
fulltext - DiVA Portal
Doctoral Thesis in Biochemistry, Stockholm University Sweden Integration and topology of membrane proteins related to diseases Patricia Lara Vasquez Cover illustration “Lipids and membrane proteins in the membrane”. The lipids are represented as dancers and the membrane proteins as colourful boxes. ©Patricia Lara Vasquez, Stockholm University 2015 ISBN 978-91-7649-094-5 Printed in Sweden by Publit Sweden AB, Stockholm 2015 Distributor: Department of Biochemistry and Biophysics, Stockholm University Integration and topology of membrane proteins related to diseases Patricia Lara Vasquez A mis padres, hermanos y sobrinos List of publications I. Wanngren J*, Lara P*, Öjemalm K, Maioli S, Moradi N, Chen L, Tjernberg LO, Lundkvist J, Nilsson I, Karlström H. (2014) Changed membrane integration and catalytic site conformation are two mechanisms behind the increased Ab42/Ab40 ratio by presenilin 1 familial Alzheimer-linked mutations. FEBS Open Bio 4:393-406 II. Ostuni A*, Lara P*, Armentano MF, Miglionico R, Salvia AM, Mönnich M, Carmosino M, Lasorsa FM, Monné M, Nilsson I, Bissaccia F. (2013) The hepatitis B x antigen anti-apoptotic effector URG7 is localized to the endoplasmic reticulum membrane. FEBS lett. 587(18):3058-62 III. Saenz A, Presto J*, Lara P*, Akinyl OL, Nilsson I, Johansson J, Casals C. Folding and intramembraneous BRICHOS binding of the proSP-C transmembrane segment. Submitted IV. Lee H*, Lara P*, Ostuni A, Presto J, Johansson J, Nilsson I, Kim H. (2014) Live-cell topology assessment of URG7, MRP6102 and SP-C using glycosylatable green fluorescent protein in mammalian cells. Biochem. Biophys. Res. Commun. 450(4):1587-92 * These authors contributed equally Additional publications V. Gadalla SE, Öjemalm K, Lara P, Nilsson I, Ericsson C, Zhao J, Nistér M. (2013) EpCAM associates with endoplasmic reticulum aminopeptidase 2 (ERAP2) in breast cancer cells Biochem Biophys Res Commun 439(2): 203-208 VI. Goel, S, Palmkvist M, Moll K, Joannin N, Lara P, Akhouri R, Moradi N, Öjemalm K, Westman M, Angeletti D, Kjellin H, Lehtiö J, Hult AK, Olsson ML, von Heijne G, Nilsson I, and Wahlgren M RIFINs are Adhesins Implicated in Severe Plasmodium falciparum Malaria Nature Medicine just accepted VII. Nilsson I, Lara P, Hessa T, Johnson AE, von Heijne G, Karamyshev AL. The code for directing proteins for translocation across ER membrane: SRP cotranslationally recognizes specific features of a signal sequence. J Mol Biol in press Abstract Membranes are boundaries that separate the cell from the external environment. Membrane proteins can function as e.g. receptors and channels, al- lowing cells to communicate with the exterior and molecules to pass through the membrane. The biogenesis of membrane proteins involves a proteinconducting channel that aids the hydrophobic segments to partition into the membrane and translocate the hydrophilic loops. Membrane proteins need to fold to its native conformation including post-translational modifications and assembly with other proteins and/or cofactors. If this regulated pathway goes wrong the degradation machinery degrades the protein. If the system is failing can result in serious disorders. The main focus in this thesis is membrane proteins associated to diseases. We have studied mutations in the gene of presenilin 1, which is involved in Alzheimer’s disease. We found that some mutations affect the structure and other the function of the PS1. URG7 is an unknown protein associated with liver cancer. We suggest it is localized and targeted to the ER membrane, having an NoutCin topology. SP-C is important for our lungs to function. Mutations can cause the protein to aggregate. We have studied the highly Valrich transmembrane segment (poly-Val) and its analogue (poly-Leu) and show that poly-Leu folds into a more compact conformation than poly-Val. We show that the C-terminal chaperon-like BRICHOS domain interacts with the ER membrane, suggesting an involvement in poly-Val folding. We have also confirmed the topology of URG7, MRP6 and SP-C poly-Val/Leu using gGFP that is fused to the C-terminal of the protein. Populärvetenskaplig sammanfattning En cell är den minsta byggstenen i alla levande organismer. Människor består av flera miljarder celler medan t.ex. en bakterie bara består av en enda cell. Cellen innehåller arvsmassan, DNA:t som kodar för proteiner. Proteiner har livsvikta uppgifter i cellen, man kan säga att proteiner är cellens maskineri. Cellen är omslutet av ett membran som består av lipider (fett) och membranproteiner som skiljer cellens inre från yttre miljön. Membranproteiner har flera olika funktioner, som t.ex. kommunicera med omvärlden och tillåta molekyler passera genom membranet som annars är hel impermeabel. För att membranproteiner ska kunna utföra sina uppgifter behöver de anta sin korrekta struktur. Processen från att proteinet tillverkas till att den är funktionsduglig är väldig komplex och kan ibland gå fel. Cellen kan snabbt reparera alla misstag, däremot om systemet inte fungerar som det ska, kan defekta proteiner uppstå och orsaka sjukdomar. Denna avhandling handlar om membranproteiner och deras roll i sjukdomar. Vi har undersökt mutationer, som är kopplade till den ärftliga, så kallad familjära Alzheimers sjukdom, som har en åldersmässigt tidig debut. Mutationerna i genen som kodar för presenilin 1 (PS1) kan påverka strukturen och även aktiviteten av PS1. URG7 är ett okänt protein, som är kopplad till levercancer. Vi har visat att det är ett membranprotein, som sitter i endoplasmatiska nätverket. Surfactant C (SP-C) är ett protein som kan aggregera, vilket resulterar i lungsjukdomar. Vi har undersökt den transmembrana regionen av SP-C, som visar sig behöva hjälp av en annan domän i proteinet, den så kallade BRICHOS, för att anta sin korrekta struktur. Hur detta sker behövs undersöka ytterligare, men vi visar att BRICHOS domänen kan interagera med membran lipiderna. Vi har även bekräftat topologin av URG7 och SP-C genom att använda oss av en metod som kan glykosylera ett grönt fluorescerande protein i celler. Resumen en español Voy a tratar de explicar con lo que he trabajado durante estos 5 años de doctorado en español, para que mis seres queridos puedan entender aunque sea un poquito mas: Una célula es la parte más pequeña de la que están formados todo ser vivo. El numero de células puede variar, nosotros los humanos tenemos mas de billones células, en cambio la bacteria solo tiene una. Todas las células están rodeadas de una envoltura, una membrana de fosfolípidos y proteínas, que mantiene el medio interno diferenciado del medio externo. Las proteínas de membrana se encargan de comunicarse con el exterior. Además pueden funcionar como canales o transportadoras, permitiendo que sustancias pueden entrar o salir de la célula. Para que las proteínas de membrana puedan funcionar correctamente necesitan adoptar su estructura especifica. Este proceso, en la cual la proteína es sintetizada hasta que adopte su estructura en la membrana, es enormemente complejo y en ocasiones puede fallar. Sin embargo, la celula esta preparada y puede rápidamente arreglar la falla. En cambio si el sistema no esta funcionando como debe, puede resultar en enfermedades. El objetivo de la tesis es estudiar proteínas que están relacionadas con enfermedades. La enfermedad de Alzheimer es la forma más común de demencia. Mutaciones en el gene de la proteína presenilin 1 (PS1) puede resultar en una forma mas agresiva de Alzheimer, que aparece a menor edad. Hemos estudiado las mutaciones y pudimos ver que afectan la estructura y la actividad de PS1. URG7 es una proteína desconocida que esta asociada a cáncer al hígado. Hemos demostrado que es una proteína de membrana y esta localizada en el retículu endoplasmático. Surfactant C (SP-C) es una proteína que se puede agregar resultando en enfermedades en el pulmón. Hemos estudiado la transmembrana de SP-C y el carboxilo terminal, que contiene un dominio, BRICHOS, que ayuda la proteína a adoptar su estructura. Hemos confirmado la topología de URG7 y SP-C usando un método donde una proteína verde fluorescente es glicosilada. Contents List of publications ..................................................................................... vi Additional publications ............................................................................. vii Abstract ...................................................................................................... viii Populärvetenskaplig sammanfattning .................................................... ix Resumen en español .................................................................................. x Contents ....................................................................................................... xi Abbreviations ............................................................................................ 14 Amino acids ............................................................................................ 15 1. Introduction .......................................................................................... 16 2. Biological membranes ........................................................................ 18 2.1 General features of biological membranes .............................. 18 2.2 Lipids in the membrane ............................................................... 19 2.3 The endoplasmic reticulum ......................................................... 22 3. Membrane proteins ............................................................................. 23 3.1 Membrane protein architecture .................................................. 24 3.2 General structural features of membrane proteins ................ 25 3.2.1 Hydrophobic mismatch ......................................................... 25 3.3 Properties of amino acids in membrane proteins ................... 26 3.3.1 Polar amino acids in transmembrane segments ............. 26 3.3.2 Proline and glycine in transmembrane segments ........... 27 3.3.3 Aromatic belt .......................................................................... 28 3.3.4 Snorkeling ............................................................................... 28 4. Biogenesis of membrane proteins ................................................... 29 4.1 The co-translational translocation ............................................. 29 4.2 The post-translational translocation .......................................... 31 4.3 The Sec translocon ....................................................................... 32 4.4 Accessory protein components .................................................. 34 5. Insertion of membrane proteins into the endoplasmic reticulum membrane ................................................................................................. 37 5.1 Topology of membrane proteins ................................................. 38 5.2 What factors determines the topology of membrane proteins? ..................................................................................................................... 38 5.2.1 Positive inside rule .................................................................. 40 5.2.2 Hydrophobicity ......................................................................... 40 5.2.3 Transmembrane lenght.......................................................... 41 6. Diseases related to proteins studied ............................................... 42 6.1 Alzheimer’s disease ........................................................................ 42 6.2 Interstitial lung disease ................................................................ 43 6.3 Hepatocellular carcinoma.............................................................. 44 7. Methodology ......................................................................................... 45 7.1 The model protein: Leader peptidase ........................................ 45 7.1.1 Lep H2 and Lep H3.................................................................. 46 7.2 Determining the propensity to integrate into the ER membrane ................................................................................................. 47 7.3 Determining the topology of membrane proteins ................... 47 7.3.1 Topology prediction ................................................................ 48 7.3.2 Topology mapping .................................................................. 49 7.3.2.1 Glycosylation assay ......................................................... 49 7.3.2.2 Protease protection assay .............................................. 50 7.2.3.3 Glycosylatable GFP assay............................................... 50 8. Summary of papers ............................................................................ 51 9. Conclusions and perspectives ........................................................... 54 Acknowledgements .................................................................................. 57 References ................................................................................................. 61 Abbreviations Aβ AD ER FAD GFP i-CLiPs ILD Lep MGD OST PA PC PE PI PM PS PS1 RNC SM SP SR SRP TM amyloid β-peptide Alzheimer’s disease endoplasmic reticulum familial AD green fluorescent protein intramembrane cleaving proteases interstitial lung disease leader peptidase minimal glycosylation distance oligosaccharyltransferase phosphatidic acid Phosphateidylcholine phosphateidylethanol phosphateidylinositol plasma membrane phosphateidylserine Presenilin 1 ribosome-nascent chain sphingomyelin signal peptidase SRP receptor signal recognition particle transmembrane Amino acids Ala Alanine (A) Arg Arginine (R) Asn Asparagine (N) Asp Aspartic acid (D) Cys Cysteine (C) Gln Glutamine (Q) Glu Glutamic acid (E) Gly Glycine (G) His Histidine (H) Ile Isoleucine (I) Leu Leucine (L) Lys Lysine (K) Met Methionine (M) Phe Phenylalanine (F) Pro Proline (P) Ser Serine (S) Thr Threonine (T) Trp Tryptophan (W) Tyr Tyrosine (Y) Val Valine (V) 1. Introduction All living organisms visible or non-visible are made of cells. The cell is the basic unit of life and the smallest one that is capable to independently reproduce itself. It can vary in size and shape and is bounded by a cell membrane. The membrane is composed of lipids and proteins that build up a hydrophobic barrier that separates the cell contents from the external environment. It is important for the cell to take up nutrients and other molecules, as well as excrete waste products to its surroundings in order to function, and for that reason the membrane cannot be completely impermeable. Specialized proteins embedded in the membrane, called membrane proteins serve to transport specific molecules from one side to the other. There are different types of cells, which fall in the three distinct domains of life. Two large groups, archaea that inhabit extreme environments and prokaryotes (bacteria) are microorganisms that do not have a nucleus (Fig. 1A). Cells with a membrane bounded nucleus make up the third group, called eukaryotes e.g. animal (Fig. 1B) and plant cells. Eukaryotes have besides a nucleus, other compartments, called organelles that are surrounded by membranes, such as the endoplasmic reticulum (ER), mitochondria, chloroplasts, peroxisomes, Golgi and lysosome, each with specific functions. Although, there are many differences between archaea, prokaryotes and eukaryotes, they share common features, one is the storage of their hereditary information in form of DNA. The DNA carries instructions for the production of the cell machinery. The main focus of this thesis is the study of eukaryotic membrane proteins, how they insert into the ER membrane and the understanding of membraneprotein topology that reveals how they sit in the membrane. Interestingly, the 16 specific proteins studied are connected to diseases, the first is involved in Alzheimer’s disease, the second in liver cancer and the third is crucial for our lungs and breathing. A B Fig. 1. Schematic illustration of a prokaryot cell (A) and an animal eukaryotic cell (B). 17 2. Biological membranes 2.1 General features of biological membranes Membranes are the boundaries that surround the cell and maintain the cell integrity by separating the inside milieu from its exterior environment. The two major components are lipids and proteins. These molecules are characterized by being amphiphilic, a property that constitutes the structure of the membrane in an aqueous solution. The lipids are organized in a double layer, which thermodynamically favors and stabilize the membrane by the interaction of hydrophobic acyl chains facing each other in the interior of the membrane excluding the contact with water. On the other hand, the polar head groups are oriented to the outer, aqueous space. Our understanding and view of the dynamics and structure of the biological membrane is based on the ‘fluid mosaic model’ presented by Singer and Nicholson in 19721. The membrane is seen as a two-dimensional liquid where lipids and proteins are in constant motion, freely moving laterally within the membrane matrix (Fig. 2A). However, this model has been updated as seen in Fig. 2A, few integral proteins floating in the fluid bilayer lipid phase leaving most of the lipids unperturbed. New experimental approaches in the membrane field sustain a more crowded lipid bilayer with high numbers of integral membrane proteins limiting the free lateral diffusion of the molecules (Fig. 2B)2; 3. Moreover, the surface of the bilayer is often crowded with peripheral proteins that sometimes can be transiently attached. Oligosaccharides, sugar molecules also protrude from the membrane on the cell surface and play important 18 roles in cell-to-cell recognition and signaling. They are often attached covalently to lipids or proteins. A B Fig. 2. Membrane models. Model structure of the cell membrane based on the Singer Nicolson model. Lipids and proteins are freely diffusing laterally1 (A). An update version of a more crowded membrane according to Engelman2 (B). 2.2 Lipids in the membrane As mentioned above, membrane lipids are considered as amphipathic structures, they have a hydrophobic tail and polar head region (Fig 3). In an aqueous solution they spontaneously form a bilayer structure, shielding their hydrophobic moieties, facing them to each other and their polar structures are exposed towards the water environment. This ‘hydrophobic effect’ is the foundation of the biological membrane structure. The hydrophilic head group determines the charge of the lipid. They can be neutral, zwitterionic, with a positive and negative dipole, resulting in no overall charge, or they can be negatively charged. The three major kinds of lipids in a biological membrane are phospholipids, glycolipids and cholesterol. Considering the properties of the two first lipids, such as the diversity of chain length of the fatty acids, and the varying de- 19 gree of saturation, including the different head groups that exist, one can come up to a high number of different lipid species. There are more than hundred different lipid species present in a membrane of a prokaryotic cell such as Escherichia coli4, and more than 1 000 different types in a eukaryotic cell5. The features of a lipid contribute to its shape, so lipids can be divided into three groups based on their structure: cylindrical, conical, and inverted conical, which display a no curvature, negative curvature respectively positive curvature nature (Fig. 3), when they are accumulated locally. A B Fig. 3. Membrane lipids. Schematic structure of a phospholipid, in this special case, phosphateidylcholine (A). The polar head group and the hydrophobic tail can vary in size resulting in different lipid shapes, such as cylindrical, conical and inverted conical. Adapted from Suetsugu et al.6 (B). 20 Phosphateidylcholine (PC), phosphateidylethanol (PE), phosphateidylserine (PS) and sphingomyelin (SM) are the most common lipids in the eukaryotic plasma membrane (PM). Both PC and PS are cylindrical lipids that form flat bilayers, while PE contributes with a negative curvature structure. PS possesses a negatively charge head group. Additionally, two other lipids also exist but to lower extent, that contribute to negative charges, they are phosphatidic acid (PA) and phosphatidylinositol (PI) as well as, its phosphorylated derivatives, phosphoinositides6. A special type of lipid can be associated to a certain type of organelles. For example, cardiolipin is mainly found in the inner mitochondrial membrane, while the PM is enriched in sphingolipids and sterols7; 8. Membranes are asymmetric; the lipids are asymmetrically distributed in the inner and outer leaflet of the lipid bilayer9, with this, the membrane can adopt different shapes. Lipids that induce curvature (PE, PA, and PI), contributing to membrane bending, are important for vesicle budding, fusion and fission. Moreover, the asymmetry has essential regulatory features, for example, PS is present exclusively in the inner leaflet of the PM. However, when PS is transported and exposed to the cell surface, in the outer leaflet, it is a signal for apoptosis10. Furthermore, the phospholipid composition varies between cell types and membranes. The eukaryotic PM is enriched in sphingolipids and cholesterol. The first has saturated tails and the second has properties that allow it to intercalate between the lipids, generating a more tightly packed membrane8; 9 , which is characteristic for the PM in contrast to the intracellular organelle membranes. The lipid composition of the PM, including the presence of more saturated PS and PE in yeast for example11, contributes to a more rigid and stable membrane. One can suggest that this is the case for other cell types as well, as the general view of the PM is that it is tightly packed, high21 ly charged (high PS and phosphoinositides content)12 and more lipidasymmetric than other membranes9, which defines its function as the permeability barrier of cells. 2.3 The endoplasmic reticulum The eukaryotic organelle endoplasmic reticulum (ER) is highly dynamic and its membrane forms a network of tubules and cisternae that separates its interior, the lumen, from the cytoplasm13. In addition to being the major site of lipid synthesis, it is also the platform for protein production, folding and quality control. Although cholesterol is synthesized in the ER, the ER membrane displays low levels of cholesterol and also of sphingolipids. The level of cholesterol is maintained low since it is transported to other organelles. This is possible because the ER is in contact with the nucleus, mitochondria, peroxisomes, and the PM in the cell8; 13. The scarcity of sphingolipids and cholesterol, including the abundance of unsaturated fatty acids, results in looser lipid packing that is consistent with the role of this organelle in transporting and inserting newly synthesized lipids and proteins into the lipid bilayer8. In addition it has been suggested that the ER is less asymmetric than the PM and may be regulated by the presence of ATP-dependent lipid transporters, they so called flippases, floppases and scramblases9. The first one move phospholipids from the outer to cytosolic leaflet, the second moves phospholipids from the cytosolic to the outer leaflet and the third moves lipids in either direction toward equilibrium. In summary, ER is characterized by loose lipid packing and seems to be less asymmetric and electrostatic, due to the weakly charged cytosolic leaflet, because of the low content of negatively charged lipids12. So far I have only mentioned one of the major components in the biological membrane, the lipids, however, in the next chapter I will explain more about the membrane proteins and their role in the lipid bilayer. 22 3. Membrane proteins Membrane proteins are important molecules that have crucial and specific roles, and are involved in many biological functions. They make the cell membrane permeable to molecules needed for the cell to function and they allow the cell to communicate with its environment. In addition to maintaining the shape of the lipid bilayer, they act as sensors (receptors) of external signals allowing the information to cross the membrane and for the cell to respond to them. Furthermore, they function as transporters for polar molecules such as ions, water and peptides and as channels, allowing molecules to pass the membrane by diffusion14; 15. Not to forget, they are important factors of transforming energy, converting chemical energy into electrical and synthesizing ATP, the cellular energy currency of life16. There are three types of membrane proteins. Peripheral (or extrinsic) membrane proteins interact weakly and reversible with the membrane on its surface. They are distinguished by how they can be released from the membrane, by using relatively gentle treatments such as high salt or high pH without affecting the membrane structure. Peripheral proteins associate with the membrane through electrostatic interactions and hydrogen bonding with the polar head group of the lipids and hydrophilic domains of proteins1; 3. The second group, amphitropic proteins are found in the interior of the cell, in the cytosol. They can either interact non-covalently with lipids and proteins or they can have one or more lipids covalently attached to them. 23 The third group of membrane proteins is critical for structural integrity of membranes. Integral (or intrinsic) membrane proteins are buried inside the bilayer, surrounded by lipids, with their hydrophilic domains protruding into the aqueous milieu. They are strongly associated with the membrane and require harsher treatment such as detergents, organic solvent, or denaturants to remove them from the membrane. 3.1 Membrane protein architecture Determination of the three-dimensional (3D) structures of membrane proteins has revealed two types of structural motifs that dominate in membrane proteins, α-helical bundles and β-barrels16; 17; 18. The latter one found almost exclusively in the outer membranes of Gram-negative bacteria and in mitochondria and chloroplast of eukaryotes, while most proteins in cellular membranes are bundles of α-helices. The folding into these two basic secondary structures (Fig. 4) is favorable for a protein embedded in a nonaqueous environment, which are stabilized by having the hydrogen bonding of the polar backbones satisfied14. A B Fig. 4. Structures of the two major types of membrane proteins. The OmpA β-barrel protein (PDB: 1QJP)19 (A). The Rhodopsin α-helical protein (PDB: 2I35) (B). 24 Three years after the Singer and Nicholson model of the cell membrane was published, the 3D structure of the first integral membrane protein was resolved, the light driven proton pump of bacteriorhodopsin from Halobacterium halobium20. Even though, the resolution was not so high, it revealed for the first time that membrane proteins consist of α-helical transmembrane (TM) segments oriented perpendicular to the plane of the membrane (Fig 5). Years after came the first high-resolution crystal structure of the bacterial photosynthetic reaction center21 and after that until now the number of determined structures has been increasing exponentially14; 22. However, it has been, and still is a challenge to work with membrane proteins, as they are unstable outside the membrane environment. In order to stabilize them they need to be reinserted into a kind of lipid, detergent milieu, by adding ligands or inhibitors, or by introducing point mutations to generate a more intrinsically stable protein16. 3.2 General structural features of membrane proteins The high-resolution membrane protein structures have revealed important general structural features: The length of a typical α-helix is between 20 and 30 amino acids. The hydrocarbon core of the membrane is approximately 30 Å, and the membrane-water interface around 15 Å. To fit a TM helix in the core would require around 20 amino acids. Interactions between the TM segment and lipids are essential to many cellular activities. 3.2.1 Hydrophobic mismatch Mismatch can occur in which the hydrophobic length of the TM segment is longer than the core (positive mismatch) or the opposite, the core is thicker than the TM (negative mismatch). There is a variety of ways to minimize the energetically unfavorable exposure of hydrophobic groups to the hydrophilic environment and polar groups of the peptide to the lipid bilayer23. Studies 25 have indicated that both TM segments and lipids respond to mismatch. The length of a TM segment can vary a lot, when it is longer than the thickness of the membrane it tilts with respect to the bilayer or results in kinked and even interrupted α-helices24; 25. Alternatively, it can oligomerize or even aggregate to minimize the exposed hydrophobic parts. In contrast, when the hydrophobic part is too small it can result again in aggregation or changes in the side chain orientation. Furthermore, as a consequence ‘snorkerling’ could occur, discussed below. Although, it has been experimentally shown that a TM segment as short as 10 residues can be inserted into the ER membrane26, too short TMs may adopt a surface localization27. Lipids are also capable of responding to hydrophobic mismatch by stretching or disordering their fatty acid chains. Moreover, while the thickness of the membrane can vary with the lipid composition, the plasma membrane is likely to be thicker than the ER due to the high content of cholesterol and sphingomyelin. This suggests that hydrophobic mismatch may be required for specific functions in the membrane, such as affecting protein sorting to particular organelles27; 28 and seems to be involved in the functionality of several proteins29; 30. 3.3 Properties of amino acids in membrane proteins Another feature that membrane proteins share, is the amino acid distribution, which shows preferential locations within the TM helix24; 31. Not surprisingly, the hydrophobic residues such as Leu, Ile, Val. Phe and Ala are the most frequent ones in the TM helices buried in the hydrocarbon core. 3.3.1 Polar amino acids in transmembrane segments As expected polar and charged residues are less frequently in a TM segment. If present, they are conserved and often involved in important biologically functions, conformational specificity and thermodynamic stability14; 24; 32. Nevertheless, the high energetic cost can be compensated by the formation 26 of salt bridges, which can drive specific interactions between helical segments33 and even facilitate hairpin formation34. The helix-helix interaction has been widely studied35, their associations being governed by already mentioned, electrostatic and also van der Waals interactions. For example, AspAsp or Asn-Asn interactions stabilize the TM topography of helices in the bilayer36. 3.3.2 Proline and glycine in transmembrane segments Despite the fact that Gly and Pro are known as ‘helix-breakers’ they are found in TM helices, Gly is especially crucial in helix-helix interactions37. The dimeric protein, Glycophorin A (GpA) has been well studied and reveals the involvement of Gly in dimerization of membrane proteins37; 38. The many studies of GpA led to the identification of the GxxxG motif found in several membrane proteins39. The small side chain of Gly allows a very close contact between two helices. Pro is unique because its side chain lacks the proton necessary for hydrogen bonding, which results in a kink in the protein backbone40. However, the structure of Rhodopsin shows that a Pro in the middle of a TM helix does not necessary always cause bending41. Pro has also been suggested to be involved in helix-helix interactions of many membrane proteins40. Fig. 5. Model of the first 3D structure of the bacteriorhodopsin membrane protein obtained by electron microscopy at 7 Å resolution20. 27 3.3.3 ‘Aromatic belt’ The aromatic amino acids Tyr and Trp are enriched at the ends of TM helices, in the lipid-water interface. Their amphipathic character allows them to hydrogen bond to lipid head groups. One possible explanation is that it could provide extra stability to the helix, serving as an anchor. Furthermore, it has been shown that both Tyr and Trp destabilize the TM state when it is in the central part of a TM helix. On the other hand, Phe, also aromatic and fully hydrophobic shows a preference for the hydrocarbon core rather than for the interface space17; 24; 42; 43; 44. 3.3.4 ‘Snorkeling’ Charged residues are highly costly to keep within the membrane core. Interestingly, both Lys and Arg are more tolerated at the edges of the TM helix. Probably, because their long, flexible alipathic side chain buried inside the bilayer could ‘snorkel’ up into the head group region, so that the positively charged end may interact with the phosphate groups. Nevertheless, negatively charged residues have a small side chain and might be repelled by the negatively charged head groups, which would be energetically unfavorable to keep in the interface43; 44; 45. 28 4. Biogenesis of membrane proteins How do membrane proteins end up in the lipid bilayer? First, they are synthesized by the ribosome, which translates the mRNA into amino acids and catalyses the peptide bond formation. The growing polypeptide chain is released into the cytosol, where the ribosome operates. However, the hydrophobic nature of a membrane protein would cause the protein to aggregate in the polar milieu of the cytosol. To overcome this problem the nonpolar parts of the protein needs to be masked before entering the membrane, facilitated by the translocon, in order to avoid unfavourable contacts with the polar environment. There is a common evolutionarily conserved pathway for most membrane and secreted proteins, where the ribosome and the translocon work together with the signal recognition particle (SRP). Most of eukaryotic membrane proteins insert co-translationally, fold and oligomerize in the ER membrane. Moreover, those that are destined to function in other membrane compartments such as the plasma membrane are transported in vesicles to their target destination46; 47. 4.1 The co-translational translocation The SRP-dependent pathway starts when a signal sequence or the first TM of the growing nascent chain emerges from the ribosome and is recognized by the SRP. The cleavable signal sequence has a positively charged Nterminal region followed by a hydrophobic domain composed of 7-15 residues and a polar C-terminal region. In contrast, the non-cleavable hydrophobic domains that anchors the protein in the membrane are longer, usually 1927 residues48; 49. When SRP binds to the signal sequence it pauses the nas- 29 cent chain elongation50; 51 and brings the whole ribosome-nascent chain (RNC) complex to the ER membrane by interacting with the SRP receptor (SR)52. Both SRP and SR have a GTPase domain and their contact and interaction occurs through the GTPase modules. They are bound to GTP when they are associated with the RNC, however, when GTP is hydrolyzed the whole complex is disassembled and SRP and SR are recycled for the next run52. Moreover, it promotes ribosome binding to cytosolic loops of the protein conduction channel, the translocon. The docked ribosome can reinitiate the elongation of protein synthesis, while it releases the growing polypeptide into the proteinous environment of the translocon, which allows soluble domains to cross the membrane and hydrophobic TM segments to exit laterally into the lipid phase53 (Fig. 6). Fig. 6. Overview of the co-translational SRP-depedent targeting pathway of newly synthesized polypeptides54. The RNC targeting is universally conserved over all domains of life. The SRP pathway mechanism is similar between eukaryotic and prokaryotic systems, although there are small differences. In prokaryotes the RNC is 30 destined to the bacteria inner membrane (IM). The bacterial SRP contains the conserved protein component Ffh (called SRP54 in eukaryotes) and the 4.5S SRP RNA. They are mainly involved in the targeting of inner membrane proteins to the translocon, while secreted proteins utilize the posttranslational pathway55; 56. The bacterial SRP lacks the subunits that are associated with the translational arrest seen in eukaryotes. Moreover, the bacterial SR, called FtsY, is a single protein that lacks TM segments and may interact weakly with the bacterial IM55. 4.2 The post-translational translocation In the post-translational pathway, the protein is completely synthesize in the cytosol and thereafter targeted and inserted into the ER membrane. It begins with the binding of cytosolic chaperones while the nascent chain emerges from the ribosome in order to prevent premature folding before the polypeptide is transported through the channel. The substrate is later targeted to the Sec61 complex, which interacts with the Sec62/Sec63 complex. The chaperone releases the nascent chain while it is translocated through the channel46; 57 . On the lumenal side of the ER the chaperon BiP binds to the polypeptide preventing the polypeptide chain to slide back to the cytosol and instead allowing it to move forward46. BiP is an ATPase that provides energy for translocation58. The mechanism is the following: BiP in its ATP-bound state interacts with Sec63, when ATP is hydrolyzed, BiP binds to the polypeptide chain in its ADP state. When the nascent chain has moved further in the forward direction, the next BiP molecule can bind and so on. Prokaryotes have instead a cytosolic ATPase, SecA that aid long hydrophilic domains and fully synthesized polypeptides to cross the ER membrane trough the translocon46. Although, archaea lack both BiP and SecA, it has been suggested that posttranslational translocation may occur as well but how it is energized remains unclear55. 31 4.3 The Sec translocon The universal protein-conducting channel is found in all kingdoms of life, called Sec61 in eukaryotes and SecYEG in bacteria and archaea. The Sec translocon allows membrane proteins and secretory proteins to translocate and integrate into the ER membrane59. The eukaryotic Sec61 channel complex consists of three subunits, α, β and γ, both the α- and the γ-subunits are very well conserved and essential for cell viability. In 2004 van den Berg et al. determined the x-ray structure of the archaea Sec complex from Methanococcus jannaschii60 and after that additional crystal structures have been resolved with distinctive conformations61; 62. Despite the evolutionary distance between the different organisms, superimposing the structures indicates a high degree of similarity reflecting how well conserved it is63 in both prokaryotes and eukaryotes64; 65. Fig. 7. Crystal structure of the SecY complex from M. jannaschii (PBD: 1RHZ). View from the side (left) and from the top of the cytosolic side (right). The SecYsubunit (α-subunit) contains 10 TMs (in grey). The lateral gate is formed by TM2a (orange) and TM7 (blue) that allows hydrophobic segments to partition into the membrane. The β-subunit and γ-subunit are depicted in black. The blue lines indicate the approximate boundaries of the membrane 32 The 10 TM segments of Sec61α (SecY) subunit forms the pore, which has the form of an hourglass viewed from the side, with a constriction ring composed of hydrophobic residues in the cytoplasmic half of the membrane. However, studies have indicated that the pore has an aqueous environment66; 67; 68 . The α-subunit is divided into two halves (TM1-5 and TM6-10) that forms a ‘clam shell’ and has an interconnected hinge at the cytoplasmic loop between TM5 and TM6. Furthermore, from the lumenal side a short helix, TM2a also referred to as the ‘plug’ fills the center of the cavity. It maintains the channel impermeable to ions and small molecules, and it was suggested to be removed when the polypeptide enters, allowing translocation (Fig. 7). However, Gogala et al. reported that when the nascent chain is translocated the plug is only slightly displaced65. Taken together, it is important to maintain the membrane barrier, particularly for prokaryotes, since the proton gradient across the membrane is central for ATP-production and some transports69; 70. Moreover, the ribosome on the cytosolic side and chaperones such as BiP on the luminal side has been suggested to seal to the cytosol or lumen respectively depending on where the nascent chain is directed71; 72. They may also assist in the folding of the growing polypeptide chain73. The lateral gate releases TM segments to partition into the hydrophobic environment of the lipid phase. Although the mechanism is unknown it has been suggested that the presence of a hydrophobic peptide stabilizes an open conformation74. Photocross-linking experiments have shown that the signal sequence contacts and intercalates between TM2 and TM775. Additionally, this has been confirmed by the recent crystal structure of Sec61, where TM2 and TM7 move apart separating the two halves by approximately 12 Å in the presence of a hydrophobic TM segment65. The function of Secβ (SecG) is unknown, non-essential and has only minor contacts with the periphery of the α-subunit. It has been postulated to have a kinetic effect and to facilitate the cotranslational translocation60; 76. However, 33 Secγ (SecE) is essential and helps to maintain the structure of the complex by holding together the two halves of the α-subunit mentioned above60. 4.4 Accessory protein components Reconstitution of protein translocation components into proteoliposomes revealed that for efficient translocation only three proteins are necessary, the Sec61 complex, SR and the translocating chain-associated membrane protein (TRAM)77. However, for optimal and proper transport of varying substrates in the secretory pathway it is likely that other proteins involved. There are several additional membrane components in addition to the ones mentioned that have been implicated in the translocation process78, and they seem to be in a close proximity of the Sec6179. The signal peptidase (SP) complex is the protease that removes the Nterminal signal sequence from secreted proteins and some membrane proteins on the lumenal side of the membrane80. The crystal structure of the E. coli signal peptidase has revealed that it is a serine endoprotease81. SP has a substrate specificity for small and uncharged residues, such as Ala82. Following the removal of the signal sequence, the polypeptide is folded, modified, and salt bridges are formed. However, the signal sequence left in the membrane is further cleaved by a signal peptide peptidase (SPP). SPP belong to the family of intramembrane cleaving proteases (i-CLiPs) including the presenilin (PS) family of aspartyl proteases, the zinc metalloprotease site-2 protease family and the rhomboid family of serine proteases. SPP cleavage requires helix-breaking residues within the TM region of the substrate, while positive charges that flank the TM region have an inhibitory effect. Another requirement is that SP must first cleave on the lumenal side preparing the substrate for the subsequent intramembrane cleavage performed by SPP83. The aspartic proteases, PS and SPP share identical active site motifs, GxGD and the highly conserved PAL motifs, indicating that they have a common 34 catalytic mechanism84. However, their active site is positioned opposite within the plane of the membrane. This difference is due to how their substrates orient in the membrane, PS has substrates with an NoutCin topology, whereas SPP has substrates displaying an NinCout topology85; 86. In particularly PS is interesting for this thesis in its involvement in Alzheimer’s disease, as discussed later. N-linked glycosylation is one of the most common covalent protein modifications in mammals and it is essential for the folding of the protein and even for oligomerization, quality control and transport87. It is s not only restricted to eukaryotes, although it is less frequent in prokaryotes88. N-linked glycosylation occurs co-translationally by the oligosaccharyltransferase (OST) multi-subunit protein complex that is closely associated with the translocon89; 90 . It transfers the preassembled oligosaccharide, Glc3Man9GlcNAc2 in higher eukaryotes, to a specific Asn residue in the protein. The catalytic site is located on the lumenal side of the ER membrane87. TRAM spans the membrane eight times with the N- and C-terminal facing the cytoplasm91 and is involved early in the translocation and insertion of secreted and membrane proteins92. Moreover, it has been suggested to function as a chaperone for short, weak signal sequences and less hydrophobic TM segments with charges93; 94. TRAP is a tetrameric complex95. Although, its main function remains unclear, it has been proposed to facilitate the initiation of substrate transport96. In addition, it might affect the topology of membrane proteins, promoting Cterminal translocation for those that lack topogenic signals, stabilizing an NinCout orientation97. 35 In addition, it is worth to mention the ribosome-associated membrane protein 4 (RAMP4), a small single-spanning membrane protein93; 98. It seems to be involved in folding99, regulating N-linked glycosylation of proteins100 and stabilizing membranes proteins in response to stress101. Moreover, PAT10102, and BAP31103 have also been reported to interact with the translocon. 36 5. Insertion of membrane proteins into the endoplasmic reticulum membrane When the membrane protein has been effectively co- or post-translational targeted to the translocon, it needs to be recognized and inserted into the ER membrane. How this occurs is not completely understood although many studies indicate that the major factor is the overall hydrophobicity of the helix104. A broad study where each of the 20 naturally amino-acid residues were systematically tested across a model TM segment composed of Ala and Leu residues led to the well established ‘biological scale’. These results provided comprehensive information regarding membrane integration and location dependence of the individual residues in a TM helix105; 106. The experimental data from the study has been used to develop and improve algorithms for membrane topology prediction107. The ‘biological scale’ proposes that the insertion is a thermodynamically driven equilibrium process in which the partitioning into the lipid phase is dependent on the hydrophobicity of the TM domain. The TM segment positioned in the polar channel may open the lateral gate so it can access the hydrophobic milieu outside and diffuse into the membrane94. However, the process can also be kinetically controlled, where the Sec translocon plays a more active role than just providing a site in the membrane where TMs equilibrate between lipid and aqueous phases74. Substituting the hydrophobic residues in the pore ring of the yeast Sec61p affected the efficiency of TM segment integration into the lipid phase108. This suggests that the Sec translocon sets the hydrophobicity threshold for membrane integration. Moreover, marginally hydrophobic TM segments are more efficiently inserted when translocation is slower109. 37 5.1 Topology of membrane proteins During integration, the membrane protein needs to adopt the correct topology and fold into a final compact native structure, which is important for function. Membrane topology can be described as how many times a membrane protein spans the lipid bilayer and where the soluble loops that connect the TM segments are oriented relative to the plane of the membrane. This information can help us to better understand the structure and the function by revealing the localization of specific domains of a membrane protein. How do membrane proteins achieve their topology? The simplest model suggests that in a multi-spanning membrane protein, the first TM segment is the one that determines the orientation of the subsequent ones. However, this is not always the case, there are exceptions, in which the internal TM segments can reorient and change the whole topology. One example is the SecG component of the Sec translocon, which flips between different topologies during translocation of the nascent chain110. Nevertheless, there are TM segments that are not sufficiently hydrophobic to be co-translationally released into the lipid bilayer, instead they are inserted post-translationally altering the orientation of already inserted TM helices, as for aquaporin 1111. Controversially of what has been described, there are dual-topology proteins. These proteins can insert in two opposite orientations, and are fully functional when adopting both topologies, e.g. the well-studied EmrE. EmrE is a small multidrug resistance homodimer protein that has been proposed to have two oppositely orientated molecules, displaying a functional dual-topology protein112. 5.2 What factors determines the topology of a membrane protein? Membrane proteins can adopt different types of topology that result in an NoutCin or an NinCout orientation (Fig. 8) depending on various factors, such 38 as the length of the TM segment, the hydrophobicity, distribution of charged residues and other features. Some TM segments, e.g. marginally hydrophobic TM helices are unable to integrate by themselves into the membrane and need the interaction with adjacent more hydrophobic TM segments113; 114 . These poorly hydrophobic TMs are retained close to the Sec translocon by polar interactions to pack and assembly properly before they are released into the lipid bilayer115. This protein-protein interaction at the translocon has been observed for several proteins102; 116; 117; 118. Furthermore, premature dislocation of a mariginally hydrophobic TM into the membrane can cause aggregation118. Fig. 8. Different types membrane protein topologies In membrane proteins, loops with positively charge residues tend to position in the cytosol, the ‘positive-inside rule’ is a general mechanism found in all organisms119. Crystal structures of membrane proteins have revealed a number of helices, ‘reentrant loops’120; 121; 122, that do not span the whole membrane, but rather insert as hairpins that orient the N- and C-terminal end of the TM on the same side of the membrane123; 124; 125. Another important factor in membrane protein integration is the lipid composition of the lipid bilayer. The topology of lactose permease (LacY) was largely altered when inserted 39 in a membrane lacking phosphatidylethanolamine, signifying the importance of lipid influence on topology. Interestingly, by inducing PE synthesis postassembly of LacY, the TM domains could reorient to their native orientation, indicating that the process is completely reversible126. The cleavable signal sequence of the secreted proteins inserts into ER in an NinCout orientation i.e. a type I membrane protein. How the N-terminal reorients has been under debate, it has been proposed to either go in ‘head-first’ and then flip127; 128, or it inserts as a hairpin129. The N-terminus can be fixed on the lumenal side of the ER membrane by introducing a glycosylation site in the N-terminus, thus suggesting that the signal sequence can reorient within the translocon130. 5.2.1 ‘Positive inside rule’ The phenomenon of having positive charges predominantly in the hydrophilic loops on the cytoplasmic side of the membrane is well preserved131. The positive inside rule displays a strong topogenic signal104; deleted or introduced the topology is changed 132; 133 132 , as when . One explanation could be that the positive charges are arrested in the cytosol134; 135; 136 , probably because they interact with the negatively charged lipid head groups137. Nevertheless, the positive charges contribute to the insertion of TM helices into the membrane more efficiently104; 138 . In addition, the translocon may be involved in the initial orientation of the TM by following the positive inside rule; conserved charges at the Sec61p of yeast seem to interact with the nascent chain providing the driving force for signal orientation139. 5.2.2 Hydrophobicity The more hydrophobic the TM segment is the more it tends to insert with an NoutCin orientation independently of the charges of the flanking region127; 140. An explanation is that very hydrophobic helices may rapidly exit the lateral 40 gate by interacting with the hydrophobic core of the lipid bilayer94. Nevertheless, the sliding model has been proposed, which suggests that strongly hydrophobic TMs never enter the polar channel but instead slip into the lipid bilayer along the lateral gate of the translocon141. In contrast, natural cleavable signal sequences (NinCout) are less hydrophobic, and remain longer time around the translocon, which allows them to reorient during translation. 5.2.3 Transmembrane length The length of the hydrophobic sequence can also determine the orientation of the TM. Longer sequences show a preference towards an NoutCin topology, localizing the N terminal in the lumen140; 142; 143. Once again, cleavable signal sequences have shorter hydrophobic segments than TM segments adopting an NinCout topology49, confirming the TM length as a determinant in defining the orientation of the hydrophobic helix. 41 6. Diseases related to the proteins studied Proteins are the most abundant biological macromolecules in all cells, and have a variety of functions. It is difficult to imagine a world without proteins, which are involved in perhaps every process occurring in the cell. Membrane proteins constitute about 30% of all proteins. It is therefore not a surprise that malfunctioning membrane proteins can cause a wide range of diseases. Here is a presentation of the diseases that are connected to the membrane proteins studied in this thesis. 6.1 Alzheimer’s disease Alzheimer´s disease (AD) is a neurodegenerative disorder affecting millions of people worldwide, and is the most common cause of dementia in the elderly population. The majority of cases correspond to the late-onset, sporadic AD that develops after the age of 65. However, there is also the relatively rare dementia that manifests at earlier age, the early-onset familial AD (FAD). FAD is inherited in an autosomal dominant manner144; 145. Mutations in the genes encoding the β-amyloid precursor protein (APP) and presenilins (PS1 or PS2) are related to FAD146. Clinical symptoms of AD include loss of memory, declining cognitive function, personality change and decreasing physical functions147. Moreover, extracellular senile plaques and intracellular neurofibrillary tangles defines the neuropathology of AD. The plaques consist mostly of the small amyloid β-peptide (Aβ) peptide fragments derived from APP148. APP is sequentially cleaved by β- and γ-secretase generating Aβ149; 150. The γ-secretase is a membrane bound protease that is composed by 4 membrane proteins, presenilin 1 (PS1), nicastrin, anterior pharynx42 defective 1 (Aph-1) and presenilin enhancer 2 (Pen-2)151; 152. The PS1 subunit contains the catalytic site of γ-secretase153. Interestingly, γ-secretase is an aspartyl protease like the SPP, although they have different membrane orientation. Furthermore, both SPP and PS1 belong to the family of i-CLiPs, which also includes the zinc metalloprotease site-2 protease family and the rhomboid family of serine proteases154. In this thesis we focus on studying the FAD-linked substitution in PS1146. 6.2 Interstitial lung disease Interstitial lung disease (ILD) is a group of lung disorders that affect the interstitium in the lungs, which is the tissue around the air sacs. As a result the tissue becomes stiff and scarred, and the air sacs cannot expand as much impeding the uptake of oxygen. The mammalian lung is composed of millions of alveoli, which provide an extensive surface area allowing efficient gas exchange between epithelial cells and the capillaries in the alveolus155. The alveoli are coated with a thin film of pulmonary surfactant, a lipidprotein complex that reduces the surface tension at the air-liquid interface, thereby reducing the work of breathing. Deficiency in the surfactant causes severe respiratory disorders, e.g. ILD. The protein moiety of the surfactant is composed of four proteins, which are divided into two groups, the hydrophilic surfactant proteins SP-A, and SP-D, and the hydrophobic, SP-B and SP-C156. Mutations in the SP-C gene are linked to familial and sporadic ILD, where some of them cause misfolding and aggregates157; 158; 159; 160. SP-C is expressed exclusively by alveolar type II cells as a 197 amino-acid precursor protein (proSP-C) that is processed to a 34 amino-acid residue mature membrane protein161. The C-terminal of proSP-C contains a linker region and a BRICHOS domain, which is localized to the ER lumen162; 163. The TM region is Val rich, a feature that can cause folding problems, due to the low propensity of Val in forming α-helices164; 165. This was confirmed by an SPC analogue, where all the Val were replaced with Leu, which resulted into a 43 more stable helix without aggregation problems166. The BRICHOS domain has been suggested to function as a chaperone in aiding the Val rich TM segment to form an α-helix for membrane insertion, thus preventing aggregation167; 168. 6.3 Hepatocellular carcinoma The hepatitis B virus (HBV) targets the liver and replicates in hepatocytes. The infection can develop to progressive chronic liver disease (CLD), seen as hepatitis, fibrosis, cirrhosis, and finally heptacellular carcinoma (HCC). HCC is the most common cancer worldwide169. The virus encodes hepatitis B x antigen (HBxAg) that is a trans-activating protein. HBxAg seems to alter the expression of cellular host genes that promote growth, survival and tumorigenesis in the liver170. One of these is the up-regulated gene clone 7 (URG7), which is involved in blocking caspases via the TNFα pathway, thus inhibiting apoptosis171; 172. Interestingly, the URG7 protein shows a prominent similarity to the multidrug-resistance protein 6 (MRP6). Of the 99 amino acids of URG7, the N-terminal 74 residues are identical to the N-terminal of MRP6171. Although, the exact role of MRP6 is unknown it has been classified as a multidrug-resistance associated protein because it is highly homologous to MRP1173. Remarkably, mutations in the gene of MRP are associated to the pseudoxanthoma elastaticum (PXE) disorder. PXE is characterized by progressive calcification of elastic structures in the skin, eyes and cardiovascular system174. 44 7. Methodology 7.1 The model protein: Leader peptidase Leader peptidase (Lep) is a well-characterized protease that is located in the inner membrane of E. coli, where it cleaves signal sequences from secretory proteins. It has two hydrophobic domains, H1 and H2 that span the membrane, and the two soluble domains, the small N-terminal (P1) and large Cterminal (P2)175. The P2 domain faces the periplasm and contains the catalytic site and a natural glycosylation site80; 176 . Lep inserts co-translationally into the ER membrane and becomes glycosylated in the presence of microsomes, adopting its native conformation136 (Fig. 9). The H1 segment is more hydrophobic than H2 and can insert into the membrane independently of H2. In contrast, H2 needs the assistance of H1 for proper membrane integration35. Fig. 9. Topology of the E. coli model protein Lep. Lep has two TM segments and the N- and C-terminal faces the periplasm. The large P2 domain contains the catalytic site. 45 7.1.1 LepH2 and LepH3 Much of the work for this thesis was done on two modified versions of Lep, LepH2 and LepH3 (Fig 10). In LepH2 the test segment replaces the H2, and two glycosylation sites were introduced in the N-terminal and P2 domains177. In LepH3 the test segment was introduced in the P2 domain, which is flanked by two engineered glycosylation sites105. The test segment can be positioned in two orientations depending on which Lep version is used. LepH2 displays an NinCout orientation, while LepH3 inserts in an NoutCin orientation in the ER membrane (Fig. 10). Upon expression of LepH2 and LepH3 the glycosylation patterns between singly and doubly glycosylated proteins can provide information about the insertion efficiency of the test segment. Fig. 10. Lep model protein variants. The test TM segment is in red (TMD) and have two different orientation depending on which Lep variant is used. Left panel. LepH2 test segment inserts in an NinCout orientation, where both glycosylation sites (G1 and G2) are modified. Right panel. LepH3 test segment inserts in an NoutCin topology and only G2 is modified. 46 7.2 Determining the propensity to integrate into the ER membrane The glycosylated protein species can be separated by SDS-PAGE and the intensity of the bands in the gel are quantified. This is a direct measurement of the inserted status of the test segment, which can be used to calculate the apparent change in Gibbs free energy, ΔGapp. This can be seen as a simplified model where the translocon-mediated insertion process is considered purely as an equilibrium process105; 106. The extent of inserted (!! ) and noninserted (!! ) test segment can be written as a probability (!) of insertion: != !! !! + !! and as an apparent equilibrium constant (!!"" ): !!"" = !! !! that can be placed in the equation of Gibbs free energy(ΔG!"# ): Δ!!"# = −!" ln !!"" where ! is the gas constant and the ! absolute temperature. 7.3 Determining the topology of membrane proteins It is evident that membrane proteins are difficult to work with in respect to the fact that they are buried inside the membrane. In order to crystalize them they need to be isolated from the lipid bilayer, however, this is not an easy task because they tend to aggregate. Moreover, by using detergents or artifi- 47 cial membranes to solubilize them, may affect their behavior and even disturb their structure. Therefore they represent only 1% of the solved protein structures178, due to the difficulties of overexpressing, purifying and crystalize them, as discussed above. Mammalian membrane protein structures are even more difficult to obtain because of their complexity. This is why structural biologists commonly work on bacterial homologues instead14. However, solved structures need to be validated experimentally. There are a numbers of topology determinant tools available that can provide structural information experimentally. In addition, it is suitable to test membrane proteins in silico, using computational tools as a first approach when only the sequence is known of a membrane protein. 7.3.1 Topology prediction Despite the fact that membrane proteins are biologically important, very few have had their topology determined experimentally. Topology prediction algorithms may be helpful in providing a first approximation. They rely on two major topological features. Firstly the hydrophobic sequence stretches, and secondly the positive charge biases towards the intracellular side of the membrane e.g. the ‘positive inside rule’133; 179 . These algorithms provide information about how many TM segments a protein has, including the TM domain boundaries, and the localization of the loop domains180. However, the increase of the crystal structures of membrane proteins has contributed developing better prediction programs. Additionally, the number of experiments dealing with membrane protein topology has increased the accuracy of predicting topologies, which has reached the 70-80% level181. Still, the available prediction algorithms are not completely reliable and can predict wrong topology, much remains to be done to improve the output178. 48 7.3.2 Topology mapping Although there are a number of prediction programs for membrane protein topology, it is necessary to verify the topology experimentally107; 182; 183; 184; 185 . There are several techniques available that can provide membrane pro- tein structural knowledge. This often involves a reporter that is fused to the protein and reveals at which side of the membrane the fusion site resides. However, cautions need to be taken as fusing a reporter can affect the folding and additionally the targeting if it is introduced N-terminally186. Another type of technique is when identified target sites are introduced in the polypeptide, e.g. N-linked glycosylation sites, Cys residues, antibody epitopes and proteolytic sites. Again, these sites are used to determine their accessibility at one side of the membrane187. An additional approach is proteolytic cleavage of all the loops that reside outside the membrane. 7.3.2.1 Glycosylation assay In eukaryotes the N-linked glycosylation occurs in the ER lumen by the action of the OST complex, mentioned previously. Carbohydrates are covalently attached to the asparagine residue present in the consensus sequence AsnX-Thr/Ser, where X can be any amino acid except Pro188; 189; 190. The glycosylation sites are introduced by site-directed mutagenesis at specific positions in the protein gene. Addition of an oligosaccharide to the target site results in an increase of the molecular mass by approximately 2.5 kDa, which can be detected by SDS-PAGE191. For efficient glycosylation the acceptor site should not be too close to the membrane90. The distance between the active site of OST and the ER membrane was determined192. Additional studies led to the minimal glycosylation distance (MGD), which is the distance required for half-maximal glycosylation, that is approximately 14 residues upstream or 10 residues downstream of the TM domain124; 193. 49 7.3.2.2 Protease protection assay The widely used Proteinase K is a non-specific protease194. It cleaves exposed hydrophilic loops, while the membrane embedded segments are protected. In addition since proteolytic enzymes cannot cross the membrane the inside loops are also protected against cleavage. The protected fragments can be detected by SDS-PAGE, thus revealing or confirming topologies of membrane proteins. This technique has an advantage of the fact that the studied protein does not need any kind of modification. However, the limitation of inaccessibility of the loops because of steric hindrance can give negative results187. 7.3.2.3 Glycosylatable GFP assay The Green fluorescent protein (GFP) originally comes from jellyfish Aequorea victoria195. This protein is widely used as a C-terminal fused reporter. It displays fluorescence when it is localized in the cytoplasm due to folding, in contrast when it is translocated to the periplasm it will not196, thus providing information about the localization of the C-terminal197; 198 . Extended studies have provided the use of a glycosylatable GFP (gGFP) for eukaryotic cells, which is a variant of GFP that contains an engineered N-linked glycosylation site199. Hence, when gGFP is localized in the ER membrane and glycosylated the fluorescence is abolished. In contrast, the gGFP displays fluorescence in the cytoplasm. 50 8. Summary of papers Paper I In this paper we studied PS1 that is part of the γ-secretase, an intramembrane aspartyl protease. PS1 is involved in the processing of APP that results in Aβ peptides, which are found as aggregates in the brain of people suffering from Alzheimer’s disease. In addition, mutations have been found in the PS1 gene, which are connected to the more severe form of AD that appears at earlier age, i.e. early-onset AD. It is of interest to elucidate the effect of the FAD-linked mutations in terms of function and structure of PS1. With the Nlinked glycosylation assay we are able to address questions concerning membrane integration. For this reason we chose a substitution located in the TM region that can be detectable by this method. The insertion efficiency differences between the wild type (WT) and the TM domain containing a substitution were measured. We found that very well inserted TM segments did not show any differences in insertion (TM1-5, TM8-9). Interestingly, TM6, H7 and TM7, which are the hydrophobic domains that constitute the catalytic site, were more susceptible for substitutions. As expected, hydrophobic to polar residue substitution resulted in membrane integration decrease, the opposite effect was obtained when polar residues were substituted to hydrophobic ones. Moreover, the substitution that showed effect were expressed and tested in vivo for functionality. The ratio between the Aβ42 and Aβ40 was measured. We observed that changes in the membrane integration did not correlate well with the increase in the Aβ42/ Aβ40 ratio but did show conformational changes in the active site. The FAD-linked muta- 51 tions may affect the PS1 molecule in various ways and there is not only one mechanism that is behind the AD pathogenesis. Paper II Here we study the novel protein URG7, which is one of the proteins that is up regulated during the development of liver cancer. It has been suggested to be involved in blocking caspases, which leads to the inhibition of apoptosis. URG7 has a natural glycosylation site on the N-terminal. In order to assess the topology additional acceptor sites were introduced. We propose that it anchors the membrane in an NoutCin manner, consistent with the orientation of MRP6. The truncated MRP6 (TM1-TM2) with known topology was used as a control for ER targeting. URG7 has the N-terminal 74 residues identical to the N-terminal of MRP6. Moreover, URG7 was expressed in HepG2 cells and we found that it is glycosylated and localized to the ER in agree with the in vitro studies. Paper III In this paper we study the SP-C, which is part of the lung surfactant that is involved in reducing the surface tension at the air-liquid interface in the lung. SP-C is associated with lung diseases; mutations found in the gene of SP-C can result in amyloid deposits in the lung. The SP-C is synthesized as a 197 residues proprotein (proSPC-C) and is proteolytically processed to a hydrophobic 35 amino-acid peptide. The TM domain of this protein is mainly composed of Val residues, which has a high propensity to form β-strands. The C-terminal contains a chaperon-like domain, the BRICHOS domain that is thought to aid the TM segment to fold correctly and insert into the membrane. In contrast, if the TM segment is not properly folded it will instead convert into β-sheet aggregates and amyoid-like fibrils. A synthetic SP-C analogue, where all the Val residues were substituted to Leu, resulted in a 52 more stable TM domain. Here we study the folding of SP-C (poly-Val) and its analogue (poly-Leu) by introducing N-linked glycosylation sites in the Cterminal of the protein at different distances from the TM segment. We found that poly-Val gets more efficiently glycosylated at closer distances from the membrane than poly-Leu. This suggests that poly-Val is in a more extended conformation, while poly-Leu adopts a more compact, folded conformation. We also study the interaction of membranes with the WT BRICHOS and an ILD-associated variant (L188Q) in order to better understand how BRICHOS execute its role as a chaperon. We could see that WT BRICHOS can insert into the membrane, probably into the head group region, while no insertion was observed for L188Q. This suggests that membrane insertion of the BRICHOS domain is important for the binding to the misfolded proSP-C. Moreover, the insertion may change the property of the membrane locally, preventing β-sheet formation. Paper IV In this paper we developed the use of gGFP in mammalian cells as a membrane protein topology reporter. The gGFP is C-terminally fused to preferable single-spanning membrane proteins. The engineered gGFP contains two glycosylation sites that upon modification in the ER loose the fluorescence. On the other hand, if located in the cytosol the proteins becomes fluorescent and non-glycosylated. By measuring the fluorescence and assessing the glycosylated state of a gGFP fusion protein, the localization of the C-terminal part of the protein can be determined. Here we confirm the topology of the four clinically important membrane proteins analysed in paper II and III. The URG7 and MRP6 were fluorescent and non-glycosylated respectively nonfluorescent and glycosylated, displaying an NoutCin and an NinCout topology. In addition both SP-C and its analogue were glycosylated but did not exhibit fluorescence, having the C-terminal in the lumen. gGFP can easily be used to assess topologies of membrane proteins in mammalian cells. 53 9. Conclusions and perspectives Membrane protein is the main focus in this thesis, especially those that are connected to diseases are particularly interesting for us. It is common to hear researchers say that membrane proteins are difficult to work with, the reasons are discussed above, and many avoid working with them for that. However, they are extremely important and the interest for them arises further when realizing their involvement in diseases. They become malfunctioned by for example acquiring a mutation in their gene that lead to loss of function. In addition the membrane protein can in worse case aggregate and infect other proteins, causing them to aggregate as well. This reaction can lead to cell death, and later on to tissue lost that can become life threatening. A critical step toward elucidation of their roles in the complex processes is the understanding of their structure and function. In the first paper we study PS1, which is involved in AD. This disorder is spread all over the world and is one of the most financially costly diseases. The well-studied PS1 has been accepted to play a key role in the disease, mutations has been found in the gene of PS1 that cause worse cases of AD. In this case the topology of PS1 is known, so we focus in studying what effect the mutations have in terms of structure and function. We learnt that in many cases they do not correlate to each other. However, there are limitations in the method used, the FAD-linked mutations where studied in individual TM segments, lacking the rest of the protein. We can suggest what effect the specific substitution will have on the specific TM segments where it sits. However, the TM segment containing the substitution is in reality surrounded by the other PS1 TM segments, which may shield and lowered 54 the effect. In addition, they might cause rearrangements in the whole protein that we cannot detect, causing it to loose its function. However, these changes can be very small to perceive, even though the effect is immense. In the case of URG7 protein, both the topology and function is unknown, but it has been suggested to be involved in the progress of liver cancer. Liver cancer is one of the most frequent cancer globally that cause death. We suggest that URG7 is targeted to the ER, and it spans the membrane once with the N-terminal in the lumen. In this case we work with the full-length protein that has a natural glycosylation site in the N-terminal, which is modified by the OST complex. In order to verify these results we introduce further sites that confirm the NoutCin topology of the protein. Moreover, the protein is expressed in vivo to find its localization, which is in the ER. The importance of synthesizing, inserting, folding and sorting the membrane protein correctly is crucial for its function. Misfolding and aggregation can occur during the process of maturation that chaperons and protein degradation system present in cells can take care of. However, this can be fatal if the system is not completely working and is overloaded with protein aggregates. An example of membrane protein that has the ability to aggregate is the SPC, which is involved in lung diseases. This protein is essential for our lungs to work properly. Mutations found in the gene of this membrane protein can result in aggregation and respiratory failure. Interestingly, this protein contains a chaperon-like domain, the BRICHOS domain that might be required for correct folding of the TM segment. The SP-C TM domain is special in the sense of that it is composed of mostly Val and Ile, residues not commonly found in α-helices. The Val rich TM domain might need assistance from the BRICHOS domain to fold correctly and insert into the membrane. We studied the biogenesis of proSP-C and its analogue, which has all the Val replaced by Leu. As expected, we could show that the poly-Leu is better 55 suited to fold than poly-Val. We could also show that the BRICHOS domain interacts with the lipid bilayer suggesting its involvement as a chaperone aiding the TM segment to fold. In the fourth paper, we use gGFP as a reporter protein to confirm the topologies of URG7, MRP6102, proSP-C (poly-Val) and proSP-C (poly-Leu) in vivo. Since the gGFP is fused C-terminally it can only report the localization of the C-terminal, which is highly useful for single-spanning membrane proteins. However, for multi-spanning it is not as suitable, as for PS1 that has nine TM segments. Knowing the C-terminal location of a multi-spanning membrane protein is not enough to know its whole topology. One way to overcome this problem is to truncate the protein from the C-terminal, but it is always preferentially to use the full-length protein instead of truncated ones. 56 Acknowledgements I would like to thank all the people that have made this thesis possible and that have been present during these 5 years of PhD studies. Even though it has been like a roller coaster, it is the great moments that will be memorized. First of all I have to thank my supervisor IngMarie Nilsson. Tack så mycket för all hjälp som jag har fått utav dig. Det har varit prisvärt att få ta del av din erfarenhet. Tack för alla mysiga och goda luncher J. Jag är även tacksam för chansen att ha fått jobba utomlands, vilket har varit väldigt lärorikt. Min andra handledare Gunnar von Heijne, för att du har gjort det möjligt för mig att doktorera i DBB och för att få mig känna som en del av din grupp. Tack Stefan för alla dina inputs, för att du finns alltid där för oss doktorander. Tack Elzbieta, Joe, Peter mina entusiastiska utvärderade! All co-authors for sharing their knowledge, Johanna Wanngren and Helena Karlström, tack för alla diskussionerna angående PS1. Magnus Monné för att ha fått chansen att jobba med det spännande URG7. Janne, tack för alla förklaringar vad gäller SP-C. Jenny, alltid positiv, ditt intresse för science är verkligen inspirerande och smittar av sig J. Joy I’m really thankful for the time in South Korea, for letting me work in your lab, and also for the time spent outside the lab. And as I told you, you have a wonderful group! Hunsang, thank you for being such a nice lab mate and also a friend! I’m so happy that our work could be published while we had lots of fun in and outside the lab J. The people from SouthKorea: Jake, first of all thank you for introducing me to bachata J. It was great to have you here in Stockholm, and thanks for all the help in Korea, for showing me the fun side of Seoul. Sungjun, my sweetie! Thank you for always being so helpful and always with a smile, you are amazing! Sungminaaa and Hayoung, my best teachers in Korea kkkk. My 57 girls: Hani, Chewon, Ramla and Kyungyeun, thank you for showing me Seoul!! Johannes, you know whatever happened in Korea stays in KoreaJ. Many thanks to the people in the lab and office mates from the past and present. Carolina, Ola, Nir! Åsa, tack för att du läste min avhandling. Karin tack för allt du har lärt mig, och alla samtal. Jag kommer aldrig glömma vår resa till Japan med KatrinJ. Jätte kul att vi blir klara samtidigt. Lycka till med allt! Nina, vi har undervisat, rest och spenderat många luncher tillsammans det enda som fattades var att jobba ihop. Men vem vet, kanske i framtiden J tack för alla goda minnen, speciellt roadtripen! Florian thank you for always sharing papers and your knowledge! My dear Salome I miss you so much but even though you’re far away I can still talk to you about everything, thank you for always listening, te quiero mucho <3! Susanna, ditt sätt att prata science har alltid varit inspirerande vilket man har saknat! Tack för alla dina tips. The GvH people from the past and present: Thank you for the all the good times!! Luigi, Grant, Renuka!! Rickard, tack för alla roliga stunder vi har haft i och utanför DBB. Och för våra långa konversationer som kan handla om vad som helst J. Bill, muchas gracias för din eviga uppmuntran och för den sköna musiken som hördes ända in i vårt labb. Carmen gracias por tu compañia que mucho se aprecia! Pili!! Gracias chica por el apoyo en especial este año de estrés.!! Thank you so much Nurzian for all this time!! First for introducing me to salsa, not the one that I was used to, and also for introducing me to all your nice Singaporean friends: BJ, Kyle, Mariam, Sharon, will never forget the trip to Beijing and the Great Wall, it was amazing! Jimmie!! The pole-dancing queen!! Lol To my students I wish you all the best! Manuel, Laura, Lu. Nassim lycka till med jobbsökandet!. Aurora, mi niña!! Te tome un monton de cariño por ser tan especial J. And to people that worked in our lab for shorter period: Carlos fue demasiado entretenido tenerte con nosotros! Sweet Flavia you are soon leaving! Good luck! Maikel, my crazy friend J I will visit you soon! The GvH community: Jan-Willem group that is always so nice! Zhe, Grietje. Anna, kommer aldrig glömma alla fina minnen från och utanför DBB. Hälsosamma minnen som vårruset men även roliga som spybarJ. Thomas you always look so happy, keep up with the good energy! The cool group of Dan 58 D!! Rageia. Claudio, it’s always fun to talk to you and also danceJ. Kiavash, blir glad av att se hur mycket du har utvecklats från Biokemi 1 till nu!! Stephen, alltid lugn och snäll J. Mikaela tack för du delar med dig av din erfarenhet. Kimmo always so kind!! Söta Hena! Alltid lugn och ordentligJ. David Drew group: Mathieu, Emmanuel, Abdul, Povilas. Neighbours in DBB!!! Rob Daniels, thank you for letting me work in your lab and for nice discussions!! Johan tack för tålamodet att visa mig vad allt ligger i ert labb! Diogo, Hao, Dan you guys are always so nice!! Candan my friend! Thank you for your support and for our fika times, the Turkish coffee that I loveJ! Catarina you are really missed! Your kindness and energy is irreplaceable! Erika tack för alla dina kloka ord när man behövde de som mest! The cute couple Pedro and Beata!! Emelie, din glädje i DBBs korridorer är verkligen uppfriskande, du får mig alltid att le J. Martin Ott and group, for always being so kind! Lisa tack för alla samtal!! Sylvia och Patrik, kommer aldrig glömma SpetsesJ Nikolas, for the nice talks in the corridor! Mina tjejer från stora tentan pluggandet och undervisning! Narges, Gabriella, Minttu! Det är alltid så härligt att umgås med er, allt ni delar med er men även för att ha lyssnat på mina issues. Det har alltid varit skönt att kunna fly till er om jag behöver det J. Många kramar, ni är bäst <3!! Till alla på sekretariatet, tack för att ni har varit så hjälpsamma! Ann, Lotta, Maria, Alex, Anita och Haidi! Får inte heller glömma, Håkan, Peter och Torbjörn för att alltid ställa upp om det är något man behöver!! Människor utanför DBB som nog aldrig har riktigt förstått vad jag har jobbat medJ! Kalle för att du trodde på mig och gav mig stödet jag behövde för att påbörja det här, fick lära mig otroligt mycket av dig. Önskar dig all lycka! Fatma, una de las personas mas linda que he conocido! Gracias por tus consejos cuando mas lo necesitaba, BESO! Rojda, min kloka vän, vet inte vad jag hade gjort utan dig! Älskar våra samtal!! Love YOU <3! Mina barndomsvänner Evelyn och Pabla, ni har alltid funnits där J. Mina fina brudar Claudia och Hevi! puss på er! Mi amiga de siempre y prima Claudia TQM! Mis amigas del baile, que siempre me alegran y me hacen pensar en otras cosas fuera del trabajo J Yasmin, mi amore siempre tan alegre y linda! Te quiero mucho! Sihomara, la India fue increible, gracias por haber compartido ese viaje conmigo. Veronica! Shirley! Lissan! Pamela! las quiero mucho 59 y aprecio mucho las conversaciones con uds! Mina bachata killar, Hebbe, Felipe, Johan, Oskar y Luis - gracias por haber estado ahi cuando lo necesitaba y mi primito que mucho quiero Miguel! Sammy för att du glädjer min vardag! Mis pastelesJ!! Jocelyn y Kenia gracias por todos los buenos momentos, uds saben ;)! La familia Lara y Vasquez aqui en Suecia y tambien en Chile!! Tios, tias y primitos e hijos de los primos! No puedo nombrarlos a todos porque son muchos J!! Pero los quiero a todos!! Mi familia que lo son todo! Mis hermanitos Ivan y Marite, estoy muy orgullasa de uds y los amo! Muchas gracias por mis sobrinos preciosos Jamilia y Matthias J y sus papitos Sinan y Alicia. Mamita y papito, gracias por haber siempre confiado en mi! Sin uds no estaria aqui! Los amo mucho!! Mi padre celestial que me da la sabiduria y fuerza <3! 60 References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. Singer, S. J. & Nicolson, G. L. (1972). The fluid mosaic model of the structure of cell membranes. Science 175, 720-31. Engelman, D. M. (2005). Membranes are more mosaic than fluid. Nature 438, 578-80. Goni, F. M. (2014). The basic structure and dynamics of cell membranes: an update of the Singer-Nicolson model. Biochim Biophys Acta 1838, 1467-76. Raetz, C. R. & Dowhan, W. (1990). Biosynthesis and function of phospholipids in Escherichia coli. J Biol Chem 265, 1235-8. Sud, M., Fahy, E., Cotter, D., Brown, A., Dennis, E. A., Glass, C. K., Merrill, A. H., Jr., Murphy, R. C., Raetz, C. R., Russell, D. W. & Subramaniam, S. (2007). LMSD: LIPID MAPS structure database. Nucleic Acids Res 35, D527-32. Suetsugu, S., Kurisu, S. & Takenawa, T. (2014). Dynamic shaping of cellular membranes by phospholipids and membrane-deforming proteins. Physiol Rev 94, 1219-48. Pomorski, T. G., Nylander, T. & Cardenas, M. (2014). Model cell membranes: discerning lipid and protein contributions in shaping the cell. Adv Colloid Interface Sci 205, 207-20. van Meer, G., Voelker, D. R. & Feigenson, G. W. (2008). Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9, 112-24. Daleke, D. L. (2003). Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res 44, 233-42. Fairn, G. D., Schieber, N. L., Ariotti, N., Murphy, S., Kuerschner, L., Webb, R. I., Grinstein, S. & Parton, R. G. (2011). Highresolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J Cell Biol 194, 257-75. Schneiter, R., Brugger, B., Sandhoff, R., Zellnig, G., Leber, A., Lampl, M., Athenstaedt, K., Hrastnik, C., Eder, S., Daum, G., Paltauf, F., Wieland, F. T. & Kohlwein, S. D. (1999). Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J Cell Biol 146, 741-54. Bigay, J. & Antonny, B. (2012). Curvature, lipid packing, and electrostatics of membrane organelles: defining cellular territories in determining specificity. Dev Cell 23, 886-95. 61 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 62 Hawes, C., Kiviniemi, P. & Kriechbaumer, V. (2014). The Endoplasmic reticulum: A dynamic and well connected organelle. J Integr Plant Biol. von Heijne, G. (2007). The membrane protein universe: what's out there and why bother? J Intern Med 261, 543-57. Lacapere, J. J., Pebay-Peyroula, E., Neumann, J. M. & Etchebest, C. (2007). Determining membrane protein structures: still a challenge! Trends Biochem Sci 32, 259-70. Vinothkumar, K. R. & Henderson, R. (2010). Structures of membrane proteins. Q Rev Biophys 43, 65-158. von Heijne, G. (1994). Membrane proteins: from sequence to structure. Annu Rev Biophys Biomol Struct 23, 167-92. White, S. H. & Wimley, W. C. (1999). Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct 28, 319-65. Pautsch, A. & Schulz, G. E. (2000). High-resolution structure of the OmpA membrane domain. J Mol Biol 298, 273-82. Henderson, R. & Unwin, P. N. (1975). Three-dimensional model of purple membrane obtained by electron microscopy. Nature 257, 2832. Deisenhofer, J., Epp, O., Miki, K., Huber, R. & Michel, H. (1985). Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3A resolution. Nature 318, 618-24. White, S. H. (2009). Biophysical dissection of membrane proteins. Nature 459, 344-6. Dumas, F., Lebrun, M. C. & Tocanne, J. F. (1999). Is the protein/lipid hydrophobic matching principle relevant to membrane organization and functions? FEBS Lett 458, 271-7. Baeza-Delgado, C., Marti-Renom, M. A. & Mingarro, I. (2013). Structure-based statistical analysis of transmembrane helices. Eur Biophys J 42, 199-207. Bowie, J. U. (1997). Helix packing in membrane proteins. J Mol Biol 272, 780-9. Jaud, S., Fernandez-Vidal, M., Nilsson, I., Meindl-Beinker, N. M., Hubner, N. C., Tobias, D. J., von Heijne, G. & White, S. H. (2009). Insertion of short transmembrane helices by the Sec61 translocon. Proc Natl Acad Sci U S A 106, 11588-93. Killian, J. A. (1998). Hydrophobic mismatch between proteins and lipids in membranes. Biochim Biophys Acta 1376, 401-15. Sharpe, H. J., Stevens, T. J. & Munro, S. (2010). A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142, 158-69. Johannsson, A., Smith, G. A. & Metcalfe, J. C. (1981). The effect of bilayer thickness on the activity of (Na+ + K+)-ATPase. Biochim Biophys Acta 641, 416-21. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. Cornelius, F. (2001). Modulation of Na,K-ATPase and Na-ATPase activity by phospholipids and cholesterol. I. Steady-state kinetics. Biochemistry 40, 8842-51. Wallin, E., Tsukihara, T., Yoshikawa, S., von Heijne, G. & Elofsson, A. (1997). Architecture of helix bundle membrane proteins: an analysis of cytochrome c oxidase from bovine mitochondria. Protein Sci 6, 808-15. Gratkowski, H., Lear, J. D. & DeGrado, W. F. (2001). Polar side chains drive the association of model transmembrane peptides. Proc Natl Acad Sci U S A 98, 880-5. Walther, T. H. & Ulrich, A. S. (2014). Transmembrane helix assembly and the role of salt bridges. Curr Opin Struct Biol 27, 638. Hermansson, M. & von Heijne, G. (2003). Inter-helical hydrogen bond formation during membrane protein integration into the ER membrane. J Mol Biol 334, 803-9. Heinrich, S. U. & Rapoport, T. A. (2003). Cooperation of transmembrane segments during the integration of a doublespanning protein into the ER membrane. EMBO J 22, 3654-63. Meindl-Beinker, N. M., Lundin, C., Nilsson, I., White, S. H. & von Heijne, G. (2006). Asn- and Asp-mediated interactions between transmembrane helices during translocon-mediated membrane protein assembly. EMBO Rep 7, 1111-6. Lemmon, M. A., Flanagan, J. M., Treutlein, H. R., Zhang, J. & Engelman, D. M. (1992). Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry 31, 12719-25. Lemmon, M. A., Flanagan, J. M., Hunt, J. F., Adair, B. D., Bormann, B. J., Dempsey, C. E. & Engelman, D. M. (1992). Glycophorin A dimerization is driven by specific interactions between transmembrane alpha-helices. J Biol Chem 267, 7683-9. Russ, W. P. & Engelman, D. M. (2000). The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol 296, 911-9. Sal-Man, N., Gerber, D. & Shai, Y. (2014). Proline localized to the interaction interface can mediate self-association of transmembrane domains. Biochim Biophys Acta 1838, 2313-8. Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., Le Trong, I., Teller, D. C., Okada, T., Stenkamp, R. E., Yamamoto, M. & Miyano, M. (2000). Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739-45. London, E. & Shahidullah, K. (2009). Transmembrane vs. nontransmembrane hydrophobic helix topography in model and natural membranes. Curr Opin Struct Biol 19, 464-72. von Heijne, G. (1999). Recent advances in the understanding of membrane protein assembly and structure. Q Rev Biophys 32, 285307. 63 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 64 Killian, J. A. & von Heijne, G. (2000). How proteins adapt to a membrane-water interface. Trends Biochem Sci 25, 429-34. Monne, M., Nilsson, I., Johansson, M., Elmhed, N. & von Heijne, G. (1998). Positively and negatively charged residues have different effects on the position in the membrane of a model transmembrane helix. J Mol Biol 284, 1177-83. Park, E. & Rapoport, T. A. (2012). Mechanisms of Sec61/SecYMediated Protein Translocation Across Membranes. Annu Rev Biophys 41, 21-40. Martinez-Gil, L., Sauri, A., Marti-Renom, M. A. & Mingarro, I. (2011). Membrane protein integration into the endoplasmic reticulum. FEBS J 278, 3846-58. von Heijne, G. (1985). Signal sequences. The limits of variation. J Mol Biol 184, 99-105. Nilsson, I., Whitley, P. & von Heijne, G. (1994). The COOHterminal ends of internal signal and signal-anchor sequences are positioned differently in the ER translocase. J Cell Biol 126, 112732. Walter, P. & Blobel, G. (1981). Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol 91, 557-61. Mary, C., Scherrer, A., Huck, L., Lakkaraju, A. K., Thomas, Y., Johnson, A. E. & Strub, K. (2010). Residues in SRP9/14 essential for elongation arrest activity of the signal recognition particle define a positively charged functional domain on one side of the protein. RNA 16, 969-79. Akopian, D., Shen, K., Zhang, X. & Shan, S. O. (2013). Signal Recognition Particle: An Essential Protein-Targeting Machine. Annu Rev Biochem. Nyathi, Y., Wilkinson, B. M. & Pool, M. R. (2013). Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim Biophys Acta. Guerriero, C. J. & Brodsky, J. L. (2012). The delicate balance between secreted protein folding and endoplasmic reticulumassociated degradation in human physiology. Physiol Rev 92, 53776. du Plessis, D. J., Nouwen, N. & Driessen, A. J. (2011). The Sec translocase. Biochim Biophys Acta 1808, 851-65. Ulbrandt, N. D., Newitt, J. A. & Bernstein, H. D. (1997). The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell 88, 187-96. Plath, K. & Rapoport, T. A. (2000). Spontaneous release of cytosolic proteins from posttranslational substrates before their transport into the endoplasmic reticulum. J Cell Biol 151, 167-78. 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. Panzner, S., Dreier, L., Hartmann, E., Kostka, S. & Rapoport, T. A. (1995). Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell 81, 561-70. High, S., Andersen, S. S., Gorlich, D., Hartmann, E., Prehn, S., Rapoport, T. A. & Dobberstein, B. (1993). Sec61p is adjacent to nascent type I and type II signal-anchor proteins during their membrane insertion. J Cell Biol 121, 743-50. Van den Berg, B., Clemons, W. M., Jr., Collinson, I., Modis, Y., Hartmann, E., Harrison, S. C. & Rapoport, T. A. (2004). X-ray structure of a protein-conducting channel. Nature 427, 36-44. Tsukazaki, T., Mori, H., Fukai, S., Ishitani, R., Mori, T., Dohmae, N., Perederina, A., Sugita, Y., Vassylyev, D. G., Ito, K. & Nureki, O. (2008). Conformational transition of Sec machinery inferred from bacterial SecYE structures. Nature 455, 988-91. Egea, P. F. & Stroud, R. M. (2010). Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proc Natl Acad Sci U S A 107, 17182-7. Pohlschroder, M., Hartmann, E., Hand, N. J., Dilks, K. & Haddad, A. (2005). Diversity and evolution of protein translocation. Annu Rev Microbiol 59, 91-111. Voorhees, R. M., Fernandez, I. S., Scheres, S. H. & Hegde, R. S. (2014). Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell 157, 1632-43. Gogala, M., Becker, T., Beatrix, B., Armache, J. P., Barrio-Garcia, C., Berninghausen, O. & Beckmann, R. (2014). Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506, 107-10. Simon, S. M. & Blobel, G. (1991). A protein-conducting channel in the endoplasmic reticulum. Cell 65, 371-80. Crowley, K. S., Liao, S., Worrell, V. E., Reinhart, G. D. & Johnson, A. E. (1994). Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell 78, 461-71. Kida, Y., Kume, C., Hirano, M. & Sakaguchi, M. (2010). Environmental transition of signal-anchor sequences during membrane insertion via the endoplasmic reticulum translocon. Mol Biol Cell 21, 418-29. Rapoport, T. A. (2007). Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663-9. Park, E. & Rapoport, T. A. (2011). Preserving the membrane barrier for small molecules during bacterial protein translocation. Nature 473, 239-42. Liao, S., Lin, J., Do, H. & Johnson, A. E. (1997). Both lumenal and cytosolic gating of the aqueous ER translocon pore are regulated from inside the ribosome during membrane protein integration. Cell 90, 31-41. 65 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 66 Hamman, B. D., Hendershot, L. M. & Johnson, A. E. (1998). BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell 92, 747-58. Saparov, S. M., Erlandson, K., Cannon, K., Schaletzky, J., Schulman, S., Rapoport, T. A. & Pohl, P. (2007). Determining the conductance of the SecY protein translocation channel for small molecules. Mol Cell 26, 501-9. Zhang, B. & Miller, T. F., 3rd. (2010). Hydrophobically stabilized open state for the lateral gate of the Sec translocon. Proc Natl Acad Sci U S A 107, 5399-404. Plath, K., Mothes, W., Wilkinson, B. M., Stirling, C. J. & Rapoport, T. A. (1998). Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell 94, 795-807. Kalies, K. U., Rapoport, T. A. & Hartmann, E. (1998). The beta subunit of the Sec61 complex facilitates cotranslational protein transport and interacts with the signal peptidase during translocation. J Cell Biol 141, 887-94. Gorlich, D. & Rapoport, T. A. (1993). Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell 75, 615-30. Meacock, S. L., Lecomte, F. J., Crawshaw, S. G. & High, S. (2002). Different transmembrane domains associate with distinct endoplasmic reticulum components during membrane integration of a polytopic protein. Mol Biol Cell 13, 4114-29. Dejgaard, K., Theberge, J. F., Heath-Engel, H., Chevet, E., Tremblay, M. L. & Thomas, D. Y. (2010). Organization of the Sec61 translocon, studied by high resolution native electrophoresis. J Proteome Res 9, 1763-71. Paetzel, M., Dalbey, R. E. & Strynadka, N. C. (2002). Crystal structure of a bacterial signal peptidase apoenzyme: implications for signal peptide binding and the Ser-Lys dyad mechanism. J Biol Chem 277, 9512-9. Paetzel, M., Dalbey, R. E. & Strynadka, N. C. (1998). Crystal structure of a bacterial signal peptidase in complex with a betalactam inhibitor. Nature 396, 186-90. Dalbey, R. E. & Von Heijne, G. (1992). Signal peptidases in prokaryotes and eukaryotes--a new protease family. Trends Biochem Sci 17, 474-8. Lemberg, M. K. & Martoglio, B. (2002). Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol Cell 10, 735-44. Tomita, T. & Iwatsubo, T. (2013). Structural biology of presenilins and signal peptide peptidases. J Biol Chem 288, 14673-80. Martoglio, B. & Golde, T. E. (2003). Intramembrane-cleaving aspartic proteases and disease: presenilins, signal peptide peptidase and their homologs. Hum Mol Genet 12 Spec No 2, R201-6. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. Golde, T. E., Wolfe, M. S. & Greenbaum, D. C. (2009). Signal peptide peptidases: a family of intramembrane-cleaving proteases that cleave type 2 transmembrane proteins. Semin Cell Dev Biol 20, 225-30. Igura, M., Maita, N., Kamishikiryo, J., Yamada, M., Obita, T., Maenaka, K. & Kohda, D. (2008). Structure-guided identification of a new catalytic motif of oligosaccharyltransferase. EMBO J 27, 23443. Kelleher, D. J. & Gilmore, R. (2006). An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R-62R. Chavan, M., Yan, A. & Lennarz, W. J. (2005). Subunits of the translocon interact with components of the oligosaccharyl transferase complex. J Biol Chem 280, 22917-24. Pfeffer, S., Dudek, J., Gogala, M., Schorr, S., Linxweiler, J., Lang, S., Becker, T., Beckmann, R., Zimmermann, R. & Forster, F. (2014). Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat Commun 5, 3072. Tamborero, S., Vilar, M., Martinez-Gil, L., Johnson, A. E. & Mingarro, I. (2011). Membrane insertion and topology of the translocating chain-associating membrane protein (TRAM). J Mol Biol 406, 571-82. Voigt, S., Jungnickel, B., Hartmann, E. & Rapoport, T. A. (1996). Signal sequence-dependent function of the TRAM protein during early phases of protein transport across the endoplasmic reticulum membrane. J Cell Biol 134, 25-35. Gorlich, D., Hartmann, E., Prehn, S. & Rapoport, T. A. (1992). A protein of the endoplasmic reticulum involved early in polypeptide translocation. Nature 357, 47-52. Heinrich, S. U., Mothes, W., Brunner, J. & Rapoport, T. A. (2000). The Sec61p complex mediates the integration of a membrane protein by allowing lipid partitioning of the transmembrane domain. Cell 102, 233-44. Hartmann, E., Gorlich, D., Kostka, S., Otto, A., Kraft, R., Knespel, S., Burger, E., Rapoport, T. A. & Prehn, S. (1993). A tetrameric complex of membrane proteins in the endoplasmic reticulum. Eur J Biochem 214, 375-81. Fons, R. D., Bogert, B. A. & Hegde, R. S. (2003). Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol 160, 529-39. Sommer, N., Junne, T., Kalies, K. U., Spiess, M. & Hartmann, E. (2013). TRAP assists membrane protein topogenesis at the mammalian ER membrane. Biochim Biophys Acta 1833, 3104-11. Wang, L. & Dobberstein, B. (1999). Oligomeric complexes involved in translocation of proteins across the membrane of the endoplasmic reticulum. FEBS Lett 457, 316-22. 67 99. 100. 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. 68 Lee, A. H., Iwakoshi, N. N. & Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23, 7448-59. Schroder, K., Martoglio, B., Hofmann, M., Holscher, C., Hartmann, E., Prehn, S., Rapoport, T. A. & Dobberstein, B. (1999). Control of glycosylation of MHC class II-associated invariant chain by translocon-associated RAMP4. EMBO J 18, 4804-15. Yamaguchi, A., Hori, O., Stern, D. M., Hartmann, E., Ogawa, S. & Tohyama, M. (1999). Stress-associated endoplasmic reticulum protein 1 (SERP1)/Ribosome-associated membrane protein 4 (RAMP4) stabilizes membrane proteins during stress and facilitates subsequent glycosylation. J Cell Biol 147, 1195-204. Ismail, N., Crawshaw, S. G., Cross, B. C., Haagsma, A. C. & High, S. (2008). Specific transmembrane segments are selectively delayed at the ER translocon during opsin biogenesis. Biochem J 411, 495506. Wang, B., Heath-Engel, H., Zhang, D., Nguyen, N., Thomas, D. Y., Hanrahan, J. W. & Shore, G. C. (2008). BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell 133, 1080-92. Kuroiwa, T., Sakaguchi, M., Mihara, K. & Omura, T. (1991). Systematic analysis of stop-transfer sequence for microsomal membrane. J Biol Chem 266, 9251-5. Hessa, T., Kim, H., Bihlmaier, K., Lundin, C., Boekel, J., Andersson, H., Nilsson, I., White, S. H. & von Heijne, G. (2005). Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377-81. Hessa, T., Meindl-Beinker, N. M., Bernsel, A., Kim, H., Sato, Y., Lerch-Bader, M., Nilsson, I., White, S. H. & von Heijne, G. (2007). Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026-30. Bernsel, A., Viklund, H., Falk, J., Lindahl, E., von Heijne, G. & Elofsson, A. (2008). Prediction of membrane-protein topology from first principles. Proc Natl Acad Sci U S A 105, 7177-81. Junne, T., Kocik, L. & Spiess, M. (2010). The hydrophobic core of the Sec61 translocon defines the hydrophobicity threshold for membrane integration. Mol Biol Cell 21, 1662-70. Duong, F. & Wickner, W. (1998). Sec-dependent membrane protein biogenesis: SecYEG, preprotein hydrophobicity and translocation kinetics control the stop-transfer function. EMBO J 17, 696-705. Nishiyama, K., Suzuki, T. & Tokuda, H. (1996). Inversion of the membrane topology of SecG coupled with SecA-dependent preprotein translocation. Cell 85, 71-81. Pitonzo, D. & Skach, W. R. (2006). Molecular mechanisms of aquaporin biogenesis by the endoplasmic reticulum Sec61 translocon. Biochim Biophys Acta 1758, 976-88. 112. 113. 114. 115. 116. 117. 118. 119. 120. 121. 122. 123. 124. Seppala, S., Slusky, J. S., Lloris-Garcera, P., Rapp, M. & von Heijne, G. (2010). Control of membrane protein topology by a single C-terminal residue. Science 328, 1698-700. Hedin, L. E., Ojemalm, K., Bernsel, A., Hennerdal, A., Illergard, K., Enquist, K., Kauko, A., Cristobal, S., von Heijne, G., Lerch-Bader, M., Nilsson, I. & Elofsson, A. (2010). Membrane insertion of marginally hydrophobic transmembrane helices depends on sequence context. J Mol Biol 396, 221-9. Ojemalm, K., Halling, K. K., Nilsson, I. & von Heijne, G. (2012). Orientational preferences of neighboring helices can drive ER insertion of a marginally hydrophobic transmembrane helix. Mol Cell 45, 529-40. Hou, B., Lin, P. J. & Johnson, A. E. (2012). Membrane protein TM segments are retained at the translocon during integration until the nascent chain cues FRET-detected release into bulk lipid. Mol Cell 48, 398-408. Sadlish, H., Pitonzo, D., Johnson, A. E. & Skach, W. R. (2005). Sequential triage of transmembrane segments by Sec61alpha during biogenesis of a native multispanning membrane protein. Nat Struct Mol Biol 12, 870-8. Pitonzo, D., Yang, Z., Matsumura, Y., Johnson, A. E. & Skach, W. R. (2009). Sequence-specific retention and regulated integration of a nascent membrane protein by the endoplasmic reticulum Sec61 translocon. Mol Biol Cell 20, 685-98. Cross, B. C. & High, S. (2009). Dissecting the physiological role of selective transmembrane-segment retention at the ER translocon. J Cell Sci 122, 1768-77. Heijne, G. (1986). The distribution of positively charged residues in bacterial inner membrane proteins correlates with the transmembrane topology. EMBO J 5, 3021-7. Walz, T., Hirai, T., Murata, K., Heymann, J. B., Mitsuoka, K., Fujiyoshi, Y., Smith, B. L., Agre, P. & Engel, A. (1997). The threedimensional structure of aquaporin-1. Nature 387, 624-7. Hedfalk, K., Tornroth-Horsefield, S., Nyblom, M., Johanson, U., Kjellbom, P. & Neutze, R. (2006). Aquaporin gating. Curr Opin Struct Biol 16, 447-56. Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., Chait, B. T. & MacKinnon, R. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69-77. Viklund, H., Granseth, E. & Elofsson, A. (2006). Structural classification and prediction of reentrant regions in alpha-helical transmembrane proteins: application to complete genomes. J Mol Biol 361, 591-603. Kauko, A., Hedin, L. E., Thebaud, E., Cristobal, S., Elofsson, A. & von Heijne, G. (2010). Repositioning of transmembrane alphahelices during membrane protein folding. J Mol Biol 397, 190-201. 69 125. 126. 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 70 Yan, C. & Luo, J. (2010). An analysis of reentrant loops. Protein J 29, 350-4. Bogdanov, M., Heacock, P. N. & Dowhan, W. (2002). A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J 21, 2107-16. Goder, V. & Spiess, M. (2003). Molecular mechanism of signal sequence orientation in the endoplasmic reticulum. EMBO J 22, 3645-53. Devaraneni, P. K., Conti, B., Matsumura, Y., Yang, Z., Johnson, A. E. & Skach, W. R. (2011). Stepwise insertion and inversion of a type II signal anchor sequence in the ribosome-Sec61 translocon complex. Cell 146, 134-47. Shaw, A. S., Rottier, P. J. & Rose, J. K. (1988). Evidence for the loop model of signal-sequence insertion into the endoplasmic reticulum. Proc Natl Acad Sci U S A 85, 7592-6. Goder, V., Bieri, C. & Spiess, M. (1999). Glycosylation can influence topogenesis of membrane proteins and reveals dynamic reorientation of nascent polypeptides within the translocon. J Cell Biol 147, 257-66. Wallin, E. & von Heijne, G. (1998). Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci 7, 1029-38. Beltzer, J. P., Fiedler, K., Fuhrer, C., Geffen, I., Handschin, C., Wessels, H. P. & Spiess, M. (1991). Charged residues are major determinants of the transmembrane orientation of a signal-anchor sequence. J Biol Chem 266, 973-8. von Heijne, G. (1989). Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature 341, 456-8. Fujita, H., Yamagishi, M., Kida, Y. & Sakaguchi, M. (2011). Positive charges on the translocating polypeptide chain arrest movement through the translocon. J Cell Sci 124, 4184-93. Yamagishi, M., Onishi, Y., Yoshimura, S., Fujita, H., Imai, K., Kida, Y. & Sakaguchi, M. (2014). A few positively charged residues slow movement of a polypeptide chain across the endoplasmic reticulum membrane. Biochemistry 53, 5375-83. Johansson, M., Nilsson, I. & von Heijne, G. (1993). Positively charged amino acids placed next to a signal sequence block protein translocation more efficiently in Escherichia coli than in mammalian microsomes. Mol Gen Genet 239, 251-6. van Klompenburg, W., Nilsson, I., von Heijne, G. & de Kruijff, B. (1997). Anionic phospholipids are determinants of membrane protein topology. EMBO J 16, 4261-6. Lerch-Bader, M., Lundin, C., Kim, H., Nilsson, I. & von Heijne, G. (2008). Contribution of positively charged flanking residues to the insertion of transmembrane helices into the endoplasmic reticulum. Proc Natl Acad Sci U S A 105, 4127-32. 139. 140. 141. 142. 143. 144. 145. 146. 147. 148. 149. 150. 151. 152. 153. 154. 155. Goder, V., Junne, T. & Spiess, M. (2004). Sec61p contributes to signal sequence orientation according to the positive-inside rule. Mol Biol Cell 15, 1470-8. Wahlberg, J. M. & Spiess, M. (1997). Multiple determinants direct the orientation of signal-anchor proteins: the topogenic role of the hydrophobic signal domain. J Cell Biol 137, 555-62. Cymer, F., von Heijne, G. & White, S. H. (2014). Mechanisms of Integral Membrane Protein Insertion and Folding. J Mol Biol. Sakaguchi, M., Tomiyoshi, R., Kuroiwa, T., Mihara, K. & Omura, T. (1992). Functions of signal and signal-anchor sequences are determined by the balance between the hydrophobic segment and the N-terminal charge. Proc Natl Acad Sci U S A 89, 16-9. Eusebio, A., Friedberg, T. & Spiess, M. (1998). The role of the hydrophobic domain in orienting natural signal sequences within the ER membrane. Exp Cell Res 241, 181-5. Price, D. L. & Sisodia, S. S. (1998). Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci 21, 479-505. Kowalska, A. (2004). Genetic basis of neurodegeneration in familial Alzheimer's disease. Pol J Pharmacol 56, 171-8. Kim, S. D. & Kim, J. (2008). Sequence analyses of presenilin mutations linked to familial Alzheimer's disease. Cell Stress Chaperones 13, 401-12. Czech, C., Tremp, G. & Pradier, L. (2000). Presenilins and Alzheimer's disease: biological functions and pathogenic mechanisms. Prog Neurobiol 60, 363-84. Masters, C. L., Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L. & Beyreuther, K. (1985). Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82, 4245-9. Dominguez, D. I., Hartmann, D. & De Strooper, B. (2004). BACE1 and presenilin: two unusual aspartyl proteases involved in Alzheimer's disease. Neurodegener Dis 1, 168-74. Yankner, B. A. & Lu, T. (2009). Amyloid beta-protein toxicity and the pathogenesis of Alzheimer disease. J Biol Chem 284, 4755-9. Brown, M. S., Ye, J., Rawson, R. B. & Goldstein, J. L. (2000). Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100, 391-8. Tolia, A. & De Strooper, B. (2009). Structure and function of gamma-secretase. Semin Cell Dev Biol 20, 211-8. Tandon, A. & Fraser, P. (2002). The presenilins. Genome Biol 3, reviews3014. Zhang, X., Li, Y., Xu, H. & Zhang, Y. (2014). The -secretase complex: from structure to function. Front Cell Neurosci 8, 427. El-Gendy, N., Kaviratna, A., Berkland, C. & Dhar, P. (2013). Delivery and performance of surfactant replacement therapies to treat pulmonary disorders. Ther Deliv 4, 951-80. 71 156. 157. 158. 159. 160. 161. 162. 163. 164. 165. 166. 167. 72 Haagsman, H. P. & Diemel, R. V. (2001). Surfactant-associated proteins: functions and structural variation. Comp Biochem Physiol A Mol Integr Physiol 129, 91-108. Brasch, F., Griese, M., Tredano, M., Johnen, G., Ochs, M., Rieger, C., Mulugeta, S., Muller, K. M., Bahuau, M. & Beers, M. F. (2004). Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene. Eur Respir J 24, 30-9. Thurm, T., Kaltenborn, E., Kern, S., Griese, M. & Zarbock, R. (2013). SFTPC mutations cause SP-C degradation and aggregate formation without increasing ER stress. Eur J Clin Invest 43, 791800. Willander, H., Askarieh, G., Landreh, M., Westermark, P., Nordling, K., Keranen, H., Hermansson, E., Hamvas, A., Nogee, L. M., Bergman, T., Saenz, A., Casals, C., Aqvistg, J., Jornvall, H., Berglund, H., Presto, J., Knight, S. D. & Johansson, J. (2012). Highresolution structure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lung surfactant protein C. Proc Natl Acad Sci U S A 109, 2325-9. Nogee, L. M., Dunbar, A. E., 3rd, Wert, S. E., Askin, F., Hamvas, A. & Whitsett, J. A. (2001). A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 344, 573-9. Beers, M. F. & Mulugeta, S. (2005). Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol 67, 663-96. Keller, A., Eistetter, H. R., Voss, T. & Schafer, K. P. (1991). The pulmonary surfactant protein C (SP-C) precursor is a type II transmembrane protein. Biochem J 277 ( Pt 2), 493-9. Mulugeta, S. & Beers, M. F. (2006). Surfactant protein C: its unique properties and emerging immunomodulatory role in the lung. Microbes Infect 8, 2317-23. Szyperski, T., Vandenbussche, G., Curstedt, T., Ruysschaert, J. M., Wuthrich, K. & Johansson, J. (1998). Pulmonary surfactantassociated polypeptide C in a mixed organic solvent transforms from a monomeric alpha-helical state into insoluble beta-sheet aggregates. Protein Sci 7, 2533-40. Nerelius, C., Fitzen, M. & Johansson, J. (2010). Amino acid sequence determinants and molecular chaperones in amyloid fibril formation. Biochem Biophys Res Commun 396, 2-6. Johansson, J., Some, M., Linderholm, B. M., Almlen, A., Curstedt, T. & Robertson, B. (2003). A synthetic surfactant based on a polyLeu SP-C analog and phospholipids: effects on tidal volumes and lung gas volumes in ventilated immature newborn rabbits. J Appl Physiol (1985) 95, 2055-63. Johansson, H., Nerelius, C., Nordling, K. & Johansson, J. (2009). Preventing amyloid formation by catching unfolded transmembrane segments. J Mol Biol 389, 227-9. 168. 169. 170. 171. 172. 173. 174. 175. 176. 177. 178. 179. 180. Willander, H., Hermansson, E., Johansson, J. & Presto, J. (2011). BRICHOS domain associated with lung fibrosis, dementia and cancer--a chaperone that prevents amyloid fibril formation? FEBS J 278, 3893-904. Arzumanyan, A., Reis, H. M. & Feitelson, M. A. (2013). Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 13, 123-35. Hann, H. W., Lee, J., Bussard, A., Liu, C., Jin, Y. R., Guha, K., Clayton, M. M., Ardlie, K., Pellini, M. J. & Feitelson, M. A. (2004). Preneoplastic markers of hepatitis B virus-associated hepatocellular carcinoma. Cancer Res 64, 7329-35. Lian, Z., Liu, J., Pan, J., Satiroglu Tufan, N. L., Zhu, M., Arbuthnot, P., Kew, M., Clayton, M. M. & Feitelson, M. A. (2001). A cellular gene up-regulated by hepatitis B virus-encoded X antigen promotes hepatocellular growth and survival. Hepatology 34, 146-57. Pan, J., Lian, Z., Wallett, S. & Feitelson, M. A. (2007). The hepatitis B x antigen effector, URG7, blocks tumour necrosis factor alphamediated apoptosis by activation of phosphoinositol 3-kinase and beta-catenin. J Gen Virol 88, 3275-85. Chen, Z. S. & Tiwari, A. K. (2011). Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J 278, 3226-45. Bergen, A. A., Plomp, A. S., Hu, X., de Jong, P. T. & Gorgels, T. G. (2007). ABCC6 and pseudoxanthoma elasticum. Pflugers Arch 453, 685-91. Wolfe, P. B., Silver, P. & Wickner, W. (1982). The isolation of homogeneous leader peptidase from a strain of Escherichia coli which overproduces the enzyme. J Biol Chem 257, 7898-902. Tschantz, W. R., Paetzel, M., Cao, G., Suciu, D., Inouye, M. & Dalbey, R. E. (1995). Characterization of a soluble, catalytically active form of Escherichia coli leader peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935-41. Lundin, C., Kim, H., Nilsson, I., White, S. H. & von Heijne, G. (2008). Molecular code for protein insertion in the endoplasmic reticulum membrane is similar for N(in)-C(out) and N(out)-C(in) transmembrane helices. Proc Natl Acad Sci U S A 105, 15702-7. Elofsson, A. & von Heijne, G. (2007). Membrane protein structure: prediction versus reality. Annu Rev Biochem 76, 125-40. von Heijne, G. (1992). Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol 225, 487-94. Ott, C. M. & Lingappa, V. R. (2002). Integral membrane protein biosynthesis: why topology is hard to predict. J Cell Sci 115, 20039. 73 181. 182. 183. 184. 185. 186. 187. 188. 189. 190. 191. 192. 193. 194. 195. 74 Tusnady, G. E. & Simon, I. (1998). Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol 283, 489-506. Jones, D. T. (2007). Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23, 538-44. Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305, 567-80. Bernsel, A., Viklund, H., Hennerdal, A. & Elofsson, A. (2009). TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res 37, W465-8. Viklund, H. & Elofsson, A. (2008). OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24, 1662-8. Lee, H. & Kim, H. (2014). Membrane topology of transmembrane proteins: determinants and experimental tools. Biochem Biophys Res Commun 453, 268-276. van Geest, M. & Lolkema, J. S. (2000). Membrane topology and insertion of membrane proteins: search for topogenic signals. Microbiol Mol Biol Rev 64, 13-33. Schwarz, F. & Aebi, M. (2011). Mechanisms and principles of Nlinked protein glycosylation. Curr Opin Struct Biol 21, 576-82. Shakin-Eshleman, S. H., Spitalnik, S. L. & Kasturi, L. (1996). The amino acid at the X position of an Asn-X-Ser sequon is an important determinant of N-linked core-glycosylation efficiency. J Biol Chem 271, 6363-6. Mellquist, J. L., Kasturi, L., Spitalnik, S. L. & Shakin-Eshleman, S. H. (1998). The amino acid following an asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37, 6833-7. Chavez, R. A. & Hall, Z. W. (1991). The transmembrane topology of the amino terminus of the alpha subunit of the nicotinic acetylcholine receptor. J Biol Chem 266, 15532-8. Nilsson, I. M. & von Heijne, G. (1993). Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J Biol Chem 268, 5798-801. Stefansson, A., Armulik, A., Nilsson, I., von Heijne, G. & Johansson, S. (2004). Determination of N- and C-terminal borders of the transmembrane domain of integrin subunits. J Biol Chem 279, 21200-5. Wu, C. C., MacCoss, M. J., Howell, K. E. & Yates, J. R., 3rd. (2003). A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol 21, 532-8. Shimomura, O., Johnson, F. H. & Saiga, Y. (1962). Extraction, purification and properties of aequorin, a bioluminescent protein 196. 197. 198. 199. from the luminous hydromedusan, Aequorea. J Cell Comp Physiol 59, 223-39. Feilmeier, B. J., Iseminger, G., Schroeder, D., Webber, H. & Phillips, G. J. (2000). Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol 182, 4068-76. Drew, D., Sjostrand, D., Nilsson, J., Urbig, T., Chin, C. N., de Gier, J. W. & von Heijne, G. (2002). Rapid topology mapping of Escherichia coli inner-membrane proteins by prediction and PhoA/GFP fusion analysis. Proc Natl Acad Sci U S A 99, 2690-5. Rapp, M., Drew, D., Daley, D. O., Nilsson, J., Carvalho, T., Melen, K., De Gier, J. W. & Von Heijne, G. (2004). Experimentally based topology models for E. coli inner membrane proteins. Protein Sci 13, 937-45. Lee, H., Min, J., von Heijne, G. & Kim, H. (2012). Glycosylatable GFP as a compartment-specific membrane topology reporter. Biochem Biophys Res Commun 427, 780-4. 75



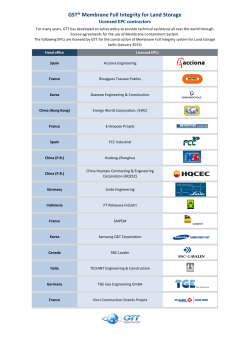






© Copyright 2026