903. Läkemedelsverket (2011) Occurrence of narcolepsy with cataplexy among children and adolescents in relation to the H1N1 pandemic and Pandemrix vaccinations‐ Results of a case inventory study by the MPA in Sweden during 2009‐2010. 1-20 p. Nohynek H, Jokinen J, Partinen M, Vaarala O, Kirjavainen T, et al. AS03 adjuvanted AH1N1 vaccine associated with an abrupt increase in the incidence of childhood narcolepsy in Finland. PLoS One 2012, 7: e33536. Partinen M, Saarenpaa-Heikkila O, Ilveskoski I, Hublin C, Linna M, et al. Increased incidence and clinical picture of childhood narcolepsy following the 2009 H1N1 pandemic vaccination campaign in Finland. PLoS One 2012, 7: e33723. Parry GJ, Steinberg JS American Academy of Neurology : Guillain-Barre Syndrome : From Diagnosis to Recovery. Demos Medical Publishing New York, NY, USA Country, 2007; pp. Baxter R, Bakshi N, Fireman B, Lewis E, Ray P, et al. Lack of association of Guillain-Barre syndrome with vaccinations. Clin Infect Dis 2013, 57: 197-204. Romio S, Weibel D, Dieleman JP, Olberg HK, de Vries CS, et al. Guillain-Barre syndrome and adjuvanted pandemic influenza A (H1N1) 2009 vaccines: a multinational self-controlled case series in Europe. PLoS One 2014, 9: e82222. Kawai AT, Li L, Kulldorff M, Vellozzi C, Weintraub E, et al. Absence of associations between influenza vaccines and increased risks of seizures, Guillain-Barre syndrome, encephalitis, or anaphylaxis in the 2012-2013 season. Pharmacoepidemiol Drug Saf 2014, 23: 548-553. Pizza M, Giuliani MM, Fontana MR, Monaci E, Douce G, et al. Mucosal vaccines: non toxic derivatives of LT and CT as mucosal adjuvants. Vaccine 2001, 19: 2534-2541. Mignot EJM History of narcolepsy at Stanford University. Immunologic Research 2014, 58: 315-339. Szakacs A, Darin N, Hallbook T Increased childhood incidence of narcolepsy in western Sweden after H1N1 influenza vaccination. Neurology 2013, 80: 1315-1321. Miller E, Andrews N, Stellitano L, Stowe J, Winstone AM, et al. Risk of narcolepsy in children and young people receiving AS03 adjuvanted pandemic A/H1N1 2009 influenza vaccine: retrospective analysis. Bmj 2013, 346: f794. Heier MS, Gautvik KM, Wannag E, Bronder KH, Midtlyng E, et al. Incidence of narcolepsy in Norwegian children and adolescents after vaccination against H1N1 influenza A. Sleep Med 2013, 14: 867-871. | 57 226. 227. 228. 229. 230. 231. 232. 233. 234. 235. 236. 237. 238. 239. 240. 241. 242. 243. 244. 245. 246. 247. Han F, Lin L, Warby SC, Faraco J, Li J, et al. Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in China. Ann Neurol 2011, 70: 410-417. Choe YJ, Bae GR, Lee DH No association between influenza A(H1N1)pdm09 vaccination and narcolepsy in South Korea: An ecological study. Vaccine 2012. Aran A, Lin L, Nevsimalova S, Plazzi G, Hong SC, et al. Elevated anti-streptococcal antibodies in patients with recent narcolepsy onset. Sleep 2009, 32: 979-983. Singh AK, Mahlios J, Mignot E Genetic association, seasonal infections and autoimmune basis of narcolepsy. J Autoimmun 2013, 43: 26-31. Tamura S, Hasegawa H, Kurata T Estimation of the effective doses of nasal-inactivated influenza vaccine in humans from mouse-model experiments. Jpn J Infect Dis 2010, 63: 8-15. JAX. The Jackson laboratory, C57BL/6J. (2014) (Accessed on. Finkelman FD, Katona IM, Urban JF, Jr., Snapper CM, Ohara J, et al. Suppression of in vivo polyclonal IgE responses by monoclonal antibody to the lymphokine B-cell stimulatory factor 1. Proc Natl Acad Sci U S A 1986, 83: 9675-9678. Finkelman FD, Katona IM, Mosmann TR, Coffman RL IFN-gamma regulates the isotypes of Ig secreted during in vivo humoral immune responses. J Immunol 1988, 140: 1022-1027. Martin RM, Lew AM Is IgG2a a good Th1 marker in mice? Immunol Today 1998, 19: 49. Martin RM, Brady JL, Lew AM The need for IgG2c specific antiserum when isotyping antibodies from C57BL/6 and NOD mice. J Immunol Methods 1998, 212: 187-192. Sabado RL, Babcock E, Kavanagh DG, Tjomsland V, Walker BD, et al. Pathways utilized by dendritic cells for binding, uptake, processing and presentation of antigens derived from HIV1. Eur J Immunol 2007, 37: 1752-1763. Duerr A, Huang Y, Buchbinder S, Coombs RW, Sanchez J, et al. Extended Follow-up Confirms Early Vaccine-Enhanced Risk of HIV Acquisition and Demonstrates Waning Effect Over Time Among Participants in a Randomized Trial of Recombinant Adenovirus HIV Vaccine (Step Study). The Journal of Infectious Diseases 2012, 206: 258-266. Sandstrom E, Nilsson C, Hejdeman B, Brave A, Bratt G, et al. Broad immunogenicity of a multigene, multiclade HIV-1 DNA vaccine boosted with heterologous HIV-1 recombinant modified vaccinia virus Ankara. J Infect Dis 2008, 198: 1482-1490. Flurkey K, M. Currer J, Harrison DE (2007) Chapter 20 - Mouse Models in Aging Research. In: James G. Fox MTD, Fred W. Quim, Stephen W. Barthold, Christian E. Newcomer and Abigail L. Smith, editor. The Mouse in Biomedical Research (Second Edition). Burlington: Academic Press. pp. 637-672. Bender BS, Johnson MP, Small PA Influenza in senescent mice: impaired cytotoxic Tlymphocyte activity is correlated with prolonged infection. Immunology 1991, 72: 514-519. Taylor SF, Cottey RJ, Zander DS, Bender BS Influenza infection of beta 2-microglobulindeficient (beta 2m-/-) mice reveals a loss of CD4+ T cell functions with aging. J Immunol 1997, 159: 3453-3459. Williams-Bey Y, Jiang J, Murasko DM Expansion of regulatory T cells in aged mice following influenza infection. Mech Ageing Dev 2011, 132: 163-170. Lanzer KG, Johnson LL, Woodland DL, Blackman MA Impact of ageing on the response and repertoire of influenza virus-specific CD4 T cells. Immun Ageing 2014, 11: 9. Jiang N, He J, Weinstein JA, Penland L, Sasaki S, et al. Lineage structure of the human antibody repertoire in response to influenza vaccination. Sci Transl Med 2013, 5: 171ra119. Schroder U, Svenson SB Nasal and parenteral immunizations with diphtheria toxoid using monoglyceride/fatty acid lipid suspensions as adjuvants. Vaccine 1999, 17: 2096-2103. Devito C, Zuber B, Schroder U, Benthin R, Okuda K, et al. Intranasal HIV-1-gp160-DNA/gp41 peptide prime-boost immunization regimen in mice results in long-term HIV-1 neutralizing humoral mucosal and systemic immunity. J Immunol 2004, 173: 7078-7089. Haile M, Schroder U, Hamasur B, Pawlowski A, Jaxmar T, et al. Immunization with heat-killed Mycobacterium bovis bacille Calmette-Guerin (BCG) in Eurocine L3 adjuvant protects against tuberculosis. Vaccine 2004, 22: 1498-1508. Papers The articles associated with this thesis have been removed for copyright reasons. For more details about these see: http://urn.kb.se/resolve?urn=urn:nbn:se:liu:diva-117981
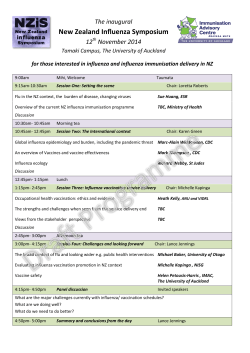









© Copyright 2026