
C
CONTENT EDITORIAL Dilated cardiomyopathy G. Jackson 3 BASIC ARTICLE Myocardial energy metabolism in dilated cardiomyopathy W. C. Stanley 5 MAIN CLINICAL ARTICLE Drugs and/or devices in dilated cardiomyopathy H. P. Schultheiss and U. Kühl 9 METABOLIC IMAGING Metabolic imaging in dilated cardiomyopathy S. Neubauer 13 NEW THERAPEUTIC APPROACHES Metabolic modulation in dilated cardiomyopathy H. Tuunanen and J. Knuuti 17 FOCUS ON TRIMETAZIDINE (VASTAREL MR) The clinical benefits provided by trimetazidine in left ventricular dysfunction patients G Marazzi, G. Caminiti and M. Voterrani 21 CASE REPORT Response to cardiac resynchronization therapy resulting from an upgrade to dual-site left ventricular pacing M. R. Ginks, S. G. Duckett, G. S. Carr-White and C. A. Rinaldi 25 REFRESHER CORNER Cardiac amyloidosis P. Schmidheiny and O. M. Hess 29 HOT TOPICS Low diagnostic yield of coronary angiography or not catching up with heart disease pathophysiology? A Huqi 33 GLOSSARY G. D. Lopaschuk 37 Editorial Dilated cardiomyopathy Graham Jackson Honorary Consultant Cardiologist, Guy’s and St Thomas’ Hospitals NHS Trust, London, UK Correspondence: Graham Jackson, Suite 301 Emblem House, London Bridge Hospital, 27 Tooley Street, London SE1 2PR, UK. Tel: +44 207 407 5887; e-mail: [email protected] Dilated cardiomyopathy (DCM) refers to chronic disease of the heart muscle leading to cavity enlargement and reduced systolic function of the left or both ventricles. The diagnosis follows on from the exclusion of specific causes including hypertension, coronary artery disease, rapid supraventricular arrhythmias, congenital heart disease, pericardial disease, cor pulmonale, and systemic disease such as sarcoidosis. In addition, though alcohol excess can lead to a DCM, abstinence may lead to reversal, so it is only included if a minimum of six months of complete abstinence still leaves a DCM at echocardiography. The prevalence of DCM in the United States (adjusted for age) is 36 per 100,000 of the population. The etiology includes genetic transmission (estimated at 30–40%) indentifying familial DCM, cytotoxic agents (e.g., anthracycline derivatives), malnutrition (e.g., protein deficiency), myocarditis (viral etiology), and autoimmune disease. The five-year survival averages 30–40% and has been improved by modern heart failure therapy, but 20% will die within a year of the diagnosis due to sudden death in most instances (64%) and heart failure deterioration in the remainder. Therefore not everyone responds and some patients rapidly deteriorate no matter the therapeutic efforts, and for them, if suitable, heart transplantation is the only option. Beta blockade and angiotensin-converting enzyme (ACE) inhibitors or angiotensin II antagonists improve left ventricular function in 50% of cases, and in 16% it returns to normal, but the remaining 34% suffer longterm disease progression. In this issue we focus on DCM, opening up the door to metabolic mechanisms and potential therapies that Heart Metab. 2011; 49:3 may improve symptoms and prognosis. Metabolic agents such as trimetazidine can optimize myocardial energy metabolism, leading to increased energy from glucose rather than free fatty acids. The link between understanding energy metabolism and selecting therapy using pharmacology or devices is thoroughly explored and offers therapeutic options that may be complimentary rather than alternatives. The value of imaging in diagnosis and monitoring therapy reveals once more a potential benefit from the metabolic agent trimetazidine (a partial free fatty acid betaoxidation inhibitor) both symptomatically and potentially prognostically, and the need for large-scale multicenter outcome studies. Cardiac amyloidosis is almost certainly under diagnosed so it is timely that it is reviewed as part of this DCM issue. Though the prognosis is poor, determining the precise etiology can lead to targeted therapy and improved survival. Throughout this issue of Heart and Metabolism, DCM is recognized as a challenging condition. By combining an understanding of the etiology, metabolic mechanisms, detection and therapeutic options, we explore the means of rising to the challenge Further reading Key references are provided with each article. A comprehensive review is provided in Hess OM, McKenna W, Schultheiss H-P (2009) Myocardial Disease. In: Camm AJ, Luscher TF, Serruys PW (eds.) ESC Textbook of Cardiovascular Medicine. Oxford University Press, Oxford, pp 665–716. 3 Basic article Myocardial energy metabolism in dilated cardiomyopathy William C. Stanley Division of Cardiology, Department of Medicine, School of Medicine, University of Maryland, Baltimore, Maryland, USA Correspondence: Professor William C. Stanley, Division of Cardiology, Department of Medicine, School of Medicine, University of Maryland, 20 Penn St, HSF-II, Room S022, Baltimore, MD 21201, USA. Tel: +1 410 706 3585; fax: +1 410 706 3583; e-mail: [email protected] Abstract Dilated cardiomyopathy can be broadly defined as heart failure with an enlarged left ventricular chamber, but with no evidence of ischemic heart disease or myocardial infarction. Recent studies from patients and animal models show that dilated cardiomyopathy results in a decrease in myocardial content of adenosine-50 -triphosphate (ATP) and phosphocreatine and a decrease in the capacity for mitochondrial oxidative metabolism and aerobic ATP production. There is a switch in the source of fuel, with a decrease in the rate of myocardial fatty acid oxidation and greater glucose oxidation. Pharmacologically preventing the decrease in fatty acid oxidation with a peroxisome proliferatoractivated receptor-a agonist does not appear to be beneficial. On the other hand, treatment with partial inhibitors of myocardial fatty acid oxidation has favorable effects on cardiac energy metabolism, contractile function, and structure. Heart Metab. 2011;49:5–8. Keywords: fatty acids, glucose, heart failure, mitochondria Introduction Cardiac power generation requires a high rate of conversion of chemical energy from bloodborne substrates (fatty acids, glucose, and lactate) to contractile power (Figure 1). Inability to meet the energy requirement results in heart failure, which is most clearly manifest by an inability to perform physical exercise. Most forms of heart failure are associated with a history of cardiac ischemia, myocardial infarction, hypertension, and/or left ventricular (LV) hypertrophy, with either a normal or decreased ejection fraction. Heart failure can also present with a dilated LV and low ejection fraction, but without the classic history of ischemia, infarction, or hypertension. This condition is broadly referred to as dilated cardiomyopathy (or ‘‘idiopathic dilated cardiomyopathy’’); it has no clear cause, yet a poor prognosis similar to other forms of heart failure. Dilated cardiomyopathy can be caused by inherited metabolic defects, specifically those in Heart Metab. 2011; 49:5–8 mitochondrial oxidative metabolism, or may be acquired, such as with chronic tachycardia, pregnancy, exposure to toxins, alcoholism, or diabetes. Several adaptations in myocardial substrate metabolism have been described in response to dilated cardiomyopathy. This brief overview will highlight recent findings related to myocardial energy metabolism in dilated cardiomyopathy. Cardiac energy metabolism in dilated cardiomyopathy The energy for cardiac mechanical work and relaxation comes from adenosine-5’-triphosphate (ATP), which is broken down to adenosine diphosphate (ADP) and inorganic phosphate to fuel contraction, and Ca2þ uptake into the sarcoplasmic reticulum to initiate diastolic relaxation (Figure 1). Oxidative phosphorylation rapidly resynthesizes ATP in the 5 Basic article William C. Stanley Contraction SR Ca2+ pump Ion homeostatis ATPases CK ADP + P i PCr O2 down of phosphocreatine (PCr) by creatine kinase (CK) can rapidly synthesize ATP (Figure 1). Hence ATP content stays relatively constant even with abrupt increases in LV power output. Long-chain fatty acids (primarily oleate, palmitate, and linoleate) are a healthy human heart’s main fuels, supplying approximately 50% to 80% of the energy requirement under post-absorptive conditions. The balance is from pyruvate oxidation, which is derived in roughly equal proportions from glucose and lactate. Under normal conditions the myocardium has tremendous metabolic flexibility, and is able to generate a maximal rate of oxygen consumption, ATP formation, and cardiac power using either fatty acids or carbohydrates as the sole substrate [1]. Patients with advanced dilated cardiomyopathy have poor systolic and diastolic function, and switch the fuel for cardiac metabolism away from fatty acid oxidation toward greater glucose oxidation [2,3]. This is illustrated in Figure 2, which shows a reduction in myocardial free fatty acid oxidation, and greater glucose uptake by the myocardium in patients with dilated cardiomyopathy. Substrate metabolism was assessed directly using 3H-oleate to measure fatty acid oxidation, and arterial and coronary sinus catheterization to assess transmyocardial substrate uptake. Similar results were found using noninvasive positron emission tomography [3], and also in the canine tachycardia model of dilated cardiomyopathy [4– 6]. Moreover, the decline in fatty acid oxidation was inversely related to the extent of LV dilation (Figure 3). When the hearts of patients with dilated cardiomyopathy were stressed by an acute bout of atrial pacing at 130 beats/min (Figure 2), there was not the normal increase in myocardial glucose uptake seen in the controls, suggesting that there is a loss of ‘‘metabolic flexibility’’ in these patients. There is growing evidence to suggest that defective mitochondrial ATP generation contributes to cardiac Cytosol ATP Cr + P i Oxidative phosphorylation NAD + NADH Mitochondrial metabolism Fatty acids pyruvate CO2 Mitochondria Glucose lactate Figure 1. Schematic depiction of cardiac energy metabolism. Adenosine-50 -triphosphate (ATP) is broken down in the cytosole to fuel the contractile work of systole, Ca2þ uptake into the sarcoplasmic reticulum, which ends systole and initiates relaxation and diastolic filling, and for ion pumps on the sarcolemma. ATP is resynthesized in the mitochondria by oxidative phosphorylation, which is fueled by electrons that are transferred to NADH with the metabolism of fatty acid and pyruvate in the mitochondrial matrix (ADP adenosine diphosphate, CK creatine kinase, Cr creatine, PCr phosphocreatine, NADH nicotinamide adenine dinucleotide). mitochondria. This process is fueled by electrons generated by metabolism of carbon substrates, namely fatty acids and pyruvate, in the mitochondrial matrix, which passes electrons to nicotinamide adenine dinucleotide (NADH) and the electron transport chain. Here oxygen is consumed and oxidative phosphorylation resynthesizes ATP (Figure 1). Normally the healthy myocardium sets the blood flow, oxygen delivery, mitochondrial metabolism, NADH formation, and ATP synthesis to match need for ATP breakdown. When there is a brief mismatch (i.e., during the transition from rest to exercise), the break- Dilated cardiomyopathy Control patients 0.20 Free fatty acid oxidation 0.20 # # 0.10 0.05 0.05 0.00 * 0.15 Baseline Pacing 0.00 Lactate uptake 0.20 µmol g −1 min −1 0.10 µmol g −1 min −1 µmol g −1 min −1 # 0.15 0.25 Glucose uptake 0.15 0.10 0.05 Baseline Pacing 0.00 Baseline Pacing Figure 2. Myocardial substrate metabolism in control patients and patients with dilated cardiomyopathy. Figure drawn from data presented in Neglia et al (2007). Am J Physiol Heart Circ Physiol 293:H3270–H3278 [2]. 6 Heart Metab. 2011; 49:5–8 Basic article Myocardial energy metabolism in dilated cardiomyopathy Free fatty acid uptake (µmol*min−1*g −1) 0.25 Control patients Dilated cardiomyopathy 0.20 0.15 0.10 0.05 R = − 0.70 P < 0.01 0.00 45 50 55 60 65 70 75 LV end diastolic diameter (mm) Figure 3. Myocardial fatty acid uptake plotted as a function of left ventricular end diastolic diameter. Figure drawn from data presented in Neglia et al (2007). Am J Physiol Heart Circ Physiol 293:H3270–H3278 [2]. dysfunction in patients with dilated cardiomyopathy. In the canine model, there is a decrease in myocardial ATP, PCr and creatine (Cr) during the progression to severe dilated end-stage heart failure [7]. Many studies found that heart failure results in a decrease in the capacity for mitochondrial ATP generation at the level of oxidative phosphorylation, particularly seen in a decline in the electron transport chain’s integrative function [1,8,9]. Unfortunately, it is very difficult to assess mitochondrial function in samples from normal humans and patients with dilated cardiomyopathy, thus we do not have a clear understanding of the precise molecular mechanisms responsible for the decrease is ATP and impaired oxidative phosphorylation in this condition. At present, it is not clear whether these alterations are the cause or consequence of dilated cardiomyopathy. Clearly there are relatively rare inherited defects in mitochondrial metabolism that present as dilated cardiomyopathy [10], however this does not explain the vast majority of cases. In general, it appears that the mitochondrial defects and the switch in substrate metabolism are acquired over the course of the disorder’s development and progression. While it is clear that it is important to maintain mitochondrial ATPgenerating capacity in dilated cardiomyopathy, it is less clear if it is important to maintain the normal high rate of fatty acid oxidation. Maintaining myocardial fatty acid oxidation over the progression of tachycardia-induced dilated cardiomyopathy using fenofibrate, an agonist for peroxisome proliferatoractivated receptor-a, did not improve cardiac function or slow the progression of heart failure [11]. On the other hand, treatment with oxfenicine, an inhibitor of myocardial fatty acid oxidation at the level of carnitine palmitoyl transferase I, prevented LV dilation and delayed decompensated heart failure in this model [12]. This approach has not been evaluated specifically in patients with dilated cardiomyopathy, Heart Metab. 2011; 49:5–8 but small clinical studies in heart-failure patients have shown promising results with the partial fatty acid oxidation inhibitors perhexiline [13] or trimetazidine [14–17]. These finding suggest that the decline in fatty acid oxidation in dilated cardiomyopathy is not detrimental, and inhibition of fatty acid oxidation has potential as a therapy for this condition. In summary, dilated cardiomyopathy results in a decreased capacity for mitochondrial oxidative metabolism and aerobic ATP production, and a decrease in the rate of myocardial fatty acid oxidation and increased glucose oxidation. Pharmacologically preventing the decrease in fatty acid oxidation with peroxisome proliferator-activated receptor-a agonist does not appear to be beneficial, but treatment with partial inhibitors of myocardial fatty acid oxidation has favorable effects on cardiac energy metabolism, contractile function, and structure. REFERENCES 1. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85 (3):1093–1129. 2. Neglia D, De CA, Marraccini P, et al. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2007;293 (6):H3270–H3278. 3. Davila-Roman VG, Vedala G, Herrero P, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40 (2):271– 277. 4. Recchia FA, McConnell PI, Bernstein RD, Vogel TR, Xu X, Hintze TH. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacinginduced heart failure in the conscious dog. Circ Res. 1998;83 (10):969–979. 5. Osorio JC, Stanley WC, Linke A, et al. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation. 2002;106 (5):606–612. 6. Lei B, Lionetti V, Young ME, et al. Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J Mol Cell Cardiol. 2004;36 (4):567– 576. 7. Shen W, Asai K, Uechi M, et al. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation. 1999;100 (20):2113–2118. 8. Rosca MG, Vazquez EC, Kerner J, et al. Cardiac mitochondria in coronary microembolization-induced heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80:30–39. 9. Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000;32 (12):2361–2367. 10. Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330 (13):913–919. 11. Labinskyy V, Bellomo M, Chandler MP, et al. Chronic activation of PPAR{alpha} with fenofibrate prevents alterations in cardiac metabolic phenotype without changing the onset of decompensation in pacing-induced heart failure. J Pharmacol Exp Ther. 2007;321 (1):165–171. 12. Lionetti V, Linke A, Chandler MP, et al. Carnitine palmitoyl transferase-I inhibition prevents ventricular remodeling and delays decompensation in pacing-induced heart failure. Cardiovasc Res. 2005;66 (3):454–461. 7 Basic article William C. Stanley 13. Lee L, Campbell R, Scheuermann-Freestone M, et al. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation. 2005;112 (21):3280–3288. 14. Belardinelli R, Purcaro A. Effects of trimetazidine on the contractile response of chronically dysfunctional myocardium to low-dose dobutamine in ischaemic cardiomyopathy. Eur Heart J. 2001;22 (23):2164–2170. 15. Rosano GM, Vitale C, Sposato B, Mercuro G, Fini M. Trimetazidine improves left ventricular function in diabetic patients 8 with coronary artery disease: a double-blind placebocontrolled study. Cardiovasc Diabetol. 2003;2 (1):16. 16. Vitale C, Wajngaten M, Sposato B, et al. Trimetazidine improves left ventricular function and quality of life in elderly patients with coronary artery disease. Eur Heart J. 2004;25 (20):1814–1821. 17. Fragasso G, Palloshi A, Puccetti P, et al. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol. 2006;48 (5):992–998. Heart Metab. 2011; 49:5–8 Main clinical article Drugs and/or devices in dilated cardiomyopathy Heinz-Peter Schultheiss and Uwe Kühl Medizinische Klinik für Kardiologie und Pulmologie, Charité – Universitätsmedizin Berlin, Campus Benjamin Franklin, Hindenburgdamm 30, D-12200 Berlin, Germany Correspondence: Dr Uwe Kühl, Medizinische Klinik II, Cardiology and Pneumonology, Charité – Universitätsmedizin Berlin, Campus Benjamin Franklin, Hindenburgdamm 30 D-12200 Berlin, Germany. Tel: +49 30 8445 4219; fax: +49 30 8445 4219; e-mail: [email protected] Abstract Dilated cardiomyopathy (DCM) is the most common type of nonischemic heart muscle disease, with a prevalence of 36 cases per 100,000 people. In contrast to other heart diseases it often affects the young and presents with reduced systolic function and ventricular dilatation due to infectious, toxic, metabolic, or genetic causes. Regardless of the underlying cause, patients are given standard medication for heart failure in order to improve cardiac function, treat symptoms, and prevent complications. Subgroups of patients with acquired infectious or postinfectious autoimmune diseases can be treated successfully if underlying causes have been characterized carefully. Heart Metab. 2011;49:9–12. Keywords: Anti-viral or immunomodulatory treatment, autoimmunity, dilated cardiomyopathy, heart failure therapy, ICD, immunosuppression, myocarditis Introduction Dilated cardiomyopathy (DCM) is a cardiac muscle disease characterized by reduced contractile function and dilatation of the left or both ventricular chambers [1]. Aside from genetic inheritance and etiologically undefined ‘‘idiopathic’’ cases, DCM is often acquired and caused by infections or toxic agents (e.g., alcoholic cardiomyopathies), or associated with autoimmune diseases, pregnancy, systemic, and other cardiovascular disorders. Since the acquired causes constitute a sizeable proportion of DCM, the observed frequency of the undefined so-called ‘‘idiopathic’’ entities, last but not least, depends on the effort and technical means applied to come to a specific and reliable diagnosis [2,3]. This important issue notably concerns immune, autoimmune and infectious causes due to the fact that subgroups of patients may gain benefit from specific treatment strategies [4]. Uncovering such treatable causes of DCM demands, however, direct analysis of tissue samples obtained by biopsy, because non-invasive clinical tools do not Heart Metab. 2011; 49:9–12 usually provide the required diagnostic accuracy to allow successful intervention (Fig. 1) [5,6]. Symptomatic treatment of heart failure Although conventional pharmacotherapy does not influence specific causes of acquired diseases of viral or immune origin, it remains the hallmark of any heart failure therapy [7]. It does not depend on the disease’s etiology and remains primarily supportive. Despite routine use of angiotensin-converting enzyme (ACE) inhibitors or angiotensine II receptor (ABR) blockers, beta blockers, diuretics, and spironolactone in patients with heart failure due to DCM, these patients still have a considerable annual mortality rate of 5– 10%. A survival benefit has been demonstrated for ACEinhibitors, beta blockers, and spironolactone, which should be administered routinely starting with low doses in all patients with New York Heart Association (NYHA) class II to IV heart failure [8]. Diuretics may improve heart failure symptoms without 9 Main clinical article Heinz-Peter Schultheiss and Uwe Kühl Figure 1. Histology of acute and chronic dilated cardiomyopathy (DCM)/inflammatory cardiomyopathy (DCMi) influencing treatment decisions. (a) Regular myocardial tissue without infiltrating inflammatory cells, fibroses or scars. (b) Active myocarditis. (c) Chronic DCMi without relevant fibrosis or scars. (d) DCM with myocyte hypertrophy and replacement fibrosis (scars). A significant number of patients without irreversible myocardial injury may improve spontaneously (a) or upon immunosuppressive treatment (b, c) if ventricular dysfunction is caused by an acute or chronic inflammatory process. In the later stage of DCM (d) specific treatment at best can prevent inflammation-associated progression of heart failure, but ventricular function will not recover. significant effect on long-term outcome. Despite improvements in the treatment of heart failure in the last 15 years, clinical outcome following the onset of symptoms has not substantially changed. Cardiac transplantation has been considered the last treatment for end-stage heart failure, with DCM as one of the leading causes diagnosed at the time of the operation. Since the necessary number of organs is stagnating or has even declined in some countries over the past years, mechanical left ventricular assist device (LVAD) has been introduced as one promising option to improve prognosis until recovery from acute fulminant disease or to bridge to transplantation in end-stage disease (Fig. 1). Antiarrhythmic treatment Due to their negative inotropic effects, most antiarrhythmic drugs, including newer substances such as dronedarone should be avoided in patients with severe systolic heart failure and an ejection fraction below 35% and acute or worsened heart failure (NYHA IV), respectively. Beta blockers or amiodarone can be used but have to be handled with care in these patients and may prevent sinus tachycardia and different forms of supraventricular or ventricular arrhythmias, respectively. In the long run, however, 10 drugs do not significantly reduce increased mortality due to sudden cardiac death (SCD) caused by tachyarrhythmias, ventricular tachycardia (VT), or ventricular fibrillation (VF). An implantable cardioverter-defibrillator (ICD) is established as the most reliable therapeutic tool and indicated for documented VT/VF or aborted SCD for secondary prevention. If patients with irreversibly impaired ventricles and left ventricular branch block are supplied with an ICD, it can be combined with cardiac resynchronization therapy (CRT) e.g., by biventricular chamber pacing. In early stages of acute disease or DCM due to acquired causes, a wearable defibrillator (LifeVest1) rather than a permanent device is a rational choice for patients with a temporary risk of sudden cardiac arrest to bridge the first critical period if spontaneous or treatment-associated recovery is expected. Biopsy-based specific treatment of acquired disease The gold standard of diagnosing the underlying causes of infectious myocarditis and inflammatory cardiomyopathy (DCMi) is the histological, immunohistological and polymerase chain reaction (PCR)-based analysis of endomyocardial biopsies if its time-dependent and Heart Metab. 2011; 49:9–12 Main clinical article Drugs and/or devices in dilated cardiomyopathy methodological limitations are kept in mind [9]. Both persistent viral infections and infection-associated or postinfectious inflammatory processes of the myocardium have been recognized as independent prognostic parameters—in addition to left and right ventricular dysfunction—in myocarditis and DCM [3,10]. Therefore, in addition to conventional pharmacotherapy specific treatment options are required that directly address the underlying viral or inflammatory causes of the disease (Fig. 1). Immunoadsorption Persisting virus infections and postinfectious myocardial injury may produce antibodies that crossreact with myocardial antigens and the myocyte Fc-receptors thereby contributing to impairment of cardiac contractility [11,12]. Long-term reduction of such cardio-depressive antibodies from sera of patients with refractory heart failure as a result of DCM by immunoadsorption significantly improves ventricular function in many but not all patients [13,14]. The effect of immunoadsorption on longterm outcome is not currently known; a controlled treatment trial is currently being performed. Immunomodulatory treatment Myocardial inflammatory processes or autoimmunity may survive myocardial virus elimination and warrant immunosuppressive treatment in order to prevent later immune-mediated myocardial injury, particularly in the aggressive forms of myocarditis such as giant cell granulomatous myocarditis (Table 1). Frequently administered anti-inflammatory drugs are immunoglobulins, corticosteroids, azathioprine and cyclosporine, which are administered on top of regular heart-failure medication. a-Methylprednisolone is generally given, at a rate of 1 mg/kg body weight, initially for 4 weeks. Depending on the body weight, 100 to 150 mg azathioprine daily can be administered in addition to the steroid medication. The steroid dosage is titrated biweekly in increments of 10 mg until a maintenance dose of 10 mg is reached. The treatment should last for 3 to 6 months. Current data from initial randomized trials confirm efficacy of those treatment regimens in carefully selected patients [15–17]. Both treatment of early disease with frequent spontaneous recovery and the lack of information on the actual virus state due to incomplete diagnostic accuracy have blurred the results of the initial randomized trials [18–20]. More recent studies with exactly characterized cohorts of patients have confirmed earlier data that patients with inflammatory cardiomyopathy respond well to immunosuppression if persistent viral infection is excluded before treatment [15–17]. Anti-infectious treatment Interferons (IFNs) serve as a natural defense against many viral infections. Their innate production is associated with clinical recovery from viral infection and subsequent sequelae while exogenous administration is protective. Type I interferons therefore constitute a promising choice for treatment for selected infectious agents. Data from an IFN-b phase II study provided first evidence that antiviral IFN-b 1a therapy effectively clears cardiac enterovirus and adenovirus infections in patients with chronic heart failure when given subcutaneously every other day in addition to constant heart failure medication [21]. The dosage 4 106 IU and 6 106 IU IFN-b was well tolerated. After 24 weeks of treatment complete elimination of the viral genome was proven in follow-up biopsies in Table 1. Immunosuppressive treatment of patients with myocarditis and inflammatory cardiomyopathy. Muromonab (anti-CD3-antibodies)1 Methylprednisolon Pantoprazole 5 mg/day iv for 7 days 10 mg/kg bw (first 3 days) than 1 mg/kg bw (see below) Trough level: 100–150 mg 50–150 mg/day5 1 mg/kg bw (4 weeks), tapering 10 mg for 6 to 12 months 20 mg/day Calcimagon D31 1 1/day Ciclosporin2 Azathioprine4 (optional) Methylprednisolon3 GCM GCM, Myocarditis: lymphocytic (auto-)immune eosinophilic granulomateous Patients with giant cell myocarditis (GCM) should be treated with anti-CD3-antibodies, cortisone, and ciclosporin immediately after established diagnosis. Virusnegative DCMi or autoimmune myocardites, eosinophilic myocarditis, and granulomatous myocarditis such as sarcoidosis often respond to cortisone alone. Cortisone treatment may have to be replaced by other immunosuppressive drugs in case of side effects or may be extended by azathioprine, if necessary. In every case, therapy should be dispensed for about 6 months. 1Attend initial hypotension and allergic reactions; 2Control liver and kidney parameters; 3Control blood count, glucose; 4Control blood count, liver and kidney parameters; 5Reduce or terminate if leukocyte counts <1000/nl or liver enzymes >3-fold normal value. Heart Metab. 2011; 49:9–12 11 Main clinical article Heinz-Peter Schultheiss and Uwe Kühl all patients. This was paralleled by a significant improvement in left ventricular function and ventricular size and amelioration of heart failure symptoms. None of the patients deteriorated. While entero- and adenoviruses seem to respond well to the interferon-b treatment, subsequent open-labeled studies have shown that other viruses such as erythroviruses, including parvovirus B19, respond less well in terms of virus clearance, although erythrovirus load decreased and patients improved clinically. These data were confirmed by a first recently presented randomized IFN-b treatment study (BICC-trial) [22]. The other viruses might respond in a better way to distinct anti-viral treatment regimens, but optimal treatment conditions for viruses other than enterovirus and adenovirus have not yet been defined. At present, antiviral therapy is still a matter of clinical trials in specialized centers. Conclusion In acute DCM or DCMi, management is first of all supportive and does not aim at the causative agent. Rapid aggressive support of cardiac function including inotropic therapy, mechanical circulatory support, and arrhythmia management may be lifesaving by bridging the interval for return to improved or native ventricular function or providing a bridge to transplantation. Generally a biopsy-based diagnosis is mandatory to initiate any specific treatment, in addition to standard heart failure therapy, in order to improve prognosis in patients with acute and chronic infectious and/or inflammatory disease. REFERENCES 1. Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841– 842. 2. Mangnani JW, Suk Danik HJ, Dec GW, DiSalvo TG. Survival in biopsy-proven myocarditis: A long-term retrospective analysis of the histopathological, clinical, and hemodynamic predictors. Am Heart J. 2006;151:463–470. 3. Caforio AL, Calabrese F, Angelini A, et al. A prospective study of biopsy-proven myocarditis: prognostic relevance of clinical and aetiopathogenetic features at diagnosis. Eur Heart J. 2007;28:1326–1333. 4. Kuhl U, Schultheiss HP. Viral myocarditis: diagnosis, aetiology and management. Drugs. 2009;69:1287–1302. 12 5. Liu PP, Schultheiss HP. Myocarditis. In: Baunwald, ed. Heart Disease. 8 ed. Philadelphia: WB Saunders Co; 2008. pp. 1775–1792. 6. Mangnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation. 2006;113:876–890. 7. Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112:e154–e235. 8. Felker GM, Thompson RE, Hare JM, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–1084. 9. Schultheiss HP, Kuehl U. Cardiovascular Viral Infections. In: Lennette’s Laboratory Diagnosis of Viral Infections, 4 ed. Edited by Lennette . New York: Informa Healthcare USA Inc; 2010. pp. 304–318. 10. Kühl U, Pauschinger M, Seeberg B, et al. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. 2005;112:1965–1970. 11. Caforio AL, Tona F, Bottaro S, et al. Clinical implications of anti-heart autoantibodies in myocarditis and dilated cardiomyopathy. Autoimmunity. 2008;41:35–45. 12. Staudt A, Eichler P, Trimpert C, Felix SB, Greinacher A. Fc(gamma) receptors IIa on cardiomyocytes and their potential functional relevance in dilated cardiomyopathy. J Am Coll Cardiol. 2007;49:1684–1692. 13. Staudt A, Hummel A, Ruppert J, et al. Immunoadsorption in dilated cardiomyopathy: 6-month results from a randomized study. Am Heart J. 2006;152:712.e1–712.e6. 14. Trimpert C, Herda LR, Eckerle LG, et al. Immunoadsorption in DCM: long-term reduction of cardiodepressant antibodies. Eur J Clin Invest. 2010;40:685–691. 15. Frustaci A, Russo MA, Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virusnegative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J. 2009; doi: 10.1093/eurheartj/ehp249. 16. Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: twoyear follow-up results. Circulation. 2001;104:39–45. 17. Kühl U, Strauer BE, Schultheiss HP. Methylprednisolone in chronic myocarditis. Postgrad Med J. 1994;70 (Suppl 1):S35– S42. 18. Mason JW, O’Connell JB, Herskowitz A, et al. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med. 1995;333:269– 275. 19. Frustaci A, Chimenti C, Calabrese F, Pieroni M, Thiene G, Maseri A. Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation. 2003;107:857–863. 20. Stanton C, Mookadam F, Cha S, et al. Greater symptom duration predicts response to immunomodulatory therapy in dilated cardiomyopathy. Int J Cardiol. 2008;128:38–41. 21. Kühl U, Pauschinger M, Schwimmbeck PL, et al. Interferonbeta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. 2003;107:2793–2798. 22. Schultheiss HP, Piper C, Sowade K, et al. The effect of subcutaneous treatment with interferon-beta-1b over 24 weeks on safety, virus elimination and clinical outcome in patients with chronic viral cardiomyopathy. Circulation. 2008;(118):Abstract 3322. Heart Metab. 2011; 49:9–12 Metabolic imaging Metabolic imaging in dilated cardiomyopathy Stefan Neubauer University of Oxford Centre for Clinical Magnetic Resonance Research, Department of Cardiovascular Medicine, John Radcliffe Hospital, Oxford, United Kingdom Correspondence: Professor Stefan Neubauer, Department of Cardiovascular Medicine, John Radcliffe Hospital, Oxford OX3 9DU, UK. Tel: +44 0 1865 851085; fax: +44 0 1865 222077; e-mail: [email protected] Sponsorship: The author is funded by the British Heart Foundation, the Medical Research Council, the Wellcome Trust, and the National Institute for Health Research Biomedical Research Centre Programme. Abstract Cardiac metabolism is deranged in dilated cardiomyopathy (DCM). Phosphorus-31 (31P) and hydrogen1 (1H) magnetic resonance spectroscopy (MRS) allow for the non-invasive assessment of various aspects of cardiac metabolism, showing deranged cardiac energetics and lipid accumulation in DCM. In the future, MRS may become an important biomarker tool for monitoring the effects of novel heart-failure treatments in DCM. Heart Metab. 2011;49:13–16. Keywords: Dilated cardiomyopathy, energetics, magnetic resonance spectroscopy, metabolism, steatosis Introduction Dilated cardiomyopathy (DCM) is a common cause of heart failure, and may be due to many different etiologies (genetic, post myocarditis, toxic, etc.). A common feature of DCM is a derangement of cardiac metabolism. As reviewed elsewhere, metabolic alterations are likely to be a key factor in the pathophysiology of contractile dysfunction [1]. Accordingly, metabolic modulation currently appears as one of the most promising new strategies for chronic drug therapy with the potential to further improve long-term prognosis, as an additive to established therapy with angiotensin-converting enzyme (ACE) inhibitors, beta blockers, and diuretics. In light of this, non-invasive assessment of the metabolic state of the myocardium in DCM has been an important goal in cardiology for many decades. In principle, two techniques allow for this: magnetic resonance spectroscopy (MRS) and positron emission tomography (PET). The largest number of studies has Heart Metab. 2011; 49:13–16 been performed with MRS [2], and early proofofprinciple investigations point to the value of metabolic MRS parameters as biomarkers of disease and prognosis, and for monitoring therapy. Methodological considerations MRS can be performed on the same magnetic resonance systems used for imaging, typically at a field strength of 1.5–3 tesla (T). There are, however, additional hardware and software requirements, such as a phosphorus-31 (31P) surface coil, a broadband radiofrequency transmitter, spectroscopy pulse sequences, and post-processing software [2,3]. After shimming the magnetic field, an MR spectrum is obtained using a spectral localization technique, which makes it possible to obtain signal from a voxel element within the heart [2]. Because of the inherent low resolution of MRS, a large number of acquisitions have to be signal averaged in order to obtain a 13 Metabolic imaging Stefan Neubauer Figure 1. In vivo human cardiac phosphorus-31 (31P) magnetic resonance spectroscopy, 3-dimensional chemical shift imaging sequence. Left: Short-axis hydrogen-1 (1H) magnetic resonance image of the heart with a superimposed grid of spectroscopic voxels. The interrogated cardiac voxel (blue square) is placed in the interventricular septum to avoid contamination from skeletal muscle. Saturation bands are placed over the chest wall skeletal muscle to suppress further any skeletal muscle signal. Right: Example of a cardiac 31P magnetic resonance spectrum in a healthy individual. Resonances for 2,3-diphosphoglycerate (2,3-DGP), phosphodiesters (PDE), phosphocreatine (PCr), and the three phosphorus atoms of adenosine-50 -triphosphate (ATP) (from left to right: g, a, and b ATP) are detectable. 3T Siemens TIM-Trio system. Acquisition matrix size 16x16x8 voxels, field of view 240 240 200 mm. Reprinted from [16]. magnetic resonance spectrum with an acceptable signal-to-noise ratio. A typical 31P spectrum from a normal individual is shown in Figure 1 (Figure 1). Resonances are shown for the three phosphorus atoms of adenosine-50 -triphosphate (ATP) (a, b and g ATP), phosphocreatine (PCr), and also 2,3-diphosphoglycerate (2,3-DPG) from blood and phosphodiesters (PDE) from phospholipids. The PCr to ATP ratio is an exqui- sitely sensitive index of the energetic state of the heart [4]. Figure 2 shows a hydrogen-1 (1H) MR spectrum from the heart (Figure 2). While 1H spectra show many metabolites, quantification has so far been limited to lipids (triglycerides), which provide a measure of myocardial steatosis (another key factor in the development of contractile dysfunction), and to the total creatine peak [5]. Figure 2. In vivo human cardiac hydrogen-1 (1H) magnetic resonance spectroscopy. Spectrum consisting of 35 averages (TR 2800 ms, TE/TM ¼ 10/7 ms), from a 12.6 cm3 voxel positioned in the interventricular septum of a healthy volunteer as shown in (B) and (C). Resonance assignment: (1) residual water, 4.7 ppm; (2) total creatine CH2, 3.9 ppm; (3) Ntrimethyl groups, 3.2 ppm; (4) total creatine CH3, 3 ppm; (5) unsaturated fat, 2.0–2.3 ppm; (6) fat (CH2)n, 1.3 ppm; (7) fat terminal methyl, 0.9 ppm. Spectrum acquired by Dr. Belen Rial, Oxford Centre for Clinical Magnetic Resonance Research. 14 Heart Metab. 2011; 49:13–16 Metabolic imaging Metabolic imaging in dilated cardiomyopathy There are only two previous studies on 1H-MRS in DCM. In a mixed group of patients with non-ischemic heart failure (15 patients, 11 of whom had DCM), Nakae et al. [10] found a nearly 50% decrease in total creatine content, which also correlated inversely with plasma brain natriuretic peptide (BNP) levels. Apart from an early proof-of-principle study [11], there are no data on 1H-MRS-measured myocardial lipid levels in patients with DCM, and these studies are urgently needed. Findings in DCM Deranged cardiac energy metabolism is a hallmark of the failing heart [1], regardless of the etiology. In DCM, PCr to ATP ratios are reduced, correlating with the New York Heart Association (NYHA) functional class [4] and left ventricular ejection fraction [6]. Most importantly, they are a strong predictor of prognosis, and in one study the PCr to ATP ratio was a better predictor of long-term survival than NYHA class or left ventricular ejection fraction (Figure 3) [7]. Although PCr to ATP ratios are powerful indicators of the extent of energetic derangement in DCM, they still underestimate the true extent of metabolic derangement. Recent spectroscopy techniques have made the absolute quantification of ATP and PCr possible, showing approximately 50% reduction in PCr and 35% reduction in ATP, with a concomitant 25% decrease in the PCr to ATP ratio, in heart failure [8]. An even more sensitive indicator of deranged energetics may be the dynamic rate of turnover of ATP. Recently, ATP turnover (ATP flux through the creatine kinase reaction) was measured in volunteers and patients with heart failure; for an 18% reduction in PCr concentrations, a 50% reduction in the rate of turnover of ATP was demonstrated [9]. Thus, the measurement of ATP turnover rates holds promise as a highly sensitive indicator of energetic derangement in heart failure. Treatment studies An important area is the use of MRS for monitoring energetic changes in the heart after novel forms of treatment of DCM. We showed that conventional treatment of DCM with beta blockers, ACE inhibitors and diuretics for 3 months significantly improved the PCr to ATP ratio, together with clinical improvement [12]. Most recently, a study of the use of the investigational drug trimetazidine in patients with heart failure of mixed etiology revealed that trimetazidine was associated with a 33% increase in the PCr to ATP ratio, concomitant with improvements in NYHA class and left ventricular ejection fraction (EF) [13]. In a recent study, we investigated the effects of chronic exercise on DCM, and found an improvement on EF after 8 months of therapy, achievable DCM patient died 7 days after MR examination DCM, PCr/ATP < 1.6 DCM, PCr/ATP > 1.6 PCr γ PDE Volumeer α β-ATP Total mortality (%) 2,3 DPG 50 40 p = 0.036 30 PCr/ATP < 1.60 20 PCr/ATP > 1.60 10 0 0 10 0 −10 −20 ppm 500 1000 1500 Days of follow-up Figure 3. Phosphorus-31 (31P) magnetic resonance spectroscopy in dilated cardiomyopathy. Left: 31P magnetic resonance spectra in, from bottom upward: a healthy volunteer, a patient with mild dilated cardiomyopathy (DCM), a patient with severe DCM with reduced phosphocreatine (PCr) to adenosine-50 -triphosphate (ATP) ratio, and a patient who died 7 days after the magnetic resonance examination, showing a massive reduction in the PCr to ATP ratio. Right: Kaplan–Meyer curve analysis of total mortality in DCM. Patients were subdivided according to the initial PCr to ATP ratio (greater or less than 1.60). Substantially increased mortality was observed in patients with an initially reduced PCr to ATP ratio. Modified from Beer M et al. [8]. ß2002 American College of Cardiology Foundation. Heart Metab. 2011; 49:13–16 15 Metabolic imaging Stefan Neubauer without extra energetic cost to the myocardium, as estimated by similar PCr to ATP ratios [14]. Although currently available studies on this subject are clearly limited by their small size, the concept of monitoring novel drug therapy for heart failure with regards to its effects on cardiac energetics is an extremely appealing one. Future perspective The main challenge for MRS is the improvement of the method’s low signal to noise. Promising techniques on the horizon to achieve this are higher field strength (7T and more) and hyperpolarisation methods (for carbon13 [13C] MRS) [15], which can boost the signal by several orders of magnitude, but require intravenous application of external hyperpolarized 13C metabolites. These are exciting novel directions for future research. Summary MRS allows for the non-invasive assessment of various aspects of cardiac metabolism in DCM, and has the potential to become an important biomarker tool for diagnostic and prognostic assessment as well as for the monitoring of therapy. REFERENCES 1. Neubauer S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356:1140–1151. 2. Bottomley PA. MR spectroscopy of the human heart: the status and the challenges. Radiology. 1994;191:593–612. 3. Hudsmith LE, Neubauer S. Magnetic resonance spectroscopy in myocardial disease. JACC Cardiovasc Imaging. 2009;2:87– 96. 16 4. Clarke K, O’Connor AJ, Willis RJ. Temporal relation between energy metabolism and myocardial function during ischemia and reperfusion. Am J Physiol. 1987;253:H412–H421. 5. van der Meer RW, Doornbos J, Kozerke S, Schär M, Bax JJ, Hammer S, Smit JW, Romijn JA, Diamant M, Rijzewijk LJ, de Roos A, Lamb HJ. Metabolic imaging of myocardial triglyceride content: reproducibility of 1H MR spectroscopy with respiratory navigator gating in volunteers. Radiology. 2007;245 (1):251–257. 6. Neubauer S, Horn M, Pabst T, et al. Contributions of 31P magnetic resonance spectroscopy to the understanding of dilated heart muscle disease. Eur Heart J. 1995;16 (Suppl O):115–118. 7. Neubauer S, Horn M, Cramer M, et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. 8. Beer M, Seyfarth T, Sandstede J, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–1274. 9. Smith CS, Bottomley PA, Schulman SP, Gerstenblith G, Weiss RG. Altered creatine kinase adenosine triphosphate kinetics in failing hypertrophied human myocardium. Circulation. 2006;114:1151–1158. 10. Nakae I, Mitsunami K, Matsuo S, Matsumoto T, Morikawa S, Inubushi T, Koh T, Horie M. Assessment of myocardial creatine concentration in dysfunctional human heart by proton magnetic resonance spectroscopy. MRM. 2004;3:19–25. 11. Walecki J, Michalak MJ, Michalak E, Bilinska ZT, Ruzyllo W. Usefulness of 1H MR spectroscopy in the evaluation of myocardial metabolism in patients with dilated idiopathic cardiomyopathy: pilot study. Acad Radiol. 2003;10 (10):1187–1192. 12. Neubauer S, Krahe T, Schindler R, et al. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease. Altered cardiac high-energy phosphate metabolism in heart failure. Circulation. 1992;86:1810–1818. 13. Fragasso G, Perseghin G, De Cobelli F, et al. Effects of metabolic modulation by trimetazidine on left ventricular function and phosphocreatine/adenosine triphosphate ratio in patients with heart failure. Eur Heart J. 2006;27:942–948. 14. Beer M, Wagner D, Myers J, Sandstede J, Köstler H, Hahn D, Neubauer S, Dubach P. Effects of exercise training on myocardial energy metabolism and ventricular function assessed by quantitative phosphorus-31 magnetic resonance spectroscopy and magnetic resonance imaging in dilated cardiomyopathy. J Am Coll Cardiol. 2008;51 (19):1883–1891. 15. Schroeder MA, Cochlin LE, Heather LC, Clarke K, Radda GK, Tyler DJ. In vivo assessment of pyruvate dehydrogenase flux in the heart using hyperpolarized carbon-13 magnetic resonance. Proc Natl Acad Sci U S A. 2008;105:12051–12056. 16. Neubauer, Stefan. Metabolic imaging with cardiac magnetic resonance spectroscopy. Heart Metab. 2009;44:17–20. Heart Metab. 2011; 49:13–16 New therapeutic approaches Metabolic modulation in dilated cardiomyopathy Helena Tuunanen and Juhani Knuuti Turku PET Centre, University of Turku, Finland, Department of Medicine, Turku University Hospital, Turku, Finland Correspondence to Correspondence: Helena Tuunanen, Department of Medicine, Turku University Hospital, PO Box 51, 20521 Turku, Finland. Tel: 358 2 313 0000; e-mail: [email protected] Management of patients with dilated cardiomyopathy (DCM) and chronic heart failure (HF) remains challenging. There is a need for novel medical therapies independent of the neurohormonal axis that improve cardiac performance. Experimental and preliminary clinical studies have demonstrated that metabolic modulators enhancing myocardial glucose metabolism directly or indirectly by limiting free fatty acid (FFA) metabolism, may improve left ventricular (LV) function in HF. This paper reviews the literature on the effects of metabolic modulators on human HF (both ischemic and idiopathic). The effects of acute metabolic modulation (mainly by manipulating circulating substrate levels) and longterm metabolic modulation (trimetazidine, perhexiline, etomoxir) are discussed. Heart Metab. 2011;49:17–19. Keywords: etomoxir, Heart failure, metabolism, perhexiline, trimetazidine Introduction Metabolic modulation is linked to glucose being a more energy-efficient fuel than free fatty acids (FFAs). Cardiac FFA and glucose metabolism are tightly coupled, which means that inhibition of one results in an increment in another. Several different agents can achieve a myocardial metabolic switch from oxidation of FFAs towards that of glucose. In this paper we focus on metabolic shift achieved by 1) manipulating circulating substrate levels (nicotinic acid and its derivatives, glucose-insulin-potassium infusion), 2) FFA betaoxidation inhibitors (trimetazidine, ranolazine), 3) carnitine palmitoyltransferase (CPT)-1 blockers (perhexiline, etomoxir), and 4) dichloroacetate (direct carbohydrate oxidation activator). Acute metabolic modulation In small groups of patients with heart failure (HF), intravenous infusion of dichloroacetate [1] or glucoseinsulin-potassium [2] and intracoronary infusion of Heart Metab. 2011; 49:17–19 pyruvate [3,4] has been associated with improved cardiac function. Compared with double-blind placebo, three-day treatment with trimetazidine improved ejection fraction (EF) and enhanced myocardial perfusion in patients with prior myocardial infarction and ischemic HF [5]. However, the positive effects of trimetazidine were lost in patients with severe left ventricular (LV) dysfunction, possibly due to large, metabolically inactive scar tissue in these patients. In contrast, Wiggers et al. [6] demonstrated that acute substrate availability modulation does not influence cardiac function in hibernating myocardium either at rest or after exercise. The study included substrate modulation either with insulinglucose (high insulin, low FFA) and somatostatinheparin (high FFA, low insulin). Furthermore, a previous study demonstrated that acute FFA deprivation in patients with idiopathic dilated cardio-myopathy (DCM), in contrast to healthy controls, uncouples cardiac contractile function from oxidative metabolism so that myocardial efficiency deteriorates further [7]. Thus, these preliminary studies suggest that acute manipulation of substrate levels may not be a promising form of therapy in HF. 17 New therapeutic approaches Helena Tuunanen and Juhani Knuuti 18 Baseline Follow-up 9 8 FFA uptake (umol/100g/min) A first study in the early 1990 s showed that compared with double-blind placebo, six-month treatment with trimetazidine improves EF in patients with ischemic HF medicated with digoxin and diuretics only [8]. Thereafter, trimetazidine has been shown to improve not only EF in both ischemic [9–15] and non-ischemic HF [11,16], but also regional myocardial wall motion both at rest and during dobutamine-induced stress [9,13] as well as to enhance LV diastolic function [15] and to reverse LV remodeling [10,11,15,17] in patients with ischemic HF. The clinical effects of trimetazidine include improvement in exercise tolerance, New York Heart Association (NYHA) class and quality of life [10,12,15,17,18] in patients with ischemic HF. Interestingly, the largest study [18], involving 200 patients with multivessel coronary artery disease and mildly impaired LV function (EF <50%), demonstrated surprisingly pronounced improvement in survival rates after two years of treatment with trimetazidine (92% with trimetazidine versus 62% with placebo, p < 0.0001). The metabolic effects of trimetazidine include improvement in whole-body insulin sensitivity in patients with ischemic HF and type II diabetes treated with diet only [17]. Furthermore, Monti et al. [19] showed that improvement in whole-body insulin sensitivity by trimetazidine is accompanied by a metabolic shift from FFA to glucose oxidation in skeletal muscle during euglycemic hyperinsulinemia in diabetic HF patients. Trimetazidine also improved metabolism at the myocardial level, as evidenced by increased myocardial phosphocreatine (PCr)-adenosine-5’-triphosphate (ATP) ratio in non-diabetic patients with ischemic HF [12]. Additionally, there is some evidence that trimetazidine limits inflammatory response [10] and improves endothelial function [20] in HF patients. Our recent positron emission tomography (PET) study including non-diabetic patients with idiopathic DCM demonstrated improvement in EF after three months of treatment with trimetazidine as compared with placebo (Fig. 1) [16]. Trimetazidine had no effect on myocardial FFA uptake and only minor inhibitory effects on myocardial FFA oxidation (Fig. 2), suggesting another major mechanism contributing to the benefit in the study. Rather, our data showed increased whole-body insulin sensitivity in idiopathic DCM, as also found by Fragasso et al. in diabetic ischemic HF patients [17], thus hypothetically countering the myocardial damage of insulin resistance. Furthermore, the positive effects of trimetazidine on LV function were especially evident in patients with a high degree of beta-blockade, strongly suggesting an additive effect of these two modalities of therapy. (a) 7 6 5 4 3 2 1 0 TMZ placebo (b) 0.07 Beta-oxidation rate constant (1/min) Long-term metabolic modulation 0.06 Baseline Follow-up −10% P < 0.05 0.05 0.04 0.03 0.02 0.01 0 TMZ placebo Figure 1. Myocardial A) free fatty acid (FFA) uptake and B) betaoxidation rate constant at baseline (blue bars) and after three months of treatment with trimetazidine (TMZ) or placebo (red bars) in patients with idiopathic dilated cardiomyopathy and heart failure. Reprinted with permission from Tuunanen et al. [16]. The other agents reported in long-term metabolic interventions are the carnitine palmitoyltransferase (CPT)-1 blockers etomoxir and perhexiline. A small open-label study without a control group demonstrated that three months of treatment with etomoxir might improve EF both at rest and after maximal exercise in patients with chronic HF [21]. However, due to the liver toxicity of etomoxir revealed in the Etomoxir for the Recovery of Glucose Oxidation (ERGO) study [22], it was judged not to be a candidate for further development for treatment of patients with HF. A double-blind placebo-controlled study demonstrated that eight weeks of administration of perhexiline improves EF, resting and peak dobutamine stress regional myocardial function, peak exercise oxygen consumption, and quality of life, as well as normalizes skeletal muscle phosphocreatine recovery after exercise in both ischemic and non-ischemic HF patients Heart Metab. 2011; 49:17–19 New therapeutic approaches Metabolic modulation in dilated cardiomyopathy 60 P < 0.03 Ejection fraction (%) 50 Baseline Follow-up 40 30 20 10 0 TMZ Placebo Figure 2. Ejection fraction at baseline (blue bars) and after three months treatment (red bars) with trimetazidine (TMZ) or placebo in patients with idiopathic dilated cardiomyopathy and heart failure. Reprinted with permission from Tuunanen et al. [16]. [23]. Furthermore, a retrospective multi-center database analysis in the United Kingdom revealed that perhexiline therapy is well tolerated and provides symptomatic relief in approximately 59% of patients with chronic HF and/ or refractory angina [24]. In contrast, a recent study [25] including 36 patients with post-infarction HF showed that one-year administration of perhexiline has no impact on exercise capacity and does not improve the deformation of abnormal myocardial segments in these patients. Summary The concept that metabolic agents may optimize myo-cardial energy metabolism and allow more efficient production of energy from glucose than from FFAs is appealing. However, studies on of the effects of metabolic modulation on patients with HF have yielded conflicting results. At present trimetazidine, a partial FFA betaoxidation inhibitor, appears to be the most promising agent for the metabolic approach. Larger clinical randomized multi-center studies are warranted to confirm the effects of these agents on patients with HF. REFERENCES 1. Bersin, et al. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol 1994; 23(7):1617–1624. 2. Cottin, et al. Glucose insulin potassium infusion improves systolic function in patients with chronic ischemic cardiomyopathy. Eur J Heart Fail 2002; 4(2):181–184. 3. Hermann, et al. Haemodynamic effects of intracoronary pyruvate in patients with congestive heart failure: An open study. Lancet 1999; 353(9161):1321–1323. Heart Metab. 2011; 49:17–19 4. Hermann, et al. Improved systolic and diastolic myocar- dial function with intracoronary pyruvate in patients with congestive heart failure. Eur J Heart Fail 2004; 6(2):213–218. 5. Feola, et al. The acute administration of trimetazidine modified myocardial perfusion and left ventricular function in 31 patients with ischaemic ventricular dysfunction. Int J Cardiovasc Imaging 2004; 20(4):315–320. 6. Wiggers, et al. Influence of insulin and free fatty acids on contractile function in patients with chronically stunned and hibernating myocardium. Am J Physiol Heart Circ Physiol 2005; 289(2):H938–H946. 7. Tuunanen, et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 2006; 114(20):2130–2137. 8. Brottier, et al. Therapeutic value of a cardioprotective agent in patients with severe ischaemic cardiomyopathy. Eur Heart J 1990; 11(3):207–212. 9. Belardinelli, et al. Effects of trimetazidine on the contrac tile response of chronically dysfunctional myocardium to lowdose dobutamine in ischaemic cardiomyopathy. Eur Heart J 2001; 22(23):2164–2170. 10. Di Napoli, et al. Long-term cardioprotective action of trimetazidine and potential effect on the inflammatory process in patients with ischaemic dilated cardiomyopathy. Heart 2005; 91 (2):161–165. 11. Fragasso, et al. A randomized clinical trial of trimetazi dine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol 2006; 48(5):992–998. 12. Fragasso, et al. Effects of metabolic modulation by tri metazidine on left ventricular function and phosphocreatine/adenosine triphosphate ratio in patients with heart failure. Eur Heart J 2006; 27(8):942–948. 13. Lu, et al. Effects of trimetazidine on ischemic left ventric ular dysfunction in patients with coronary artery disease. Am J Cardiol 1998; 82(7):898–901. 14. Rosano, et al. Trimetazidine improves left ventricular function in diabetic patients with coronary artery disease: A doubleblind placebo-controlled study. Cardiovasc Diabetol 2003; 2(16):1–9. 15. Vitale, et al. Trimetazidine improves left ventricular func tion and quality of life in elderly patients with coronary artery disease. Eur Heart J 2004; 25(20):1814–1821. 16. Tuunanen, et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated car diomyopathy. Circulation 2008; 118(12): 1250–1258. 17. Fragasso, et al. Short- and long-term beneficial effects of trimetazidine in patients with diabetes and ischemic cardio myopathy. Am Heart J 2003; 146(5):E18. 18. El-Kady, et al. Effects of trimetazidine on myocardial per fusion and the contractile response of chronically dysfunctional myocardium in ischemic cardiomyopathy: A 24-month study. Am J Cardiovasc Drugs 2005; 5(4):271–278. 19. Monti, et al. Metabolic and endothelial effects of trimeta zidine on forearm skeletal muscle in patients with type 2 dia betes and ischemic cardiomyopathy. Am J Physiol Endocr Metab 2006; 290(1):E54–E59. 20. Belardinelli, et al. Trimetazidine improves endothelial function in chronic heart failure: an antioxidant effect. Eur Heart J 2007; 28:1102–1108. 21. Schmidt-Schweda, et al. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci 2000; 99(1): 27–35. 22. Holubarsh, et al. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (Etomoxir for the Recovery of Glucose Oxidation) study. Clin Sci (Lond) 2007; 113(4):205–212. 23. Lee, et al. Metabolic modulation with perhexiline in chronic heart failure: A randomized, controlled trial of short- term use of a novel treatment. Circulation 2005; 112(21):3280–3288. 24. Phan, et al. Multi-centre experience on the use of perhexiline in chronic heart failure and refractory angina: old drug, new hope. Eur J Heart Fail 2009; 11(9):881–886. 25. Bansal, et al. Effects of perhexiline on myocardial defor mation in patients with ischaemic left ventricular dysfunction. Int J Cardiol 2010; 139(2): 107–112. 19 Focus on vastarelW mr The clinical benefits provided by trimetazidine (VastarelW MR) in left ventricular dysfunction patients Giuseppe Marazzi, Giuseppe Caminiti and Maurizio Volterrani Center for Clinical and Basic Researc, Cardiovascular Research Unit, Department of Medical Sciences, IRCCS San Raffaele Roma, Rome, Italy Correspondence: Giuseppe Marazzi, Centre for Clinical and Basic Research, Cardiovascular Research Unit, Department of Medical Sciences, IRCCS San Raffaele Roma, Rome, Italy. E-mail: [email protected] Abstract Metabolic treatment involves the use of drugs that improve cardiomyocyte function. Trimetazidine (Vastarel1 MR) is the most investigated drug in this group with a well-established role in the treatment of chronic angina. The available data suggest that therapy combining trimetazidine and hemodynamic drugs is effective in patients with chronic heart failure, leading to additional benefits such as improvement in left ventricular function, exercise tolerance and quality of life. However, while trimetazidine has shown beneficial effects on surrogate endpoints in several small trials, its effect on cardiovascular events in chronic heart failure subjects needs to be confirmed in further large randomized studies. Heart Metab. 2011;49:21–24. Keywords: Chronic heart failure, left ventricular function, metabolic therapy, trimetazidine Introduction Despite treatment with conventional agents, a high proportion of patients with chronic heart failure (CHF) and left ventricular (LV) dysfunction continue to have symptoms and poor quality of life (QOL). Emerging evidence suggests that chronic heart failure is an ‘‘energy-deprived’’ state in which alterations in substrate metabolism of myocardial cells contribute to the development and maintenance of LV dysfunction [1]. Trimetazidine (TMZ) (Vastarel1 MR) belong to a group of drugs called ‘‘metabolic modulators’’ that benefit CHF patients by modulating cardiac metabolism without altering hemodynamics. TMZ selectively inhibits the activity of long-chain 3-ketoacyl coenzyme A thiolase (3-KAT), the final enzyme of the fatty acid b-oxidation path. This leads to a change in substrate energy, partially inhibiting b-oxidation of fatty acids and increasing glucose oxidation, which is more efficient with regards to adenosine- 5’-triphosphate (ATP) proHeart Metab. 2011; 49:21–24 duction per molecule of oxygen consumed. This prevents intracellular acidosis and electrolyte disorders [2] and preserves energy necessary to sustain contractile function. Effects on left ventricular function Several studies published in the last decade have demonstrated significant improvements of LV function during long-term administration of TMZ [3–8]. First observations were made in diabetic patients with ischemic CHF. In a small crossover trial an increase of LV ejection fraction from 36% to 45% was observed in the TMZ group compared with placebo, while exercise time did not change between the two groups [3]. In a similar population, Rosano et al. [4] observed a significant reduction of LV diastolic and systolic diameters (from 63 mm to 58 mm and from 41 mm to 34 mm respectively) and a significant increase of LV ejection fraction (þ5.4% U) in the TMZ group, while 21 Focus on vastarelW mr Giuseppe Marazzi, Giuseppe Caminiti and Maurizio Volterrani no significant changes were detected in the placebo group. These results have been confirmed and amplified by a larger study reporting improvements in LV volumes, LV ejection fraction, and diastolic function in elderly subjects with post-ischemic CHF, most of whom had multi-vessel coronary disease and large areas of hibernating myocardium, with and without diabetes [5]. According to these data, it has been hypothesized that benefits of TMZ on LV function are related to its metabolic action leading to improved glucose utilization and optimization of myocardial energy metabolism even in the presence of chronic reduction of blood flow. Consequently, the structural and metabolic alterations of myocardial cells related to chronic ischemiamay be reversed by TMZ, with a resulting improvement of the mechanical efficiency of areas of viable myocardium. Several studies have been published supporting this hypothesis. In 38 patients with ischemic LV dysfunction, Belardinelli et al. [6] evaluated the contractile response to low-dose infusions of dobutamine at baseline and after 2 months of treatment with TMZ or placebo. At the end of the follow-up period, TMZ significantly improved the rest and peak systolic wall thickening score index, LV ejection fraction, and peak oxygen uptake. The investigators concluded that the improvement in LV contractility was due to the recruitment of stunned or hibernating cells after optimal oxygen utilization by these cells. El-Kady et al. [7] obtained a similar result using gated single photon emission computerized tomography. In this study, there was a significant improvement of summed stress and rest scores, systolic wall thickness, and wall motion score index with TMZ compared with placebo. On the other hand the benefits of TMZ on LV function seem not to be limited to the group of CHF of ischemic origin. In a study by Fragasso et al. [8], ejection fraction significantly increased in patients treated with TMZ, regardless of the etiology of CHF. Tuunanen et al. [9] evaluated 19 patients with idiopathic dilated cardiomyopathy randomized to single-blind TMZ or placebo for 3 months. Interestingly in the TMZ group, EF was increased by 15% during the treatment, whereas in the placebo group, it decreased by 17%. Exercise tolerance The usefulness of TMZ to improve the functional status and the exercise capacity of patients with ischemic cardiomyopathy has been assessed in several studies [7,9–12]. Improvements in exercise capacity have been evaluated by a six-minute walking test, change in New York Heart Association (NYHA) functional class, or an ergometric test. In the study of Brottier et al. [3], 20 patients were randomized to either placebo or TMZ. At 6-month 22 follow-up, all patients on TMZ reported a considerable clinical improvement in symptoms such as angina and dyspnea, together with a 9% increase of LV ejection fraction. More recently, an Italian study obtained a similar result [5], with a reduced incidence of angina episodes and improvement in NYHA class after 6 month in the TMZ group compared with placebo. Another trial enlisted 61 patients with a past history of myocardial infarction, a depressed LV ejection fraction (<40%), and coronary anatomy unsuitable for revascularization [10]. The authors demonstrated a significant improvement in functional status (evaluated by NYHA functional class) after 18 months of treatment with TMZ administered at standard doses. El-Kady et al. further confirmed these findings [7] in a larger study involving 200 patients with multi-vessel coronary artery disease and impaired LV function (LV ejection fraction <50%). Patients were randomized to TMZ or placebo in an open-label design, and after 24 months of treatment, the patients receiving TMZ had a significant reduction in the frequency of anginal episodes per week, a reduction in the weekly consumption of nitrate tablets, and a significant increase in treadmill exercise test duration (þ75 s with TMZ versus þ25 s with placebo, p < 0.01). In the study by Fragasso et al. [8], TMZ was associated with significant improvements in functional capacity assessed by ergometric test. The authors demonstrated a significant increase of peak metabolic equivalent system (from 7.4 to 8.8, p ¼ 0.04) and total exercise time (from 314 s to 402 s, p ¼ 0.04) in the TMZ group. Conversely, these parameters remained stable in the conventional therapy group. At the same time in the TMZ group there was a significant reduction in plasma natriuretic peptide levels compared with conventional therapy. Interestingly these improvements were equally apparent in patients with non-ischemic and ischemic cardiomyopathy. In another study [11], conducted in patients with stable ischemic CHF, the distance walked in 6 minutes significantly improved in the TMZ group, from 355 m at baseline to 417 m at the end of follow-up. Conversely, the distance walked decreased in the placebo group. The improvement in exercise tolerance with TMZ may be secondary to its effects on LV systolic and diastolic performance. However, in the study of Di Napoli et al. [11], there was no significant change on LV diastolic and systolic diameter after 6 months of treatment in the TMZ group compared to placebo. Therefore, an additional mechanism of action such as a direct effect of TMZ on skeletal muscle should be taken into account. A recent study by Monti et al. [12] demonstrated that TMZ treatment in diabetic patients with ischemic cardiomyopathy improves forearm skeletal muscle metabolism. Heart Metab. 2011; 49:21–24 Focus on vastarelW mr The clinical benefits provided by trimetazidine (Vastarel j MR) in left ventricular dysfunction patients 8 Baseline Visual analog scale 7 6 months 6 5 4 3 2 1 0 Trimetazidine Placebo Figure 1. Changes (6 months vs. baseline) in visual analog scale observed in the TMZ and placebo groups. Quality of life Currently, few data are available on the impact of TMZ on QOL. In the study of Vitale et al. [5], QOL, assessed by the visual analogue scale, significantly improved in all patients treated with TMZ, while it remained unchanged in those allocated to placebo. Fragasso et al. [8] assessed QOL with two tests: a visual analogue scale measuring the general wellbeing and an LV dysfunction questionnaire (LVD36) in order to measure the impact of LV dysfunction on daily life. The authors showed a significant decrease in LVD-36 score (from 18 to 15, p ¼ 0.038) and no significant increase of visual analogue scale, which went from 63% to 71% ( p ¼ 0.07). More recently, we investigated the effects of TMZ on different areas of QOL in elderly patients with ischemic dilated cardiomyopathy by using a selfadministered questionnaire, the MacNew Quality of Life After Myocardial Infarction. We showed a significant improvement in physical and social areas in patients randomized to TMZ, but not in those allocated to placebo (Fig. 1) [13]. Benefits of TMZ on QOL could be related to several factors. First, in patients with ischemic CHF and recurrent angina, the increased well-being could be a consequence of its anti-angina effects. Moreover, the improvement in exercise tolerance and a direct action on skeletal muscle mass could play a role [14]. Additional benefits According to some recent studies, TMZ seems to have a broader spectrum of action at cardiac level than previously thought. TMZ improves several parameters related with the outcome of CHF patients. Gunes et al. [15] demonstrated that the addition of TMZ to optimal medical therapy in patients with CHF of ischemic Heart Metab. 2011; 49:21–24 origin might improve heart-rate variability that is related to the improvement of LV ejection fraction. Cera et al. [16] showed that long-term treatment with TMZ can yield a significant reduction of Tpeak– Tend-d index, a noninvasive marker of dispersion of ventricular repolarization in post-ischemic CHF patients. A study by Belardinelli et al. [17] demonstrated that TMZ improved endothelium-dependent relaxation (EDR) in patients with ischemic cardiomyopathy. The authors postulated that EDR improvement was significantly related to an antioxidant effect of TMZ because they observed a concomitant reduction in plasma levels of some malondialdehyde and lipid hydroperoxides. Taken together these studies suggest that TMZ has additional mechanisms of action in CHF subjects. Conclusion The available data suggest that therapy combining TMZ and hemodynamic drugs is effective in patients with CHF, leading to additional benefits such as improvement in LV function, exercise tolerance, and QOL. TMZ has shown beneficial effects on surrogate endpoints in several small trials. However, further large randomized studies are needed to answer the question of whether these effects could translate into decreased morbidity and mortality. REFERENCES 1. Stanley WC, Recchi FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. 2. Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial longchain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86:580–588. 3. Brottier L, Barat JL, Combe C, Boussens B, Bonnet J, Bricaud H. Therapeutic value of a cardioprotective agent in patients with severe ischemic cardiomyopathy. Eur Heart J. 1990;11:207– 212. 4. Rosano GM, Vitale C, Sposato B, et al. Trimetazidine improves left ventricular function in diabetic patients with coronary artery disease. A double-blind, placebo-controlled study. Cardiovasc Diabetol. 2003;2:1–9. 5. Vitale C, Wajngaten M, Sposato B, Gebara O, Rossini P, Fini M, Volterrani M, Rosano GM. Trimetazidine improves left ventricular function and quality of life in elderly patients with coronary artery disease. Eur Heart J. 2004;25:1814– 1821. 6. Belardinelli R, Cianci G, Gigli M, et al. Effects of trimetazidine on myocardial perfusion and left ventricular systolic function in type 2 diabetic patients with ischemic cardiomyopathy. Cardiovasc Pharmacol. 2008;51:611–615. 7. El-Kady T, El Sabban K, Gabali M, et al. Effects of trimetazidine on myocardial perfusion and the contractile response of chronically dysfunctional myocardium in ischemic cardiomyopathy: a 24-month study. Am J Cardiovasc Drugs. 2005;5:271–278. 8. Fragasso G, Palloshi A, Puccetti P, et al. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with systolic dysfunction heart failure. J Am Coll Cardiol. 2006;48:992–998. 23 Focus on vastarelW mr Giuseppe Marazzi, Giuseppe Caminiti and Maurizio Volterrani 9. Tuunanen H, Engblom E, Naum A, et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118: 1250–1258. 10. Di Napoli P, Taccardi AA, Barsotti A. Long-term cardioprotective action of trimetazidine and potential effect on the inflammatory process in patients with ischaemic dilated cardiomyopathy. Heart. 2005;91:161–165. 11. Di Napoli P, Di Giovanni P, Gaeta MA, D’Apolito G, Barsotti A. Beneficial effects of trimetazidine treatment on exercise tolerance and B-type natriuretic peptide and troponin T plasma levels in patients with stable ischemic cardiomyopathy. Am Heart J. 2007;154:602e1–602e5. 12. Monti LD, Setola E, Fragasso G, et al. Metabolic and endothelial effects of trimetazidine on forearm skeletal muscle in patients with type 2 diabetes and ischemic cardiomyopathy. Am J Physiol Endocrinol Metab. 2006;290:E54–E59. 24 13. Marazzi G, Gebara O, Vitale C, Caminiti G, Wajngarten M, Volterrani M, Ramires JA, Rosano G, Fini M. Effect of trimetazidine on quality of life in elderly patients with ischemic dilated cardiomyopathy. Adv Ther. 2009;26:455–461. 14. Emre M, Karayaylali I, San M, Demirkazik A, Kavak S. The acute effect of trimetazidine on the high frequency fatigue in the isolated rat diaphragm muscle. Arch Pharm Res. 2004;27: 646–652. 15. Gunes Y, Guntekin U, Tuncer M, Sahin M. The effects of trimetazidine on heart rate variability in patients with heart failure. Arq Bras Cardiol. 2009;93:154–158. 16. Cera M, Salerno A, Fragasso G, et al. Beneficial electrophysiological effects of trimetazidine in patients with post-ischemic chronic heart failure. J Cardiovasc Pharm Ther. 2010;15:24–30. 17. Belardinelli R, Solenghi M, Volpe L, Purcaro A. Trimetazidine improves endothelial dysfunction in chronic heart failure: an antioxidant effect. Eur Heart J. 2007;28 (9):1102–1108. Heart Metab. 2011; 49:21–24 Case report Response to cardiac resynchronization therapy resulting from an upgrade to dual-site left ventricular pacing M.R. Ginks, S.G. Duckett, G.S. Carr-White and C.A. Rinaldi Guy’s and St Thomas’ Hospitals NHS Foundation Trust, London, UK Correspondence: Dr. C.A. Rinaldi, Cardiac Department, St Thomas’ Hospital, London SE1 7EH, UK. Tel: +44 207 188 9257; fax: +44 207 188 2354; e-mail: [email protected] Abstract A 56-year-old man with ischemic dilated cardiomyopathy underwent a secondary prevention biventricular implantable cardioverter defibrillator (ICD) implant. He did not respond symptomatically despite a good radiographic position of the left ventricular (LV) lead. On the basis of an acute hemodynamic study, a second LV lead was implanted, resulting in a good clinical response from dual-site LV pacing. Heart Metab. 2011;49:25–28. Keywords: Cardiac resynchronization therapy, multi-site pacing, non-responder Introduction Cardiac resynchronization therapy (CRT) has been shown in large randomized trials to confer benefit in both symptoms and prognosis in selected patients with heart failure [1–5]. However, approximately one third of patients do not derive clear clinical benefit (while a similar proportion of matched controls improve without CRT [1]). One reason for this is positioning of the left ventricular (LV) lead in a region of myocardial scar [6–8]. Here we present a case of a patient who did not respond to CRT initially but who improved symptomatically following the implantation of a second LV lead and multi-site LV pacing. History A 56-year-old male presented with a non-ST segment elevation myocardial infarction. Coronary angiography showed occlusions in the left anterior descending and circumflex coronary arteries and a patent right Heart Metab. 2011; 49:25–28 coronary artery. A delayed enhancement cardiac magnetic resonance imaging (MRI) scan showed significant LV systolic impairment and a complex pattern of largely subendocardial infarction (Figure 1). Left ventricular ejection fraction (LVEF) was 16%. He underwent coronary artery bypass grafting with three saphenous vein grafts. Following a cardiac arrest secondary to ventricular fibrillation one month postoperatively, he received a biventricular implantable cardioverter defi-brillator (ICD). When seen in the outpatient clinic two months later, he remained short of breath with minimal exertion, with no improvement since device implantation. He remained on maximal tolerated doses of appropriate heart failure medication. Echocardiographic optimization of atrioventricular (AV) and inter-ventricular (VV) delays was performed at one and three months. At this point the device was delivering biventricular pacing 98% of the time. LVEF was noted to be 18%. However, he remained in New York Heart Association (NYHA) class III. He went on to have assessment with three-dimensional echocardiography, which 25 Case report Ginks et al. Figure 1. Short axis stack of delayed enhancement cardiac magnetic resonance images from apex (top left) to base (bottom right). (ii) exclusion of other conditions that may be underlying symptoms, (iii) optimization of device settings has been performed, and (iv) that a high percentage of biventricular pacing is being delivered. This case raises several interesting questions. First, why did this patient not respond to CRT? What is the mechanism of benefit from the implantation of a second LV lead? Is this a function of multi-site pacing or has the second lead better avoided the subendocardial scar? Should this approach be considered at the initial procedure? Should it be applied to all non- 20 15 % Change dP/dt from intrinsic gave a systolic dyssynchrony index of 11%, with basal infero-posterior and lateral LV segments showing the latest volume change. Cardio-pulmonary exercise testing (CPET) was performed and the maximal myocardial oxygen consumption (MVO2) was 9.6 ml/kg/ minute (41 % predicted). The decision was made to perform an acute hemodynamic assessment with LV stimulation at different sites within the coronary veins, with a view to implant a second LV lead should this give rise to significant acute hemodynamic improvement. This was performed 10 months after the initial CRT defibrillator implant. A RadiTM wire (Radi Medical Systems, Uppsala, Sweden) was passed to the LV cavity using a 5 French femoral arterial sheath. The acute hemodynamic response to different pacing modes is shown in Figure 2. On the basis of these findings, a second LV lead was implanted in a posterior vein and was connected to a new device along with the existing LV lead using a bifurcating adapter to the LV port (Figure 3). At six-month review he had improved symptomatically, from NYHA class III to NYHA class II. CPET was repeated with improvement in MVO2 to 49% predicted. LVEF had improved to 23%. 10 5 0 −5 −10 −15 −20 −25 Discussion We have presented a case of a clinical non-responder to CRT who improved following the addition of a second LVlead, in line with the findings of acute hemodynamic assessment. The optimal strategy for clinical non-responders is not clear. Initial steps should ensure: (i) compliance with appropriate heart failure medications and optimization of fluid status, 26 −30 RV LV old BIV old LV new BIV new 2 site LV Pacing configuration Figure 2. Percentage change in acute hemodynamics from intrinsic rhythm in different pacing configurations. RV right ventricular pacing, LV old pre-existing left ventricular (LV) lead, BIV old conventional cardiac resynchronization therapy (CRT) using previously implanted leads, LV new newly sited LV lead, BIV new conventional CRT using newly sited LV lead, 2 site LV dual-site pacing using old and new LV leads together. All modes used DDD pacing. Heart Metab. 2011; 49:25–28 Case report Response to cardiac resynchronization therapy resulting from an upgrade to dual-site left ventricular pacing Figure 3. Upper images: Posterior anterior (PA) and lateral chest X-rays of lead positions following initial implant procedure. Lower images: PA and lateral chest X-rays following upgrade with addition of second left ventricular lead. responders? What is the role of acute hemodynamic assessment? In this case it appears that the initial LV lead may have been overlying a region of subendocardial scar in the postero-lateral wall. This may not be apparent at the time of the implant procedure, as capture threshold may still be normal. If a lead is overlying an area of subendocardial scar, it may capture but give rise to a lesser degree of clinical benefit. Overlay techniques and image-guided implant technologies are in development and may have a role in the future, to facilitate LV lead delivery away from areas of myocardial scar. Intuitively, a second LV lead is more likely to be beneficial if the initial lead was not optimally positioned [9]. Multi-site pacing may enable us to pace more effectively around areas of scar. Furthermore, in a dilated left ventricle, it may be that pacing from several different loci will facilitate greater reversal of dyssyn-chrony. However, studies examining the acute hemo-dynamic response to single versus dual-site LV pacing in CRT patients have given conflicting results Heart Metab. 2011; 49:25–28 [9,10]. Furthermore, despite the fact that acute hemodynamic response has been widely used as a marker of response to CRT, the evidence that this correlates with long-term response is very limited [11]. Two groups have published data regarding longterm response to dual-site LV pacing in CRT. The TRIP-HF study assessed the effects of dual-site LV pacing in patients with atrial fibrillation, diminished LVEF and a bradycardia-related pacing indication [12]. In this cohort, dual-site LV pacing in conjunction with CRT gave rise to significant improvements over conventional CRT in reverse LV remodeling, albeit not in symptomatic response or exercise capacity. Lenarczyk et al. have also shown higher response rates to multi-site LV pacing, but this was not in the form of a randomized trial [13]. The implantation of two LV leads is feasible but technically challenging, and in the case of patients in sinus rhythm necessitates the use of a bifurcating adapter, which leads to elevated capture thresholds [14] and hence reduced device longevity. However, dual-site pacing represents an alternative approach in the context of clinical 27 Case report Ginks et al. non-response to CRT. Alternative strategies include the surgical implantation of an epicardial LV lead via mini-thoracotomy [15], or endocardial LV stimulation via a trans-septal [16,17] or apical approach [18]. Conclusion In this case, an upgrade from single-site to dual-site LV pacing resulted in improved clinical response to CRT. This represents one approach to CRT non-responders. Randomized studies are needed to establish the best approach to this challenge. REFERENCES 1. Abraham WT, Fisher WG, Smith AL, Delurgio DB, Leon AR, Loh E, Kocovic DZ, et al. Cardiac resynchronization in chronic heart failure. N Engl J Med. 2002;346 (24):1845–1853. 2. Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, et al. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med. 2004;350 (21):2140– 2150. 3. Young JB, Abraham WT, Smith AL, Leon AR, Lieberman R, Wilkoff B, Canby RC, et al. Combined cardiac resynchronization and implantable cardioversion defibrillation in advanced chronic heart failure: the MIRACLE ICD trial. JAMA. 2003;289 (20):2685–2694. 4. Cleland JGF, Daubert J-C, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L, et al. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med. 2005;352 (15):1539–1549. 5. Linde C, Abraham WT, Gold MR, John Sutton St, Ghio M, Daubert C S. Randomized trial of cardiac resynchronization in mildly symptomatic heart failure patients and in asymptomatic patients with left ventricular dysfunction and previous heart failure symptoms. J Am Coll Cardiol. 2008;52 (23):1834–1843. 6. White JA, Yee R, Yuan X, Krahn A, Skanes A, Parker M, Klein G, et al. Delayed enhancement magnetic resonance imaging predicts response to cardiac resynchronization therapy in patients with intraventricular dyssynchrony. J Am Coll Cardiol. 2006;48 (10):1953–1960. 28 7. Chalil S, Stegemann B, Muhyaldeen SA, Khadjooi K, Foley PW, Smith RE, Leyva F. Effect of posterolateral left ventricular scar on mortality and morbidity following cardiac resynchronization therapy. Pacing Clin Electrophysiol. 2007;30 (10):1201–1209. 8. Bleeker GB, Kaandorp TAM, Lamb HJ, Boersma E, Steendijk P, de Roos A, van der Wall EE, et al. Effect of posterolateral scar tissue on clinical and echocardiographic improvement after cardiac resynchronization therapy. Circulation. 2006;113 (7):969–976. 9. Padeletti L, Colella A, Michelucci A, Pieragnoli P, Ricciardi G, Porciani MC, Tronconi F, et al. Dual-site left ventricular cardiac resynchronization therapy. Am J Cardiol. 2008;102 (12):1687–1692. 10. Pappone C, Rosanio S, Oreto G, Tocchi M, Gulletta S, Salvati A, Dicandia C, et al. Cardiac pacing in heart failure patients with left bundle branch block: impact of pacing site for optimizing left ventricular resynchronization. Ital Heart J. 2000;1 (7):464–469. 11. Tournoux FB, Alabiad C, Fan D, Chen AA, Chaput M, Heist EK, Mela T, et al. Echocardiographic measures of acute haemodynamic response after cardiac resynchroniza-tion therapy predict long-term clinical outcome. Eur Heart J. 2007;28 (9):1143–1148. 12. Leclercq C, Gadler F, Kranig W, Ellery S, Gras D, Lazarus A, Clémenty J, et al. A randomized comparison of triple-site versus dual-site ventricular stimulation in patients with congestive heart failure. J Am Coll Cardiol. 2008;51 (15):1455–1462. 13. Lenarczyk R, Kowalski O, Kukulski T, Pruszkowska-Skrzep P, Sokal A, Szulik M, Zielinska T, et al. Mid-term outcomes of triple-site vs. conventional cardiac resynchronization therapy: A preliminary study. Int J Cardiol. 2009;133 (1):87–94. 14. Rho RW, Patel VV, Gerstenfeld EP, Dixit S, Poku JW, Ross HM, Callans D, et al. Elevations in ventricular pacing threshold with the use of the y adaptor. Pacing Clin Electrophysiol. 2003;26 (3):747–751. 15. Lehr EJ, Ye C, Wang S. Left ventricular epicardial lead implantation via left minithoracotomy. Thorac cardiovasc Surg. 2009;57 (06):329–332. 16. Jais P, Douard H, Shah DC, Barold S, Barat JL, Clementy J. Endocardial biventricular pacing. Pacing Clin Electrophysiol. 1998;21 (11 Pt 1):2128–2131. 17. Garrigue S, Jaı̈s P, Espil G, Labeque J-N, Hocini M, Shah DC, Haı̈ssaguerre M, et al. Comparison of chronic biventricular pacing between epicardial and endocardial left ventricular stimulation using Doppler tissue imaging in patients with heart failure. Am J Cardiol. 2001;88 (8):858–862. 18. Kassai I, Foldesi C, Szekely A, Szili-Torok T. New method for cardiac resynchronization therapy: transapical endocardial lead implantation for left ventricular free wall pacing. Europace. 2008;10 (7):882–883. Heart Metab. 2011; 49:25–28 Refresher corner Cardiac amyloidosis Pascal Schmidheiny and Otto M. Hess Swiss Cardiovascular Center, University Hospital, Bern, Switzerland Correspondence: Otto M. Hess, Professor of Cardiology, Swiss Cardiovascular Center, University Hospital, CH-3010 Bern, Switzerland. Tel.: +41 31 632 9653; fax: +41 31 632 4771 Abstract Amyloidosis is a family of disorders of the immune system in which one or more organs in the body accumulate amyloid. There are four different forms of amyloidosis: systemic amyloid light chain (AL) amyloidosis, amyloid A (AA) amyloidosis, hereditary, and senile amyloidosis. The abnormal proteins can be found as Bence-Jones proteins in urine mainly in patients with light chain (AL) amyloidosis. Cardiac amyloidosis is a myocardial disease characterized by extracellular amyloid infiltration throughout the heart and is an important cause of progressive heart failure (restrictive cardiomyopathy) with a wide spectrum of clinical manifestations. Typically, cardiac arrhythmias and diastolic heart failure occur, followed by sudden cardiac death in many cases. The diagnosis of cardiac amyloidosis is a combination of clinical, electrocardiographic and imaging methods, but definite diagnosis is based on endomyocardial biopsy. Prognosis of AL amyloidosis is poor and treatment is often a challenge, although a combination of high-dose chemotherapy, autologous stem cell transplantation and heart transplantation have shown survival benefits. Reactive AA amyloidosis may respond to anti-inflammatory and immunosuppressive drugs. Heart Metab. 2011;49:29–31. Keywords: Autologous stem cell transplantation, cardiac amyloidosis, cardiac arrhythmias, endomyocardial biopsy, restrictive cardiomyopathy Background Amyloidosis is a family of disorders of the immune system in which one or more organ systems in the body accumulate deposits of abnormal proteins [1]. The name ‘‘amyloidosis’’ was first used more than 100 years ago, yet only within the past 25 years have physicians understood the specific make-up and structure of amyloid protein. Amyloidosis represents a diverse group of diseases characterized by the common factor of deposition of twisted ß-pleated sheet fibrils (amyloid) formed as a result of the misfolding of various proteins produced in several different pathological states [1]. The various forms of amyloidosis are classified by the composition of the amyloid fibril. The three major types of amyloidosis are different from each other. Amyloid light chain (AL) amyloidosis (also known as primary amyloidosis) is a plasma cell disorder that originates in the bone marrow. It is the most common Heart Metab. 2011; 49:29–31 type of amyloidosis and occasionally occurs with multiple myeloma. The deposits in this type of the disease are made up of immunoglobulin light chain proteins, which may be deposited in any bodily tissues or organs. The disease results when enough amyloid protein builds up in one or more organs to cause the organ(s) to malfunction. The heart, kidneys, nervous system, and gastrointestinal tract are most often affected. Secondary amyloidosis is caused by a chronic infection or inflammatory disease such as rheumatoid arthritis, familial Mediterranean fever, osteomyelitis, or granulomatous ilei-tis. The deposits in this type of the disease are made up of a protein called the amyloid A (AA) protein. Familial amyloidosis is the only type of amyloidosis that is inherited. The deposits in this type are most commonly made up of the transthyretin protein, which is manufactured in the liver [1]. In elderly persons, cardiac amyloidosis is an important cause of progressive heart failure and refractory 29 Refresher corner Pascal Schmidheiny &Otto M. Hess arrhythmia of obscure origin [2]. The average survival time of amyloid heart disease after the onset of symptoms is less than three years. Clinically, amyloid heart disease may mimic constrictive pericarditis, coronary artery disease, valvular heart disease, and hypertrophic or congestive cardiomyopathy. Cardiac amyloidosis belongs to the family of restrictive cardiomyopathies. Restrictive cardiomyopathy is characterized by abnormal diastolic function with either thickened or rigid ventricular walls leading to elevated filling pressures of the left- or right-sided cardiac chambers. The classification of restrictive cardiomyopathy is based on etiological and clinical findings. Primary forms are Löffler’s endocarditis and endomyocardial fibrosis. To the secondary forms belong infiltrative diseases, storage diseases, and post-radiation disease. Secondary forms are classified by the specific type of material deposition (infiltration), storage, or replacement. conduction system, and/or vessels, with consequent clinical manifestations. Clinical presentation of cardiac amyloidosis Clinical suspicion of cardiac amyloidosis arises in the following circumstances: cardiac disease in the setting of established AL amyloidosis and/or plasma cell dyscrasia; ventricular dysfunction or arrhythmia and longstanding connective tissue disease or other chronic inflammatory disorder; any patient with restrictive cardiomyopathy of unknown etiology; left ventricular (LV) hypertrophy on echocardiography but a low-voltage electrocardiogram (ECG); and congestive heart failure of unknown origin refractory to standard medical therapy. Diagnosis of cardiac amyloidosis Acquired amyloidosis In primary (or AL) amyloidosis, the fibrillar protein is composed of K and l immunoglobulin light chains, produced by a proliferating clone of plasma cells [3]. These immunoglobulins can be found in urine as Bence-Jones proteins. A systemic disease that tends to follow a rapidly progressive course, primary amyloidosis is often seen in conjunction with plasma cell dyscrasia such as multiple myeloma or monoclonal gammopathies. Secondary (or AA) amyloidosis occurs in association with chronic inflammatory disorders such as rheumatoid arthritis, ankylosing spondylitis, and familial Mediterranean fever. Nephrotic syndrome and renal failure are common at presentation. The preliminary work-up for a patient with suspected cardiac amyloidosis includes a 12-lead ECG and twodimensional echocardiography, with or without Holter monitoring [5]. A low-voltage ECG with increased sep-tal and posterior LV wall thickness on echocardiography is specific for cardiac amyloidosis on non-invasive testing [5]. Other investigations that are of value include protein electrophoresis and genetic testing. A confirmatory biopsy (multiple specimens) is needed for diagnosis (Figure 1), since cardiac amyloidosis has no pathognomonic symptoms and signs, nor diagnostic findings. However, with advances in imaging, cardiovascular magnetic resonance (Figure 2) may prove to have value in diagnosis, treatment, and follow-up in cardiac amyloidosis [6]. Hereditary amyloidosis Management and outcome Hereditary amyloidosis is the result of a mutation in one of a number of fibril precursor proteins, including transthyretin, apolipoproteinA-I or A-II, lysozyme,fibrinogen Aa chain, gelsolin, and cystatin C. The phenotype varies according to the protein affected. Autosomal-dominant inheritance is typical. Treatment is directed at both the underlying disease process and the cardiac complications. Systemic therapy is type-specific (i.e., chemotherapy), underscoring the importance of determining the precise etiology of amyloidosis [7]. Prognosis of AL amyloidosis is poor; however, in subgroups of patients, combined treatment with high-dose melphalan and autologous stem cell transplantation has resulted in hematological remission, improved five-year survival, and reversal of amyloid-related disease [5]. In contrast, senile systemic amyloidosis is characterized by slow progression not requiring alkylating agents [8]. Reactive AA amyloidosis may respond to anti-inflammatory and immunosuppressive drugs that reduce production of acute-phase reactant serum amyloid A protein. Ventricular dysfunction secondary to amyloidosis may be difficult to treat. Diuretics and Cardiovascular involvement Amyloidosis frequently affects the heart. The propensity for cardiovascular involvement is greatest in primary, senile, and certain hereditary forms of the disease. Cardiac disease is relatively less common in AA amyloidosis, but is a marker of unfavorable prognosis when present [4]. Amyloid fibrils may be deposited in the myocardium, papillary muscles, valves, 30 Heart Metab. 2011; 49:29–31 Refresher corner Cardiac amyloidosis Figure 1. Subendocardial biopsy with Kongo red-positive extracellular amorphous deposits (arrow, panel a), disrupting the organisation of the heart muscle. On the right (panel b) with optical double refraction, amyloid deposits partly with a characteristic blue color (arrowhead). Photograph provided by Waschkowski G, MD, Institute of Pathology, University of Bern. vasodilators are used judiciously owing to the risk of hypotension with excessive falls in preload. Digoxin is contraindicated because it binds to amyloid fibrils and toxicity may develop at ordinary therapeutic doses. Complex ventricular arrhythmia has been documented in patients with cardiac amyloidosis and may be a predictor of sudden cardiac death. Beta blockers are administered with caution as they may promote atrio- Figure 2. Short-axis view: diffuse, circumferential enhancement of the myocardium (asterisks) in late gadolinium CMR picture. Photograph provided by Wahl A, MD, Department of Cardiology, University of Bern. Heart Metab. 2011; 49:29–31 ventricular (AV) block and their negative inotropism is often poorly tolerated. Implantation of a permanent pacemaker is indicated in patients with symptomatic bradyarrhyth-mias or complete AV block. REFERENCES 1. Hess OM, McKenna W, Schultheiss HP. (2009). Myocardial Disease (Chapter 18). In: Camm AJ, Lüscher TF, Serruys PW (eds), ESC Textbook of Cardiovascular Medicine, 2nd edn. Oxford University Press, Oxford, 665–716 2. Gertz MA, Rajkumar SV. Primary systemic amyloidosis. Curr Treat Options Oncol. 2002;3:261–271. 3. Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, Pepys MB, Hawkins PN. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786–1791. 4. Tanaka F, Migita K, Honda S, Fukuda T, Mine M, Nakamura T, Yamasaki S, Ida H, Kawakami A, Origuchi T, Eguchi K. Clinical outcome and survival of secondary (AA) amyloidosis. Clin Exp Rheumatol. 2003;21:343–346. 5. Rahman JE, Helou EF, Gelzer-Bell R, Thompson RE, Kuo C, Rodriguez R, Hare JM, Baughmann KL, Kasper EK. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43:410–415. 6. Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, Sheppard MN, Poole-Wilson PA, Hawkins PN, Pennell DJ. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111:186–193. 7. Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology (2004) Guidelines on the diagnosis and management of AL amyloidosis. Br J Haematol 125:681– 700 8. Kyle RA, Spittell PC, Gertz MA, Li CY, Edwards WD, Olson LJ, Thibodeau SN. The premortem recognition of systemic senile amyloidosis with cardiac involvement. Am J Med. 1996;101: 395–400. 31 Hot topics Low diagnostic yield of coronary angiography or not catching up with heart disease pathophysiology? Alda Huqi Cardiovascular Medicine Division, Cardio Thoracic Department, University of Pisa, Pisa, Italy Correspondence: Dr Alda Huqi, Cardiovascular Medicine Division, Cardio Thoracic Department, University of Pisa, Via Paradisa, 2 56100 Pisa, Italy. Tel: +39 32972 56426; e-mail: [email protected] Fifty years after its introduction, coronary angiography remains the reference technique to assess coronary anatomy. Its impact on the diagnosis and management of ischemic heart disease (IHD) is corroborated by the fact that coronary angiography is still essential for aortocoronary bypass grafting and for percutaneous coronary interventions. Given the invasiveness and the measurable risk associated with coronary angiography, noninvasive tests are widely used to select candidates for angiography and for therapy planning. The large majority of the noninvasive tests are focused on ischemia detection and left ventricular (LV) function assessment. Latest additions are more focused on coronary anatomy. In the early phase of application, all tests were extensively compared with coronary angiography as the gold standard and achieved reasonable sensitivity, specificity, and overall diagnostic accuracy. Heated debates and large trials have addressed the question of which test is the best, but final conclusions have not yet been reached and probably will never be. The main reasons for this are inconsistencies in the results of different tests in the same patient and the common assumption that coronary stenosis always implies myocardial ischemia. The frequent mismatch between noninvasive tests and coronary anatomy are justified by generic inaccuracies of the noninvasive tools or inaccurate measurement of stenosis severity. Recently, a study with a catchy title— ‘‘Low diagnostic yield of coronary angiography’’ [1]— was published in the New England Journal of Medicine. Data were obtained from a large cardiac-catheterization registry to assess the effectiveness of current practices in enhancing the yield of diagnostic cardiac catheterization as measured by the prevalence of obstructive coronary artery disease (CAD). The study enrolled 397,954 patients without known CAD undergoing cardiac catheterization and noninvasive testing was performed in 83.9% of them. A Heart Metab. 2011; 49:33–35 positive test result was recorded in 68.6% of all the patients in the cohort. Surprisingly enough, only a minority of patients (37.6%) had obstructive CAD. Patients with higher Framingham Risk Score (FRS) were more likely to have obstructive CAD, but the results of the noninvasive testing had a limited additive value for presence of obstructive CAD. In light of such results, the authors concluded that ‘‘better strategies for risk stratification, in order to increase the diagnostic yield of cardiac catheterization are needed.’’ The claim that there is ‘‘a need for better strategies to risk stratify patients’’ represents just another coupler in the scaffold around the rising use of cardiac imaging modalities. Between 1993 and 2001, stress imaging (particularly stress gated single photon emission computed tomography [SPECT] imaging and stress echocardiography) presented an annual increase rate of 6%, which is far in excess with respect to the increase in cardiac catheterization, revascularization, or acute myocardial infarction [2]. More recently, morphologic imaging techniques such as magnetic resonance imaging (MRI) and computed tomography coronary angiography (CTCA) have become a more prominent component of the increase. However, this increasing use has also generated continuous disputation, mainly related to two worrisome issues. Inconsistencies among different techniques and rate of ‘‘false positive’’ and ‘‘false negative’’ results with respect to the ‘‘gold standard’’ for coronary atherosclerosis: invasive coronary angiography (ICA) A patient with angina and documented ischemia is classified as a patient with IHD only when coronary stenosis can be documented. A test result is rated as ‘‘false positive’’ even in the presence of symptoms and 33 Hot topics signs of myocardial ischemia if no obstructive CAD can be detected at the control ICA. Conversely, a test result is rated as ‘‘false negative’’ if, in the absence of symptoms and signs of myocardial ischemia, obstructive CAD is detected at the control ICA. Despite continuous technical progress, differences in CAD detection continue to exist among the various techniques. The major gap is generally found between functional and morphological tests, often without any geographical association between the distribution of coronary plaque and perfusion defects [3]. Unfortunately, this issue does not seem to bother physicians, who are rather concentrated on improving their own technique, in order to ‘‘unmask’’ as much CAD as they can. This behavior is unfortunately driving us into a blind alley. Although there are several technical challenges related to individual testing, these cannot explain the overall rate of ‘‘false positive’’ and ‘‘false negative’’ results. In the so-called ‘‘real world’’, CAD automatically implies ischemic heart disease. However, the presence of true stress-induced ischemia in the absence of obstructive CAD is a well-established phenomenon [4,5]. Conversely, 25% of patients with normal stress SPECT images have been reported to have obstructive CAD on CTCA [6]. In line with these findings, autopsy studies of young adults dying from traffic accidents, homicides, and suicides have found that 60% between the ages of 30 and 39 years of age have left anterior descending (LAD) plaques of American Heart Association (AHA) grade 2 or higher [7], nonetheless, none of them suffered ischemic heart disease. The rate of ‘‘false positives’’ and ‘‘false negatives’’ should therefore not always be regarded as a technical challenge, but rather as a laid-back attitude to something we just do not get. Economic issues together with the ability of a diagnostic test to predict future clinical outcomes and to affect prognosis It should be admitted that, by its inherent properties as a diagnostic rather than therapeutic intervention, an imaging test merely supports clinical decision-making and does not by itself have an impact on outcomes. Moreover, additional concerns challenge the overall value of cardiologic diagnostic tests: a) dozens of studies in tens of thousands of patients have consistently shown that patients with known or suspected CAD who do not have demonstrable stress-induced ischemia have a good prognosis with very low event rates over the subsequent 3 to 7 years [8]; b) although abnormalities in perfusion imaging tests are associated with higher risk of death and myocardial infarction, this does not necessarily imply that treatment of 34 patients with abnormal tests results in reduction of events [9,10]; and c) the preponderance of CAD responsible for myocardial infarction or sudden cardiac death is due to non-obstructive coronary artery plaques [11]. Given this intricate relationship between CAD and myocardial ischemia, the limited prognostic impact of noninvasive testing should no longer come as a surprise. Nevertheless, as mentioned previously, noninvasive testing currently constitutes one of the most blooming areas of cardiology. A carefully conducted study from Canada with contemporary controls [12] demonstrated that the rate of normal invasive coronary angiograms decreased after introduction of CTCA, but the overall rate of ICA increased. Turning back to our study, is this the way authors hope to achieve a ‘‘higher’’ diagnostic yield of coronary angiography? For what purpose? Ischemic heart disease, which includes acute coronary syndromes and chronic angina, is a leading cause of death all over the world. Because disease presentation is often fatal and a number of those who die suddenly have no previously recognized symptoms, the development of imaging strategies capable of predicting and thereafter preventing such a risk has been regarded as a remedy. The first step in the evaluation of patients is a careful history and physical examination to provide an estimate of the likelihood of increased risk for coronary events. This is generally followed by a noninvasive assessment, which then determines the need for ICA. All these efforts point towards the same target: identification and local treatment of obstructive CAD. Cardiologists have known for many years that coronary angiography yields similar results in patients with both stable and acute coronary syndromes. Now we have learned that coronary angiography yields similar results in patients with myocardial ischemia and in patients without. How can we still assume that coronary anatomy dictates clinical conditions? In fact, studies aimed at determining the impact of removal of coronary stenosis have yielded disappointing results, both in terms of prognosis and symptom relief [9,13]. In line with these findings, there are no data to show that information from imaging techniques benefits patients. Morphologic imaging techniques consume large amounts of resources, yet their role, if any, appears to be in reclassifying patients at intermediate risk with traditional risk-factor models. On the other hand, it has been widely demonstrated that prevention is probably the most important part of managing heart disease [14]. It is also a definitively more economic strategy and can be based on simpler risk factor-models such as FRS, which seem to be good Heart Metab. 2011; 49:33–35 Hot topics Low (< 10%) Intermediate High (> 20%) 100 physiological link between CAD and ischemic heart disease. 90 Obstructive CAD (%) 80 REFERENCES 70 60 50 40 30 20 10 0 Positive Negative Equivocal No test Results of noninvasive tests Figure 1. Patients with obstructive coronary artery disease (CAD), according to noninvasive test results. Results are presented according to the Framingham Risk Score (low, intermediate, or high). Reprinted with permission from [1]. ß2010 Massachusetts Medical Society. All rights reserved. CAD predictors. Contrary to the authors’ intentions, Patel et al.’s paper [1] provides strong evidence that coronary risk factors, as assessed by FRS, have a clear relation with coronary atherosclerosis but are not predictive of IHD (see Fig. 1). In conclusion, CAD is a widely accepted predictor of adverse clinical outcomes and its extent and severity are considered important prognostic factors. However, its use as a surrogate marker does not always coincide with the presence of established IHD. In this regard, the ‘‘low diagnostic yield’’ of coronary angiography points to the flawed patho- Heart Metab. 2011; 49:33–35 1. Patel MR, et al. Low diagnostic yield of elective coronary angiography. N Engl J Med. 2010;362:886–895. 2. Lucas FL, et al. Temporal trends in the utilization of diagnostic testing and treatments for cardiovascular disease in the United States, 1993–2001. Circulation. 2006;113:374–379. 3. Lin F, et al. Multidetector computed tomography coronary artery plaque predictors of stress-induced myocardial ischemia by SPECT. Atherosclerosis. 2008;197:700–709. 4. Wieneke H, et al. Non-invasive characterization of cardiac microvascular disease by nuclear medicine using singlephoton emission tomography. Herz. 1999;24:515–521. 5. Soman P, et al. Vascular endothelial dysfunction is associated with reversible myocardial perfusion defects in the absence of obstructive coronary artery disease. J Nucl Cardiol. 2006;13: 756–760. 6. van Werkhoven JM, et al. Anatomic correlates of a normal perfusion scan using 64-slice computed tomographic coronary angiography. Am J Cardiol. 2008;101:40–45. 7. McGill HC Jr et al. Association of coronary heart disease risk factors with microscopic qualities of coronary atherosclerosis in youth. Circulation. 2000;102:374–379. 8. Gibbons RJ. Noninvasive diagnosis and prognosis assessment in chronic coronary artery disease: stress testing with and without imaging perspective. Circ Cardiovasc Imaging. 2008;1:257–269; discussion 269. 9. Boden WE, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med. 2007;356:1503–1516. 10. Henderson RA, et al. Seven-year outcome in the RITA-2 trial: coronary angioplasty versus medical therapy. J Am Coll Cardiol. 2003;42:1161–1170. 11. Narula J, Strauss HW. The popcorn plaques. Nat Med. 2007;13:532–534. 12. Chow BJ, et al. Diagnostic accuracy and impact of computed tomographic coronary angiography on utilization of invasive coronary angiography. Circ Cardiovasc Imaging. 2009;2:16–23. 13. Adamu U, et al. Results of interventional treatment of stress positive coronary artery disease. Am J Cardiol. 2010;105: 1535– 1539. 14. Mosca L, et al. Waist circumference predicts cardiometabolic and global Framingham risk among women screened during National Woman’s Heart Day. J Womens Health (Larchmt). 2006;15:24–34. 35 Glossary Glossary Gary D. Lopaschuk Amyloidosis A large, heterogeneous group of diseases characterized by themisfolding of extracellular protein. Misfolding occurs in parallel or as an alternative to physiological folding and generates insoluble protein aggregates (bundles of b-sheet fibrillar protein). Despite possessing heterogeneous structures and functions, fibrillar proteins are morphologically similar. Organ dysfunction results from the deposition and cytotoxic effects of insoluble amyloid proteins. Beta-oxidation inhibitors Compounds or drugs that inhibit mitochondrial fatty acid beta-oxidation. They do so primarily by either directly inhibiting mitochondrial fatty acid betaoxidation enzymes (i.e., trimetazidine, an inhibitor of 3ketoacyl CoA thiolase), or preventing the uptake of fatty acids into the mitochondria (i.e., perhexiline, an inhibitor of carnitine palmitoyl transferase). Cardiomyopathy Cardiomyopathy simply means ‘‘heart muscle disease’’, and refers to any type of deterioration of heart muscle function. If this dysfunction is due to ischemia, it is referred to as ischemic cardiomyopathy, or if it is due to underlying diabetes, it is referred to as diabetic cardiomyopathy. Carnitine palmitoyl transferase 1 (CPT-1) inhibitors Compounds or drugs that inhibit the mitochondrial outer membrane enzyme CPT-1, which is the ratelimiting enzyme for mitochondrial fatty acid uptake and subsequent beta-oxidation. Therefore, CPT-1 inhibitors inhibit mitochondrial fatty acid beta-oxidation secondary to an inhibition of its uptake into the mitochondria. Dichloroacetate (DCA) Dichloroacetate (DCA) is an inhibitor of pyruvate dehydrogenase kinase, which is the enzyme responHeart Metab. 2011; 49:37–38 sible for phosphorylating and inactivating pyruvate dehydrogenase, the rate-limiting enzyme of glucose oxidation. DCA therefore activates pyruvate dehydrogenase and increases subsequent glucose oxidation rates. Free fatty acids (FFAs) Acid moieties found in the circulation bound to albumin, or derived from triacylglycerol contained in chylomicrons or very-low density lipoprotein. Following cellular uptake, free fatty acids are activated via esterification to coenzyme A, and can be metabolized via mitochondrial fatty acid b-oxidation to generate reducing equivalents (e.g., nicotinamide adenine dinucleotide [NADH]) for the electron transport chain and oxidative phosphorylation. Immunoabsorption (affinity chromatography) A chromatographic method for the purification of a specific molecule(s) from a complex mixture(s) based on the highly specific biological interaction between twomolecules (i.e., antibody and antigen). The interaction is usually reversible, and purification is achieved by immobilizing one of the molecules (affinity ligand) onto a solid matrix, thereby creating a stationary phase, while the target ligand is in a mobile phase as part of a complex mixture. Capture of the target molecule is typically followed by washing and elution, which results in the recovery of a purified molecular species. Nicotinamide adenine dinucleotide (NADþ/NADH) A coenzyme critical to life in all living cells that consists of two nucleotides joined through a phosphate group. One of the nucleotides possesses an adenine base, whereas the other possesses nicotinamide. As a coenzyme, it is involved in numerous redox reactions that occur in metabolism. One of its major roles is to act as a reducing agent and electron donor during the electron transport chain, which is critical to adenosine-5’-triphosphate (ATP) 37 Glossary Gary D. Lopaschuk production during the process of oxidative phosphorylation. Pyruvate dehydrogenase (PDH) Pyruvate dehydrogenase (PDH) is a mitochondrial enzyme that catalyzes the committed step of pyruvate oxidation (i.e., oxidative decarboxylation), thereby generating acetyl coenzyme A (CoA) for the tricar- 38 boxylic acid cycle and nicotinamide adenine dinucleotide (NADH) for the electron transport chain. PDH is part of a multienzyme complex, consisting of PDH kinase and PDH phosphatase. Phosphorylation of PDH by PDH kinase inhibits its activity, while dephosphorylation by PDH phosphates increases its activity. PDH activity is also sensitive to inhibition by substrate/product ratios as decreased ratios of NADþ/ NADH and CoA/ acetyl-CoA decrease the rate of pyruvate oxidation. Heart Metab. 2011; 49:37–38





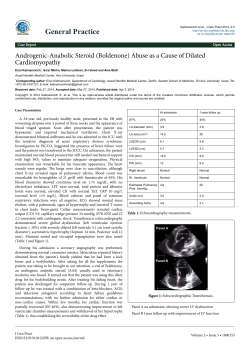






© Copyright 2026