
學生壁報摘要 - 國立中正大學
Franck-Condon simulation with damped harmonic oscillators: solvent effects for molecular electronic spectroscopy of azacarbazole and carbazole Chen-Wen Wang (王辰文) and Chaoyuan Zhu* (朱超原) Department of Applied Chemistry, National Chiao Tung University, Address: 1001, Ta-Hsueh Rd. Hsinchu 30010, Taiwan e-mail: [email protected] We have developed Franck-Condon simulation with damped harmonic oscillators to deal with solvent effects in molecular absorption and fluorescence spectra [1]. This method is applied to study AC(1-azacarbazole) and CZ(carbazole) molecules in hexane and ACN solutions [2]. Both AC and CZ are the crucial biomolecules that play an important role in the human body and thus it is great interesting to understand solvent effect of spectra. In order to simulate the absorption and fluorescence spectra in solutions observed in experiment, we first utilize ab initio quantum chemistry method at the level of density functional theory (DFT) and time-dependent DFT method, namely (TD)B3LYP, (TD)B3LYP35, (TD)B3LYP50 and (TD)B3LYP100, to optimize structures of ground- and excited-state of AC and CZ molecules. Then, we utilize the Franck-Condon simulation to study absorption and fluorescence spectra in the two solutions mentioned above. We report our simulation results in this poster. References [1] C.-W. Wang, L. Yang, C. Zhu, J.-G. Yu, and S. H. Lin, J. Chem. Phys. 141, 084106 (2014). [2] E. García-Fernández, C. Carmona, M. A. Munoz, J. Hidalgo, M. Balon, Spectrochimica Acta Part A 84 130 (2011). Gate Control of Single-Molecule Machines Liang-Yan Hsu,∗,† Chun-Yin Chen,‡ Elise Y. Li,∗,‡ and Herschel Rabitz∗,† † Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States, and ‡Department of Chemistry, National Taiwan Normal University, Taipei 11677, Taiwan [email protected] Artificial molecular machines are a growing field in nanoscience and nanotechnology. [1,2] This study proposes a new class of artificial molecular machines, the secondgeneration singlemolecule electric revolving doors (2G S-MERDs), a direct extension of our previous work [3]. We investigate destructive quantum interference with tunneling and conductance dependence upon molecular conformation in the 2G S-MERDs by using the Green’s function method together with density functional theory. The simulations show that the 2G S-MERDs have a large on-off conductance ratio (> 104), and that their open and closed door states can be operated by an experimentally feasible external electric field (1 V/nm). Conductance - gate electric field characteristics are also introduced to illustrate the operation of the 2G S-MERDs. References [1] Koumura, N.; Zijlstra, R. W. J.; Delden, R. A. v.; Harada, N.; Feringa, B. L. Light-Driven Monodirectional Molecular Rotor. Nature 1999, 401, 152–155. [2] Delden, R. A. v.; Wiel, M. K. J. t.; Pollard, M. M.; Vicario, J.; Koumura, N.; Feringa, B. L.Unidirectional Molecular Motor on a Gold Surface. Nature 2005, 437, 1337–1340 [3] Hsu, L.-Y.; Li, E. Y.; Rabitz, H. Single-Molecule Electric Revolving Door. Nano Lett. 2013,13, 5020–5025. Semi-Quantitative Assessment of Intersystem Crossing Rate: An Extension of El-Sayed Rule to the Emissive Transition Metal Complexes Elise Yu-Tzu Li1* (李祐慈),Tzung-Ying Jiang1(姜宗螢),Tsao-Pei Chou1(周晁霈), Yun Chi2(季昀),Pi-Tai Chou3(周必泰) 1 Department of Chemistry, National Taiwan Normal University, Taipei 116, Taiwan Department of Chemistry, National Tsing Hua University, Hsinchu 300, Taiwan 3 Department of Chemistry and Center for Emerging Material and Advanced Devices, 2 National Taiwan University, Taipei 106, Taiwan The main goal of this study is to provide systematic elucidation of the parameters that influence S T intersystem crossing (ISC). Particular attention is paid to: (i) the computation of Sn Tm spinorbit coupling strength based on a non-adiabatic approach, (ii) crucial factors that facilitate ISC, such as the atomic number, ligand structure, and particularly types of electronic transition, (iii) formulating a discussion on the standpoints of the fundamental photophysical theory. Combining the theoretical and empirical approaches, we propose the following empirical rules as general guidelines for assessing the ISC in a qualitative manner: (1) In general, the higher MLCT characteristic involved in the singlet and triplet excited states, the stronger the SOC integral. However, the strength of SOC also depends strongly on the detailed electronic structure of the states involved. (2) When the S1 and T1 originate from almost identical electronic transitions, the major ISC channel usually occurs via S1 to higher Tm (m > 1) state, which has both a proper orbital transition and a smaller energy gap with S1. (3) When the structure of the ligands involves extended conjugated systems and that the lowest excited states have a more dominating ππ* character, the ISC originating from higher singlet excited states can be large enough to violate the Kasha’s rule. (4) The overall SOC strength (and ISC rate) relies on a delicate balance between the one-electron SOC constant and the nature electronic structure of the involved states. In other words, the “heavy atom effect” might not be so heavily weighted after all; through a careful design, lighter elements may also exhibit similar emission efficiency with that of heavier elements. References [1] Cheng, Y. M.; Li, E. Y.; Lee, G. H.; Chou, P. T.; Lin, S. Y.; Shu, C. F.; Hwang, K. C.; Chen, Y. L.; Song, Y. H.; Chi, Y. Inorg. Chem. 2007, 46, 10276–10286. [2]Hsu, C. W.; Lin, C. C.; Chung, M. W.; Chi, Y.; Lee, G. H.; Chou, P. T.; Chang, C. H.; Chen, P. Y. J. Am. Chem. Soc. 2011, 133, 12085 [3] El-Sayed, M. J. Chem. Phys. 1963, 38, 2834. [4] Elise Yu-Tzu Li, Tzung-Ying Jiang, Yun Chi and Pi-Tai Chou. Phys. Chem. Chem. Phys., 2014, DOI: 10.1039/c4cp03540b Probing π→π* photoisomerization mechanism for cis-azobenzene by on-the-fly trajectory-based nonadiabatic molecular dynamics simulation Le Yu(于樂) and Chaoyuan Zhu*(朱超原) Department of Applied Chemistry, National Chiao Tung University, Address: 1001, Ta-Hsueh Rd. Hsinchu 30010, Taiwan e-mail: [email protected] We developed nonadiabatic trajectory surface hopping algorithm without calculating nonadiabatic coupling vector and applied this new method to the azobenzene S1 state photoisomerization dynamics for both cis-to-trans and trans-to-cis reactions [1]. Actually, the photoisomerization dynamics observed in experiment is involved in several lowing lying excited states. We probe this complicated π→π* photoisomerization reaction mechanisms from cis-azobenzene. We found there are seven conical intersections among the four low-lying singlet states and a long conical seam between S2 and S3 states. The five-state-averaged CASSCF method was utilized in the trajectory simulation to probe the cis-azobenzene photodynamic mechanisms. In both S2 and S3 states, we found significantly distinguishable fast and slow reaction processes; the fast channels usually needs several hundred fs to achieve the final product, and in the slow channels, there exists the excited state trans-azobenzene intermediate which has been confirmed in the recent transient absorption spectroscopy experiment [2]. For trajectories started from S2 state, all the mechanisms dominated by the torsion of CNNC and NNCC moiety, while for trajectories started from S3 state, the mixed torsion of CNNC and inversion of NNC motions were confirmed by the present nonadiabatic molecular dynamics simulation. References [1] L. Yu, C. Xu, , Y. Lei, C. Zhu, and Z. Wen, Phys. Chem. Chem. Phys., (in press, 2014). [2] M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting, and S. A. Kovalenko, J. Phys. Chem. B, 118, 8756 (2014). Nonadiabatic trajectory surface hopping for photochemical reaction of o-nitrophenol in the low-lying singlet and triplet excited states Chao Xu(徐超), Le Yu(于樂), Chaoyuan Zhu*(朱超原) and Jian-Guo Yu Department of Applied Chemistry, National Chiao Tung University, Address: 1001, Ta-Hsueh Rd. Hsinchu 30010, Taiwan e-mail: [email protected] We developed nonadiabatic trajectory surface hopping algorithm without calculating nonadiabatic coupling vector for nonadiabatic processes involving in singlet excited states[1], and plus calculating spin-orbital coupling we extend this method to deal with intersystem crossing for nonadiabatic processes involving in both singlet and triplet excited states. We study and analyze the most efficient photophysical and photochemical decay and isomerization pathways for the o-nitrophenol molecule [2]. By using ab initio quantum chemistry method at level of SA-CASSCF(10, 10)/6-31G (d, p) and MRCI+Q/cc-pVDZ, we optimize the S0, T1, T2 and S1 equilibrium structures, and the several transition states linking nitro with aci-nitro isomers in the T1 and S0 states. From those static points of potential energy surfaces of low-lying singlet and triplet excited states, we could qualitatively understand photochemical reaction mechanism of o-nitrophenol. However, for quantitatively understanding, we perform nonadiabatic dynamics simulation involving on-the-fly calculation of global potential energy surfaces on coupled low-lying singlet and triplet excited states. We will report simulation results in this poster and hopefully provide detailed mechanistic insight by depicting the most relevant channels for the decay and the reaction funnels. References [1] L. Yu, C. Xu, , Y. Lei, C. Zhu, and Z. Wen, Phys. Chem. Chem. Phys., (in press, 2014). [2] I. Bejan, Y. A. E., I. Barnes et al., Phys. Chem. Chem. Phys. 8, 2028 (2006). Shape resonances of H2S, H2Se, and H2Te Yin-Cheng Chen and Hsiu-Yao Cheng Department of Chemistry, Tunghai University, 1727, Taiwan Boulevard Sec. 4, Taichung 40704, Taiwan, Republic of China. e-mail: [email protected]; [email protected] The stabilized Koopmans’ theorem in long-range corrected density functional theory is used to investigate the shape resonances of H 2X molecules (X: S, Se, and Te). In this approach, stabilization is accomplished by varying the exponents of appropriate diffuse functions. The resonance energies are then identified by investigating the relationship between the resultant eigenvalues and scale parameter. The characteristics of resonance orbitals are also examined. For the lowest unfilled orbitals of H2X molecules, results indicate that they are essentially *(H-X) in character. Moreover, several shape resonances with strong X “d” characters have been identified. These results will help us understand the bonding and chemical properties of these hydrides. References: 1. Dezarnaud, C.; Tronc, M.; Modelli, A. Chem. Phys. 1991, 156, 129-140. 2. Varella, M. T. D. N.; Bettega, M. H.; Lima, M. A.; Ferreira, L. G. J. Chem. Phys. 1999, 111, 6396-6406. 3. Cheng, H.-Y.; Chen, C.-W. J. Phys. Chem. A 2012, 116, 12364-12372. Familial Mutations of Alzheimer Amyloid-Peptide Alter the Configurations of Central Hydrophobic Core within Membrane: A Replica Exchange Molecular Dynamics Study Jian-Bin Lee and Hui-Hsu Gavin Tsai* Department of Chemistry, National Central University, Jhong-Li City, Tao-Yuan County 32001, Taiwan Correspondence e-mail address: [email protected] Extracellular deposits of amyloid (A) aggregates in the brain is the hallmark of Alzheimer’s disease (AD). Ais cleaved from the amyloid precursor protein (APP) by- and -secretases. A number of mutations in the APP have been shown to associate with several familial AD. The significant pathogenic mutations occur in the region of E22 and D23 in the A sequence. For instance, E22Q (Dutch) and D23N (Iowa) mutants exhibits higher neurotoxicity and aggregation rate than wildtype A(1–40). To our best knowledge, there is not systematic study of these mutants by MD simulation in a water-membrane environment. Here, we employ replica–exchange molecular dynamics to investigate the configuration of the A E22Q-A, D23N-A and E22Q/D23N-A mutants in water-membrane environments. Based on our results, we observe that the network of residue contacts between N-terminal (residues 1-16) and central hydrophobic core (CHC) fragment (residues 17-21) is decreased on familial mutants. In addition, we observe that the configuration transition of CHC fragment in familial mutants is easier than wildtype A(1–40). It is indicated more independent N-terminal fragment and highly exposure of the hydrophobic CHC fragment of familial mutants in water-membrane environment. Therefore, the loose structure and frequently configurational transition of CHC fragments of familial Aβ within the membrane is an important role for initial amyloid peptide aggregation. In this study, the CHC fragment’s properties could explain a part of rapid amyloid aggregation of familial mutant peptides and provide the insight to rational drug design to prevent amyloid associated diseases. Scanning Alanine Mutagenesis Study of the Stability of Alzheimer’s Amyloid Fibril-like Oligomer by Molecular Dynamics Simulations Wen-Yuan Chu and Hui-Hsu Gavin Tsai* Department of Chemistry, National Central University, Jhong-Li City, Tao-Yuan County 32001, Taiwan Correspondence e-mail address: [email protected] Alzheimer’s disease (AD) is characterized by the extracellular deposit of senile plaques in the brain. Senile plaques are mainly composed of the aggregated amyloid beta (Aβ) protein. called amyloid. Amyloid fibrils are semi-ordered nanostructures as the result of self assembly of proteins when they are misfolded under critical conditions. Due to the complexity of Aβ amyloids, the underlying biophysical mechanisms of formation and toxicity are still unclear. Therefore, it is crucial to determine the key residues in stabilizing Aβ amyloid fibrils. In this study, we employed all atom molecular dynamics simulations to investigate the relative stability of Aβ-fibril like oligomer and its mutants by point mutagenesis. To investigate the intra-peptide interactions, we simulated the structures of Aβ-fibril like oligomer with one of its residue is systemically mutated to Ala by MD simulations. The secondary structure, inter-peptide backbone hydrogen-bonds, solvent-accessible surface area (SASA) of the central hydrophobic core, and RMSD from the solid-state NMR determined structure were analyzed. Our results show the following tendency. For the mutagenesis of the residues within the hydrophobic core, the simulated RMSD is increased when (a). charged and/or polar residues were mutated to hydrophobic Ala such as D23A, K28A, and N27A and (b). larger hydrophobic residues were mutated to smaller hydrophobic Ala such as F19A and V40A. The former one is due to the imbalance of electrostatic interaction and the later one is due to the loss of shape complementarity. Interestingly, the point mutations on the pointing-out residues on 1-sheet do not cause significant structural instability, while the point mutations on the pointing-out residues on 2-sheet generally lead to structural instability. These results provide clues to design drugs preventing the formation of Aβ-fibrils. Fig1. Representation the F19A is loss of shape complementarity. Fig2. Representation the charged were mutated to hydrophobic Ala. D23A is absence of the Asp23-Lys28 salt bridge. Electron Transfer of Squaraine-Derived Dyes Adsorbed on TiO2 Clusters in Dye-Sensitized Solar Cells: A Density Functional Theory Investigation Chun-Jui Tan, Wen-Hsin Tseng and Hui-Hsu Gavin Tsai* Department of Chemistry, National Central University, Jhong-Li City, Tao-Yuan County 32001, Taiwan E-mail: [email protected] Squaraine (SQ) dyes are well-known for their intense absorption in the red/near-IR spectral regions to ultilize the solar radiation. SQ-derived dyes are supposed to have effective potential applied in dye-sensitized solar cells (DSCs). In this research, we employed density functional theory (DFT) and time-dependent DFT to investigate the structural, optical, and electron transfer properties of seven recently reported SQ-derived dyes adsorbed on (TiO2)38 cluster with anatase (101) surface corresponding to DSCs. Particular efforts were given to calculate the proportions of the electron densities in the dye–(TiO2)38 systems transferred to TiO2 moieties upon photo-excitation, allowing us to investigate their electron injection mechanisms. JD10 and YR6 have two intense absorption bands with significant proportions of electron density delocalized into TiO 2 upon excitation and have driving forces for excited-state electron injection, followed direct electron injection mechanism. These results are compatible with their higher experimentally observed shortcircuit currents (Jsc) than those of the SQ dyes. In contrast, SQ12, SQ2, and SQ4 followed the indirect electron injection mechanism because of their negligible proportion of electron density injected into TiO2; in addition, SQ2 and SQ4 do not provide driving force for electron injection. The Jsc values of SQ12, SQ2, and SQ4 dyes observed in experiment were lower than that of other studied SQ dyes. The calculated probabilities of electron density being delocalized into TiO2 and driving force for excited-state electron injection from these studied SQ dyes are correlated with experimentally observed Jsc values. This study provides insight into the electron injection mechanisms of SQ-derived dyes adsorbed on TiO2 upon photo-excitation. Furthermore, our calculations and findings give clues for designing new SQ-derived sensitizers for DSC applications. A DFT Study of the Photo-Absorption and Photo-Emission Properties of two Novel D−A−π−A organic dyes Jia-Cheng Hu,a Sheng Hsiung Chang,b Kuo-Yuan Chiu,b Chun-Guey Wu,a,b and Hui-Hsu Gavin Tsaia* a. Department of Chemistry, National Central University, Jhong-Li City, Tao-Yuan County 32001, Taiwan b. Research Center for New Generation Photovoltaics, National Central University E-mail: [email protected] The working principle of DSCs is light-harvesting processes similar to those of the photosynthetic system in nature. One of the most successful and popular framework of organic dyes is a dipolar D-π-A structure based the push-pull architecture. D-π-A dyes generally have efficient intramolecular charge transfer (ICT) properties. Based on this strategy, two novel donor−acceptor−π−acceptor (D−A−π−A) organic dyes (CKY-33 and CKY-34) incorporating internal electron-withdrawing units of diketopyrrolopyrrole (DPP) have been newly synthesized and tested for DSC applications. CKY-33 and CKY-34 have similar framework, instead, CKY-33 has a phenyl ring for its π-spacer and CKY-34 a thiophene ring for its π-spacer. CKY-34 has a more red-shifted and intense absorption than that of CKY-33. In principle, CKY-34 has a better spectral match with sunlight radiation than that of CKY-33. However, CKY-34 has a slightly lower Jsc value and lower photo-to-cuurent efficiency than those of CKY-33. Broadband exciton dynamics of these two dye dissolved in THF solution shows the lifetime of “cold” exciton of CKY-33 dye is longer than that of CKY-34 dye. To understand the different lifetimes as well as different Jsc values of CKY-33 and CKY-34 dyes we performed the DFT and TD-DFT calculations to investigate the absorption and emission properties of these two dyes in THF and adosrbed on a (TiO2)38 cluster having an anatase (101) surface, as a model for corresponding DSCs. Our calculations show the CKY-34 has a stronger emission oscillator strength than that of CKY-33 indicating that the “cold” excition of CKY-34 will have a shorter lifetime. Our calculations are consistent with experimental observations. Oxidation of CO on a Carbon-based Material Composed of Nickel Hydroxide on Reduced Graphene Oxide, (Ni4(OH)3/rGO) - a FirstPrinciples Calculation Chen-Hao Yeh(葉丞豪) and Jia-Jen Ho(何嘉仁)* Department of Chemistry, National Taiwan Normal University, No. 88, Section 4, Tingchow Road, Taipei, Taiwan E-mail : [email protected] Nickel or nickel hydroxide clusters on reduced graphene oxide (rGO) are novel nanomaterials in the application of electrochemical catalysts. In this work, we calculated the energy of Ni4 adsorbed on saturated hydroxyl GO, which forms a Ni4(OH)3 cluster on the hydroxyl rGO (Ni4(OH)3/rGO) and releases 4.47 eV (5.22 eV with DFT-D3 correction). We subsequently studied the oxidation of CO on the Ni4(OH)3/rGO system via three mechanisms – LH, ER and carbonated mechanisms. Our results show that the activation energy for oxidation of the first CO molecule according to the ER mechanism is 0.14 eV (0.12 eV with DFT-D3 correction), much smaller than that with LH (Ea = 0.65 eV, 0.61 eV with DFT-D3 correction) and with carbonated (Ea = 1.28 eV, 1.20 eV with DFT-D3 correction) mechanisms. The barrier to oxidation of the second CO molecule to CO2 with the ER mechanism increases to 0.43 eV (0.37 eV with DFT-D3 correction), but still less than that via LH (Ea = 1.09 eV, 1.07 eV with DFT-D3 correction), indicating that CO could be effectively oxidized through the ER mechanism on the Ni4(OH)3/rGO catalyst. [1] Y. Matsumoto, H. Tateishi, M. Koinuma, Y. Kamei, C. Ogata, K. Gezuhara, K. Hatakeyama, S. Hayami, T. Taniguchi, A. Funatsu, J. Electroanal. Chem., 2013, 704, 233-241. [2] S. Rastgar, S. Shahrokhian, Talanta, 2014, 119, 156-163. CO Oxidation on Tungsten Carbide Surface: A Density Functional Study Yu-Jhe Tong, Hsin-Tsung Chen [email protected], [email protected] Chung Yuan Christian University, 200, Chung Pei Rd, Chung Li, Taiwan 32023, R.O.C Keywords: Tungsten carbide, gas adsorption, carbon oxide, Metal carbide, ab initio, density functional theory Abstract: Tungsten carbide (WC) has been popular discuss while its properties such like Platinum. WC is cheaper than Platinum and we can use it to replace Pt. It is a potential material that we can use for gas adsorption. Nowadays, the carbon oxide is a critical pollution in our life. We use the density functional theory calculation to find out the reaction of WC surface and carbon oxide. CO + O2 → CO2 + O CO + H2O → CO2 + H2 Following to the reaction, we discuss and find out the mechanism of carbon oxide react with other gas on WC surface. We also discuss different surface, location and direct to find the best chemical mechanism. And expect the reaction will be useful in the future. References: 1. R.B. Levy, M. Boudart, Platinum-like behavior of tungsten carbide in surface catalysis, Science 181 (1973) 547–549. 2. Zheng, W., Chen, L. , Ma, C, Density functional study of H2O adsorption and dissociation on WC(0001), Computational and Theoretical Chemistry 1039 (2014) 75-80 Nitrogen-doped carbon nanotubes as a metal-free catalyst for CO oxidation I Hsiang, Lin (林逸庠), Hsin-Tsung, Chen (陳欣聰)* Department of Chemistry, Chung Yuan Christian, University, Chung-Li, 32023, Taiwan [email protected], [email protected]* The oxidation of carbon monoxide (CO) on catalyst has attracted considerable research interest in recent years because it can remove CO from fuel cells or lower CO pollution. The effective catalyst for CO oxidation must comprise metal. We usually use noble metal catalyst for CO oxidation, but the noble metals ( Pd, Au, and Pt) are rare and expensive. So we must to find the new catalysts replacing the noble metals. In our research, We have study CO oxidation by O2 on nitrogen doped carbon nanotube (NCNT) with different diameter ((n,n)-NCNT (n= 3,4,5))by the density functional theory (DFT) calculations. We also find the reaction mechanism of CO oxidation and use NEB to map the potential energy surfaces. In our result, we show that the oxidation of CO energy barrier can reduce effectively. The result has demonstrated that NCNT is a good, low-cost, and metal-free catalyst for CO oxidation. CO2 Capture in Aluminum Metal-Organic Frameworks: A Theoretical Study Yu-Huan Lu (盧禹寰), Hsin-Tsung Chen* (陳欣聰) Department of Chemistry, Chung Yuan Christian University, Chungli 32023, Taiwan [email protected], [email protected] Keywords: DFT-D, MOF, Aluminum, TDC, FDCA Using density functional theory with van der Waals corrected functional, we study and explain trends in the binding between CO2 and two novel metal-organic frameworks (MOFs) with two kinds of ligands: 2,5-thiophenedicarboxylic acid (TDC), 2,5-furandicarboxylic acid (FDCA). The present work shows the TDC ligands exhibit six coordination modes, structure analysis reveals that 2,5-thiophenedicarboxylic acid play a versatile role toward different metal ions to form three-dimensional inorganic-organic hybrid frameworks. We use one of these modes (Figure 1) to calculate how CO2 molecule binding with TDC/FDCA-MOFs. A CO2 can form weak hydrogen bonding with electron deficient hydrogen of thiophene and furan rings. The present sulfone groups increases hydrogen bonding in CO2-TDC/FDCA complex. This study elucidates the Lewis-base interaction and hydrogen bonding may be important stabilizing interactions that merit consideration in the design of future CO2-philes. Figure 1 Adsorption and Reaction of O2 and C2H4 on Cu38 Clusters: A Computational Study Chen-Chi, Lee (李偵綺), Hsin-Tsung, Chen (陳欣聰)* Department of Chemistry, Chung Yuan Christian University, Chung-Li, 32023, Taiwan [email protected], [email protected]* Keywords: DFT, Cu38, O2, C2H4, Coadsorption The heterogeneously catalyzed epoxidation of alkenes is experimentally challenging, so we try to use theoretically method to find the better route. We had studied the adsorption of O2 and C2H4 molecules on Au38 by using density functional theory (DFT) calculations already, in this case, we try to confer the coadsorption on Cu38. Some adsorption sites were discussed in this study and characterized as top, bridge, hollow, and hcp sites. Cooperative adsorption O2 and C2H4 on Cu38 clusters were investigated. To predict the competing reaction pathways for epoxide formation versus acetaldehyde formation on Cu38 and search out a lower transition state (TS), we use NEB method to map the potential energy surfaces (PES). Results suggest that epoxidation proceeds via a surface oxametallacycle intermediate (OMME structure), such as a ring structure, Cu-CC-O-Cu, it is the key of this reaction. Figure 1. Located isomers of coadsorbed O2&C2H4 on the Au38 nanoparticle . The bond lengths are given in angstroms. References [1] Andrey, L.; Tetsuya T. J. Phys. Chem. Lett., 2009, 113, 12930-12934. [2] W. Gao; X. F. Chen; J. C. Li; Q. Jiang. J. Phys. Chem. C, 2010, 114, 11481153. [3] H. T. Chen; J. G. Chang; S. P. Ju; H. L. Chen. J. Phys. Chem. Lett., 2010, 1, 739-742. A Study of the Reaction Mechanism of Wittig Rearrangements by Ab Initio Computations Pei-Kang Tsou and Chin-Hui Yu* Department of Chemistry, National Tsing-Hua University, HsinChu 300 Taiwan E-mail : [email protected] The high regio- and stereo-selectivity of Wittig rearrangements makes them a versatile tool in organic synthesis. The [2,3]-Wittig rearrangement is a concerted reaction with a high degree of stereocontrol. On the other hand, the concerted [1,2]-Wittig rearrangement is a orbital-symmetry forbidden process and is assumed to go through a stepwise pathway. An ionic and a radical pair are proposed as possible intermediates along the stepwise path. Simple model compounds are used to analyze important reaction structures along the concerted and the stepwise reaction paths using the multireference CASSCF/CASPT2 and density function theory methods. The oxygen-lithium chelation fixes the key intermediate geometry, and controlls regio- and stereo-selectivity in the following [1,2] and [2,3] rearrangements along the stepwise pathway. The interaction between the solvent molecules and the Li+ are calculated to examine the salvation effect via DFT method. A theoretical study on the phosphorescence spectra of sulfur dioxide Kuan-Cheng Lin (林冠臣), Jia-Lin Chang(張嘉麟)* Department of Science Application and Dissemination, National Taichung University of Education, Taichung 403, Taiwan (KCL email: [email protected]; JLC email: [email protected]) Abstract The equilibrium geometries and vibrational frequencies of the singlet ground state and three triplet excited states of sulfur dioxide were computed by using the density-functional theory and the coupled-cluster theory with various basis sets. The phosphorescence spectra of SO2 were simulated by using the approach developed by our group, in which the harmonic-oscillator model with the Duschinscky effect was taken into account.1 The adiabatic excitation energies were obtained by extrapolating the CCSD(T) energies to the complete basis set (CBS) limit. The simulated phosphorescence spectra are in agreement with the experiments and provide insights into the vibrational structures. The calculated excitation energies are in excellent agreement with the experimental values. References 1. J.-L. Chang, C.-H. Huang, S.-C. Chen, T.-H. Yin, Y.-T. Chen, J. Comput. Chem. 34, 757 (2013). FePt Nanodendrites with High-Index Facets as Active Electrocatalysts for Oxygen Reduction Reaction – DFT Simulation Studies Hung-Lung Chou,1,* Di-Yan Wang,2,3 Ching-Che Cheng,2 Yu-Han Wu,2 Chin-Ming Tsai,2 HengYi Lin,2 Yuh-Lin Wang,2 Bing-Joe Hwang,4,5 and Chia-Chun Chen,2,3 1 Graduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology, Taipei, 10617, Taiwan 2 Department of Chemistry, National Taiwan Normal University, Taipei 106, Taiwan. 3 Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei 106, Taiwan Nanoelectrochemistry Laboratory, Department of Chemical Engineering, National Taiwan University of Science & Technology, Taipei 106, Taiwan 5 National Synchrotron Radiation Research Center (NSRRC), Hsinchu 300, Taiwan. Corresponding Information: [email protected] 4 Abstract In this study, three different types of alloyed FePt nanstructures, nanodendrites, nanospheres and nanocubes, were prepared and their catalytic activities for oxygen reduction reaction (ORR) were studied. The ORR catalytic activity of the nanostructures were increased in the order of E-TEK Pt/C < FePt nanospheres < FePt nanocubes < FePt nanodendrites. In particular, a cation exchanging reaction was developed for the preparation of FePt nanodendrites, consisting of a dense array of branches on a core. The FePt nanostructures were analyzed by high-resolution transmission electron microscopy (HRTEM), high angle annular dark field (HAADF), scanning transmission electron microscopy (STEM) and electron energy loss spectrum (EELS) mapping. The HRTEM images revealed that the large surface area of FePt nanodendrites with a high density of atomic steps was enclosed by high-index {311} facet. The density functional theory simulation was performed to understand the origins of the enhanced electrochemical activity of FePt nanodendrites. The enhancement could be attributed to the exposure of high-index {311} facet of the nanodendrite with high surface energy in comparison to that low-index {111} and {200} facets of FePt nanospheres and nanocubes, respectively. Our experimental and theoretical studies have opened a route toward the syntheses of new nonprecious alloyed nanostructures to replace Pt as active fuel cell catalysts. Keywords: FePt, Nanodendrites, High-index Facet, Density Functional Theory, Oxygen Reduction Reaction Acknowledgement: We thank the NCHC for providing massive computing time. Financial support from the National Science Council under Contracts number MOST 103-2221-E-011-041 is gratefully acknowledged. Investigation of Kinase-Ligand Binding Affinity Using Thermodynamic Integration MD Simulation, Docking Computation, and Kinase Assay Hsing-Chou Lee (李興洲), Wen-Chi Hsu (許文綺), An-Lun Liu (劉安倫), ChiaJen Hsu (許嘉仁), Chia-Ming Chang(張家銘)Ying-Chieh Sun (孫英傑) Department of Chemistry, National Taiwan Normal University [email protected]; [email protected] (A.L. Liu) Thermodynamic integration molecular dynamics simulation was used to investigate how TI-MD simulation preforms in reproducing relative protein-ligand binding free energy of a pair of analogous GSK3 kinase inhibitors of available experimental data, and to predict the affinity for other analogs. The computation for the pair gave a G of 1.0 kcal/mol, which was in reasonably good agreement with the experimental value of 0.1 kcal/mol. The error bar was estimated at 0.5 kcal/mol. Subsequently, we employed the same protocol to proceed with simulations to find analogous inhibitors with a stronger affinity. Four analogs with a substitution at one site inside the binding pocket were the first to be tried, but no significant enhancement in affinity was found. Subsequent simulations for another 7 analogs was focused on substitutions at the benzene ring of another site, which gave two analogs with G values of -0.6 and -0.8 kcal/mol, respectively. Both analogs had a -OH group at the meta position and another -OH group at the ortho position at the other side of the benzene ring. To explore further, another 4 analogs with this characteristic were investigated. Three analogs with G values of -2.2, -1.7 and -1.2 kcal/mol, respectively, were found. Hydrogen bond analysis suggested that the additional hydrogen bonds of the added –OH groups with Gln185 and/or Asn64 were the main contributors to an enhanced affinity. Effect of displacement of crystal water molecules at binding site will be discussed. The prediction for better inhibitors should interest experimentalists of enzyme and/or cell assays. Figure 1: Binding of the reference compound with GSK3 kinase [1] In addition, progress in using new module in Amber package, pmemd, experimental measurement for finding new GSK3 kinase inhibitors, and a study of p38 kinase-ligand complexes will be reported and discussed. [2] References [1] Lee, H.C.; Hsu, W.C.; Liu, A.L.; Hsu, C.J.; Sun, Y.C., J. Mol. Graphics & Model. 2014, 51, 37-49. [2] Hsu, W.C.; Liu, A.L.; Hsu, C.J.; Sun, Y.C. (in preparation). Dynamic Solvation Shell and Solubility of C60 in Organic Solvents Chun I Wang (王俊壹)1, Chi C. Hua (華繼中)1*, and Show A. Chen (陳壽安)2 1 Department of Chemical Engineering, National Chung Cheng University, Chia-Yi, Taiwan 2 Department of Chemical Engineering, National Tsing Hua University, Hsin-Chu, Taiwan *E-mail: [email protected] ABSTRACT The notion of (static) solvation shells has recently proved fruitful in revealing key molecular factors that dictate the solubility and aggregation properties of fullerene species in polar or ionic solvent media. Using molecular dynamics schemes, we have scrutinized both the static and the dynamic features of the solvation shells of single C 60 particle for five non-polar organic solvents (i.e., chloroform, toluene, and chlorobenzene, 1,2dichlorobenzene, 1,4-dichlorobenzene) and a range of system temperatures (i.e., T = 250330 K). The central findings have been that, while the static structures of the solvation shell remain, in general, insensitive to the effects of changing solvent type or system temperature, the dynamic behavior of solvent molecules within the shell exhibits prominent dependence on both factors. Detailed analyses led us to propose the notion of dynamically stable solvation shell, effectiveness of which can be characterized by a new physical parameter defined as the ratio of two fundamental time constants representing, respectively, the solvent relaxation (or residence) time within the first solvation shell and the characteristic time required for the fullerene particle to diffuse a distance comparable to the shell thickness. We show that, for the six (one from the literature) different solvent media and the range of system temperatures examined herein, this parameter bears a value around unity and, in particular, correlates intimately with known trends of solubility for C60 solutions. We also provide evidence revealing that, in addition to fullerene-solvent interactions, solvent-solvent interactions play an important role, too, in shaping the dynamic solvation shell, as implied by recent experimental trends. A Computational Study of the Hydrogen Adduct of 4-, 6-, 7-membered N-Heterocyclic Carbene Chin-Hung Lai School of Medical Applied Chemistry, Chung Shan Medical University, 402, Taichung, Taiwan; Department of Medical Education, Chung Shan Medical University Hospital, 402, Taichung, Taiwan. Phone: 886-4-24730022-12224 EMAIL ADDRESS: [email protected] Keywords: N-heterocyclic carbene; N,N’-diamidocarbene; Hydrogen adduct; Hydrogen storage material; ring size In our previous study,1 we found that the hydrogen adduct of a N-heterocyclic carbene is a more efficient hydrogen storage material than a N,N’-diamidocarbene is. Furthermore, the efficiency of the hydrogen adduct of a N,N-diamidocarbene to act as a hydrogen storage material is influenced by its ring size. In this study, the influence of the ring size of a N-heterocyclic carbene on the efficiency to behave as a hydrogen storage material is investigated. Reference: 1. Lai, C.-H. J. Mol. Model. 2013, 19, 5523-5532. The Capabilities of an Abnormal N-heterocyclic Carbene to Form Hetero- and Homodimer Chin-Hung Lai School of Medical Applied Chemistry, Chung Shan Medical University, 402, Taichung, Taiwan; Department of Medical Education, Chung Shan Medical University Hospital, 402, Taichung, Taiwan. Phone: 886-4-24730022-12224 EMAIL ADDRESS: [email protected] Keywords: Heterodimer; Homodimer; DFT; abnormal N-heterocyclic carbene; dimerization. In this study, we investigate the capability of imidazol-4-ylidene (aNHC) to undergo homodimerization or heterodimerization with its isomer imidazol-2-ylidene (NHC) by the M06-2X functional. Unlike its NHC isomer, the stability of aNHCs does not depend on low-barrier dimerization and they have a lower tendency toward dimerization. We tentatively ascribe this to the fact that the aromatic stabilization energy of imidazol-4-ylidene is larger than that of imidazol-2-ylidene. Adsorption of a Pt13 Cluster on Graphene Oxides at Varied Ratios of Oxygen to Carbon and Its Catalytic Reactions for CO Removal Investigated with Quantum-chemical Calculations Shiuan-Yau Wu (吳亘曜) and Jia-Jen Ho (何嘉仁)* Department of Chemistry, National Taiwan Normal University, No. 88, Section 4, Tingchow Road, Taipei, Taiwan * Corresponding author E-mail: [email protected] We applied periodic density-functional theory to investigate the interaction of a Pt13 cluster on graphene-oxide (GO) sheets at varied ratios of oxygen to carbon, and on a pristine graphene sheet. Relative to a pristine graphene sheet, the existence of oxygen atoms in an appropriate proportion in the formation of graphene oxide enhanced the adsorption capability of a Pt13 cluster. The O/C ratio of GO sheets had the following influences on the Pt13 cluster adsorption behavior: (i) for O/C ratio < 0.125, the Pt13 cluster abstracted the neighboring oxygen atom from a GO sheet to form Pt 13O, or aggregated with an adjacent cluster to form a larger cluster; (ii) for O/C ratio≧ 0.125, the Pt 13 cluster was stabilized and dispersed on the GO sheet. We calculated also the adsorption behavior of carbon monoxide on a Pt13/GO sheet; a strong interaction between the Pt13 cluster and the GO sheet modulated the electronic structure of the Pt13 cluster, thus decreasing the CO adsorption energy, which in turn decreased the combined barriers, CO + O and CO + OH, in the water-gas shift reaction (WGSR), and improved the CO tolerance of the Pt catalyst. Name: Chun-Chih Chang Email: [email protected] This abstract is for a: Poster Abstract Catalytic Activity Enhancement of CO2 Dissociation on Unzipped Graphene Oxide Supported Rhodium Nanoclusters – A DFT Study Chun-Chih Chang* and Jia-Jen Ho Department of Chemistry, National Taiwan Normal University, No. 88, Section 4, Tingchow Road, Taipei, Taiwan *presenting author, email: [email protected] The catalytic activity of rhodium nanoclusters on unzipped graphene oxide (Rhn/UGO) has been investigated to compare with that of Rh nanoclusters and that of Rh surfaces. The binding energies of Rh atoms on UGO were less than the cohesive energy (- 5.75eV) of Rh bulk, indicating that the Rh atoms adsorbed on UGO tend to collect to clusters. In our work, we systematically calculated the CO2 adsorption energies dependent on the size and shape of the Rh n nanoclusters (n = 1, 4, 5, 8, 13) on unzipped graphene oxide and found that the IcosahedralRh13/UGO had the largest one, -1.18eV with the C–O bond elongated from 1.18 to 1.29 Å . Furthermore,the dissociation barrier of CO2 on Icosahedral-Rh13/UGO is very small (Ea = 0.41eV), indicating that the Icosahedral-Rh13/UGO might be act as good catalyst to activate the scission of the C-O bond. References 1. L. C. Balba ´s,; A. Vega,; F. Aguilera-Granja J. Phys. Chem. A 2009, 113, 13483–13491. 2. G. W. Rogers,; J. Z. Liu J. Am. Chem. Soc. 2012, 134, 1250−1255. 3. T. Futschek,; M. Marsman, J. Hafner J. Phys.: Condens. Matter 2005, 17, 5927–5963. 題目: 利用線性響應理論預測分子內部訊息傳遞與異構調控效應 Title: intramolecular communications and allosteric regulations revealed by linear response theories 黃邦杰,楊立威 Bang-Chieh Huang and Lee-Wei Yang 清華大學生物資訊與結構生物研究所 Institute of bioinformatics and structural biology, National Tsing Hua University, Hsinchu, Taiwan Abstract Accumulated experimental and theoretical evidences have shown that protein functions as a physiochemically connected network. Allostery, understood in this new context, is a manifestation of residue communicating over remote sites via this network and hence a recently rising interest in identifying communication pathways mediating allosteric controls. In this study, we demonstrate that a new formulation of linear response theory (LRT) can describe a two-stage conformational relaxation- 1. ligand-induced conformational changes at a few tens to a hundred of picoseconds and 2. an early molecular ‘twitch’ that is faster than conformational relaxation by an order of magnitude. Predictions based on LRT agree with observations from site-specific UV resonance Raman, time-resolved X-ray and sound speed in a condensed medium. With the computational ease of the current implementation, we can easily perturb the protein network by thousands of times where time-resolved atomic trajectories can be tracked following each perturbation. Frequently used ‘communication centers’ are identified and it is found by experiments that mutations of these centers, many remote from the catalytic site, would greatly impact the hydride transfer rate in DHFR. Mutations on those that do not serve as communication centers impact the catalysis minimally. We also show the signal propagation is directional, highly anisotropic and need not be reciprocal. We favorably consider the method’s applicable future in probing functionally sensitive distant mutants by the physical approach herein proposed. Reference: Yang, L. W., A. Kitao, B. C. Huang, and N. Go. 2014. Ligand-induced protein responses and mechanical signal propagation described by linear response theories. Biophys J 107:1415-1425. A Theoretical Study on the Reaction of Thiyl Radicals with Nitrogen Dioxide : Biological Mechanisms Shih-Sheng Wu (吳詩盛), Ching-Han Hu (胡景瀚)* Department of Chemistry, National Changhua University of Education, Changhua 50058, Taiwan, Republic of China Keywords: cis-trans isomerization; Nitrogen dioxide; Thiyl radicals Many studies have demonstrated that fatty acid peroxidation and isomerization can occur via radicals, such as thiyl radicals (RS․) and nitrogen dioxide (NO2․).1,2 These radicals can be obtained exogenously, such as cigarette smoke; or can be generated endogenously by one-electron oxidation from hydrogen sulfide, cysteine or glutathione.3 However, the reaction about RS․ in vivo is still unclear. In this study, we used density functional theory and coupled cluster theory to investigate the mechanism. Process of forming RS․ from hydrogen abstraction mechanism of endogenous thiols with free radicals were investigated, and the environmental effects were included using the polarizable continuum model. We found a feasible ․ ․ mechanism for the formation of NO and sulfinyl radical (RSO ) by the reaction of RS․ with NO2․. The NO2․ addition mechanism through the O-terminus is more favorable than through the N-terminus. Sulfinyl nitrite (RS(O)NO) is probably formed by rearrangement of sulfenyl nitrite (RSONO), and the release of NO․ and RSO․ then take place after which the N—S bond is cleaved. Hu, C.-H.; Tzeng, Y.-Z. J. Phys. Chem. A 2014, 118, 4554−4564. 2. Balazy, M.; Chemtob, S. Pharmacol Ther 2008, 119, 275−290. 3. Tweeddale, H. J.; Kondo, M.; Gebicki, J. M. Arch Biochem Biophys 2007, 459, 151−158. 1. Novel Dipolar 5,5,10,10-tetraphenyl-5,10-dihydroindeno[2,1-a]- indene Derivatives for SM-OPV: A Combined Theoretical and Experimental Study 新穎的小分子有機光伏材料: 雙偶極衍生物 (5,5,10,10-tetraphenyl-5,10-dihydroindeno[2,1-a]-indene)理論計算和實驗探討 Chin-Kuen Tai(戴欽坤), Chun-An Hsieh(謝淳安), Kai-Lun Hsiao(蕭凱倫), Ken-Hao Chang(張根豪), Bo-ChengWang(王伯昌),Yi Wei(魏屹) Department of Chemistry, Tamkang University, Tamsui 251, Taiwan 淡江大學 化學系 In order to investigate the photo-physical, electrochemical, and optoelectronic properties of dipolar 5,5,10,10-tetraphenyl-5,10-dihydroindeno[2,1-a]-indene (TDI) derivatives, a facile synthesis has been developed to integrate arylamine (electron donor fragment, D) and aryl-2-methylenemalononitrile (electron acceptor fragment, A) into the TDI bridge. According to calculation results using the DFT/B3LYP/6-31G(d) method, the HOMO and LUMO energies of TDI derivatives are relevant to the extent of corresponding electron donating and accepting abilities, and influence the open-circuit voltage (Voc) and driving force (ΔE) in organic photovoltaics (OPV). The projected density of state (pDOS) analysis shows that the electron density distribution from the D fragment to TDI bridge in the HOMO is attributing to the electron-donating ability, whereas the electrons are mainly localized on A fragment in the LUMO. Calculations of the reorganization energy by the DFT/B3LYP/6-31G(d) method suggest these D-TDI-A derivatives are hole-transporting type materials. On the other hand, the calculated absorption spectra for these molecules in CH2Cl2 are simulated by using the TD-DFT/BHandHLYP/6-31G(d) method within the Polarizable Continuum Model (PCM) and provide the maximum absorption wavelength (λmax), which can be assigned to the HOMO to LUMO transition. HOMO is found to be the π orbital which is delocalized between the D fragment and the π-linker and LUMO is the π* orbital which is concentrated on the A fragment. The optical properties of D-TDI-A derivatives can be influenced by the D fragment and π-conjugated length. Calculated results of D-TDI-A derivatives also exhibited a large light harvesting efficiency related to the maximum absorption wavelength (RLHE)) and, according to these results, the D-TDI-A derivatives containing the Ab and Ad fragments would be useful electron donor materials for further development of new small molecular organic photovoltaic solar cell (SM-OPV) devices. Methane and Ethane Oxidative Dehydrogenation Mechanism on MoO3 and Transition Metal Doped MoO3 Surfaces Chen-Cheng Liao, Yao-Shun Zhan, Yu-Te Chan, Ming-Kang Tsai* Department of Chemistry, National Taiwan Normal University, Taipei 10677, Taiwan Massive production of shale gas becomes technologically accessible due to the development of horizontal drilling and hydraulic fracturing in the recent years, especially in United States. The global energy supply is therefore significantly reshaped in respect to the cost of liquefying shale gas for intercontinental transport. With the fact that the primary composition of shale gas is methane or ethane, converting these alkane into other derivatives play a crucial role to the current energy market. The recent experiment carried out by Al-Hazmi et al. showed the use of Mo-V-Mn-W catalyst for ethane oxidative dehydrogenation (ODH) to produce ethylene in short contact time.1 With varying the tungsten loading, different selectivity of ethylene production ratios were observed. Therefore, the present study used the first-principles calculations to compare the potential ethane dehydrogenation reaction mechanism on MoO3 surface and other V, W doped MoO3 surfaces. The minimum energy pathway of producing ethylene as well as other potential chemical product is discussed. The electronic structure effect introduced by V and W dopants are analyzed. ad2_phy_ad Reference -251.0143602 1. "Enhanced catalytic activities for the oxidative dehydrogenation of ethane by the addition of W into MoVMn mixed oxides at low temperature" T. Odedairo, Y.-S. Zhan, L. Atanda, Y. Choi, and M.H. Al-Hazmi, SABIC Technology Center, Riyadh, Saudi Arabia (private communication). Spectroscopic Study of the OH Stretching Motions in H(CH3OH)13X02; X=Ar and N2 Hsiao-Han Chuanga, Jer-Lai Kuob, Kaito Takahashib, Asuka Fujiic a Department of Chemistry, National Taiwan University, Taipei, Taiwan. Nano Science and Technology Program, Taiwan International Graduate Program. b c Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei, Taiwan. Department of Chemistry, Graduate School of Science, Tohoku University, Sendai, Japan [email protected] Tagging messengers (usually stable atoms or molecules) on ionic clusters can resolve the low-resolution infrared spectra through the vibrational pre-dissociation mechanism, and usually these messengers deliver the information of large ionic cluster well [1]. Nevertheless, in the small clusters, they perturb the system by themselves and would not act as a “messenger” anymore. Infrared Pre-Dissociation (IR-PD) spectra of the mass-selected ionic cluster H(CH3OH)13X02 (X=Ar and N2) were observed, and both Ar and N2 interfered the OH stretching motion between 2800 to 3800 cm1. The observed sharp peaks were analyzed with theoretical simulations via different vibrational calculations. In this work, expressions were derived in reduced dimension on vibrational discrete variable representation (DVR) [2], which had high accuracy potential energy surface along several selected normal mode coordinates, and had no coupling terms between kinetic operators. After concerned the anharmonicity, simulation of covalent bonds were in agreement with experimental results upon B3LYP/6-31+G(d,p) level, but overestimated the interaction between messengers and ionic protonated methanol clusters, see the figure below. Reference [1] Okumura, M.; Yeh, L.I.; Myers, J.D.; Lee, Y.T. J. Phys. Chem., 1990, 94, 34163427. [2] Light, J.C. Adv. Chem. Phys., 2000, 114, 263284. 編號 : Theoretical Study on the Polynuclear Manganese-Binding Proteins Pei-Chuan Chu (朱沛全)1, Jen-Shiang K. Yu(尤禎祥)1,2 1 Institute of Bioinformatics and System Biology, National Chiao Tung University, Hsinchu,300, Taiwan 2 Department of Biological Science and Technology, National Chiao Tung University, Hsinchu,300, Taiwan Keywords: metal-binding protein, metallothionein Metal-binding is a characteristic feature in some of the protein families. These proteins play important roles in biological functions including catalysis, activation of substrates as well as transportation and storage of metal ions1,2. Polynuclear manganese-binding proteins such as arginase and metallothionein demonstrate their coordinated effects as active sites or structural centers. The arginase is the final enzyme of the urea cycle, whereas the manganese-containing metallothionein may serve for in vivo detoxification. The coordination motif relates to the electronic structure and affects biological functions as well as the folding of metalloproteins. In this study, the electronic structure of the binding center of metallothionein is theoretically investigated using density function theories. The results show that the manganese metals form an imperfect hexagonal geometry planar to the sulfur atoms of surrounding residues. The manganese atoms exhibit antiferromagnetism according to the calculated spin density: one α electron locates at one of the manganese sites, while the β spin populates on the other manganese site in the optimized structural model of the β domain in the metallothionein crystal isolated from Rattus rattus. References 1. L.M. Kennedy, B.R. Gibney, Curr. Opin. Struct. Biol. 2001, 11, 485. 2. C. Liu, H. Xu, J. Inorg. Biochem. 2002, 88, 77. Theoretical Study of Benzoylleucine Diethyl Amides Dimer Benzoylleucine Diethyl Amides 二聚體之理論計算 Pei-Chen Lo(羅珮溱) and Yao-Yuan Chuang(莊曜遠) * Department of Applied Chemistry, National University of Kaohsiung, Kaohsiung 81148, Taiwan 本研究利用之前合成一系列具不同取代基的 Benzoylleucine Diethyl Amides (BDA)的晶體進行晶體分析與量子化學計算,我們利 用 Hirshfeld Surface 分析,可以得知大部分具有 π-π 相互作用 BDA 二 聚體為相同掌性,此與之前文獻所得結果相同,但我們以非人工方式 篩選出具有 π-π 相互作用的分子晶體。相較於之前研究,我們可利用 指紋圖定量分析多種類型原子作用力於分子間的堆疊貢獻度。為了仔 細分析二聚體分子間作用力與比較不同取代基對 π-π 相互作用的影響, 我們利用具不同分散力(Dispersion Force)模型的密度泛函理論計算具 π-π 相互作用的 BDA 二聚體,也利用 SAPT 將 BDA 二聚體相互作用 力分解成各種具有物理意義的組成能量作探討。我們希望藉由分析已 知分子晶體結構的相互作用力,了解分子晶體堆疊的方式與不同分子 間作用力的協同作用,以達到利用理論計算從事分子識別研究的目的, 進而設計出可分離掌性化合物之管柱固定相晶體。 第一原理分子動態模擬 STM-tip 誘導 CO 去吸附反應及非彈性電子穿隧光譜研究 Ab-initio molecular dynamics simulations of STM-tip induced desorption process of CO/Ag(110) : inelastic electron tunneling spectroscopy and anharmonic coupling Shao-Yu Lu(呂紹宇) and Jyh-Shing Lin(林志興) Department of Chemistry, Tamkang University, New Taipei City, Tamsui, 251, Taiwan (淡江大學化學系) E-mail : [email protected] The effects of STM-tip on adsorption dynamics are important issues to understand the detailed mechanisms in tunneling-current-induced reaction process. DFT-based molecular dynamic simulations in combination with a Fourier transform of auto-correlation function[1-2] of the derivative of local density of states (FT-ACF-δLDOS) are performed for simulating the inelastic electron tunneling spectroscopy (IETS) of CO(ads) adsorbed on the Ag(110) surface. It is found that the tunneling conductance generated based on the trajectories of LDOS is significantly increased as a Ag5 cluster STM-tip is introduced and their vibrational amplitudes of low-frequency modes are enhanced in IETS. In addition, the IETS shows the doublet feature in the regions of frustrated rotation and C-O stretching, respectively, due to the change of geometry of CO(ads). Furthermore, an anharmonic coupling between the low-frequency and high-frequency modes is investigated by using a short-time Fourier transform (STFT)[3] and single frequency pass filtter (SPFP)[4] approach. Finally, the key issue regarding to the activation of the low-frequency modes by a Ag5 cluster STM-tip to cause the desorption process of CO(ads) is addressed. Figure 1. The spectrogram obtained by STFT analysis of IETS spectrum for the Ag5(planar-tip)-CO(ads) adsorbed on the Ag(110) surface and corresponding structural changed during 6ps MD simulation. The two kind of frustrated rotation motions are shown in inserted Figure by SFPF analysis. [1] Jyh Shing Lin, Shao-Yu Lu, Po-Jung Tseng and Wen-Chi Chou, J Comp. Chem., 33, 1274-1283 (2012). [2] Shao-Yu Lu and Jyh-Shing Lin, J. Chem. Phys., 140, 024706 (2014). [3] Yung-Ting Li and Jyh-Shing Lin, J Comp. Chem, 36, 2697-2706 (2013). [4] Jen-Ping Su, Yung-Ting Lee, Shao-Yu Lu and Jyh Shing Lin, J Comp. Chem, 34, 2806-2815 (2013). The Theoretical Studies of interstitial Boron in SiGe alloy Ze-hui Yan, Yu-Wei Huang, Shyi-Long Lee* 國立中正大學暨生物化學系 621 嘉義線民雄鄉大學路一段 168 號 For years, the boron diffusion in Si and SiGe alloy have been extensively studied because it play an important issues in Si-based semiconductor device. As the size of devices components decreasing, the ultrashallow pn junction in the complementary metal-oxide semiconductor (CMOS) is required in manufactory. The critical issues in the formation of shallow junctions is the anomalous diffusion of dopant during rapid thermal annealing (RTA) after ion implantation. The phenomenon is called transient enhanced diffusion (TED). It is known that the B diffusion is dominated by the self-interstitial (Sii) defect1 and the Boron TED is suppressed by Ge. The origin of the retarded Boron diffusion is not clearly resolved, hence the formation of B-Sii complexes in Si and SiGe alloys was investigated here. In this work, the effects of Ge atoms on interstitial Boron in SiGe alloys were investigated by using the density functional theory. The formation of B-Sii complexes at neutral, positive and negative charged state in different configuration SiGe alloys were studied here. Our computational results shows that the formation energies of B-Sii complexes were slightly affected by replacing one Ge atom, even we change the distance between Boron and Ge. The differences of the B-Sii formation energies between pure Si and SiGe are all less than 0.1eV, while the positive 110-split are unstable. As the number of Ge atoms increases, the formation energies increases more significantly, when replacing four Ge atoms in the first nearest neighboring positon of Boron, the largest different value up to 0.34eV, causing the suppression of Boron transient enhanced diffusion. References: 1. A. Ural, P. B. Griffin, and J. D. Plummer, J. Appl. Phys. 1999, 85 2. Windl, W. Bunea, M. Stumpf, R. Dunham, S. Masquelier, M. Physical Review Letters 1999, 83 3. Bang, JunhyeokKang, JoongooLee, Woo-jinChang, K.Kim, Hanchul. Physical Review B 2007, 76 Theoretical Study on the Reaction of C2 with C6H2 Yi-Lun Sun and Shih-Huang Lee National Synchrotron Radiation Research Center, Hsinchu, Taiwan Email: [email protected] A product with a gross formula of C8H (m/z = 97 u) was observed in the experiment of the reaction of C2 with C6H2 using a crossed molecular-beam apparatus and synchrotron vacuum-ultraviolet ionization. In order to interpret the experimental observations, we investigated the reaction mechanisms of C2 with C6H2 by the theoretical calculations. We used B3LYP and CCSD(T) methods with aug-cc-pVDZ and aug-cc-pVTZ basis sets to calculate structures and energies of reactants, intermediates, transition states, and products located on the potential energy surface. The product C8H can be produced via H elimination from the intermediate C8H2. The energy of triplet state C2 is higher than the singlet state C2 by 3.8 kcal/mol. Thus, the reactions of both singlet and triplet state C2 were considered in this research. The calculated results show that there were four entrance channels in the singlet state surface without barriers and the product C8H was lower than the reactant by 4.4 kcal/mol. However, we found one entrance channel with a 3.3 kcal/mol barrier and three entrance channels without any barrier in the triplet state surface. The product C8H was lower than the reactant C2(T) + C8H2 by 8.2 kcal/mol. The Catalytic Cycle of Water in CO Oxidation on Ag(111): the Role of HO 2 Wen-Shyan Sheu* and Ming-Wen Chang Department of Chemistry, Fu-Jen Catholic University, Xinzhuang, New Taipei City 24205, Taiwan, R. O. C. *Corresponding author. Tel.: +886 2 29053724; Fax: +886 2 29023209. E-mail: [email protected] (W.-S. Sheu). ABSTRACT The reaction mechanism for the oxidation of CO on Ag(111) in the presence of trace amounts of water is investigated via density-functional-theory calculations. A four-step cycle for the reaction is proposed: (1) H2O + O2 → HO + HO2; (2) HO2 + CO → OH + CO2; (3) CO + OH cis-OCOH; (4) cis-OCOH + OH CO2 + H2O. In the mechanism, water is found to directly participate in the reaction as a catalyst, in addition to the previously proposed role of stabilizing the adsorbed oxygen molecules on Ag(111). Moreover, HO2 is an important reaction intermediate, which is produced via a hydrogen transfer from water to an oxygen molecule. Because the overall reaction barrier is as low as 0.20 eV, the mechanism is expected to be operative at low temperatures. 摘要 Probing water micro-solvation in proteins by water catalysed proton-transfer tautomerism 利用經由水催化之質子轉移互變異構現象探討蛋白質中微水合作用 Jiun-Yi Shen (沈俊義)1, Wei-Chih Chao (趙偉志)1, Chun Liu (劉駿)1, Hsiao-An Pan (潘校安)1, Hsiao-Ching Yang (楊小青)2, Chi-Lin Chen (陳其霖)1,2, Yi-Kang Lan (蘭 宜康)2, Li-Ju Lin (林麗如)3, Jinn-Shyan Wang (王進賢)3, Jyh-Feng Lu (盧志峰)3, Steven Chun-Wei Chou (周君偉)1, Kuo-Chun Tang (唐國鈞)1, Pi-Tai Chou (周必泰)1 1 Department of Chemistry and Center for Emerging Material and Advanced Devices, National Taiwan University, Taipei, Taiwan 台灣大學化學系及物質元件中心 2 Department of Chemistry, Fu-Jen Catholic University, New Taipei City, Taiwan 輔仁大學化學系 3 School of Medicine, Fu-Jen Catholic University, New Taipei City, Taiwan 輔仁大學醫學系 Scientists have made tremendous efforts to gain understanding of the water molecules in proteins via indirect measurements such as molecular dynamic simulation and/or probing the polarity of the local environment. Here we present a tryptophan analogue that exhibits remarkable water catalysed proton-transfer properties. The resulting multiple emissions provide unique fingerprints that can be exploited for direct sensing of a site-specific water environment in a protein without disrupting its native structure. Replacing tryptophan with the newly developed tryptophan analogue we sense different water environments surrounding the five tryptophans in human thromboxane A 2 synthase. This development may lead to future research to probe how water molecules affect the folding, structures and activities of proteins. Unique Excited-State Conformational/Electronic Responses of Saddle-Shaped N,N'Disubstituted-dihydrophenazines: Wide-Tuning Emission from Red to Deep Blue 獨特地激發態構象/電子响應之馬鞍形 N,N'-二取代二氫吩嗪: 宽调谐发射从红到深蓝色 Zhiyun Zhang 張志雲, Yu-Sin Wu 吳毓心, Chi-Lin Chen 陳其霖, and Pi-Tai Chou 周必泰* Department of Chemistry, National Taiwan University, Taipei, 10617 Taiwan, R.O.C. 國立台灣大學化學系 Understanding excited-state process of luminophore is a long-standing challenge relevant to many phenomena, such as large Stokes-shifted and/or multi- emissions, and environment-sensitive emissions. In this work, a tailored strategy is utilized to modify 5,l0-dimethylphenazine (DMP) to donor-acceptor type N,N-disubstituteddihydrodibenzo[a,c]phenazines (DMAC (N,N-dimethyl), DPAC (N,N-diphenyl) and FlPAC (N-phenyl-Nfluorenyl)), exhibiting significant and varied nonplanar distortions (i.e., saddle-shaped) as a result of steric hindrance. All compounds exhibit anomalously large Stokes-shifted emission (up to ~12000 cm-1) in solvents with slightly polarity dependence, and DPAC and FlPAC also show another very weak and largely separated normal emission with obvious solvatochromic effect. Furthermore, most of compounds but DMP exhibit prominent normal Stokes shifted emission in rigid environments, and both DPAC and FlPAC can display a wide-tuning multi-emissions from red to deep blue via white by proper control of the surrounding media. These results indicate that the symmetry rule, the key factor responsible for the large red-shifted emission in DMP (Ber. Bunsen. Phys. Chem. 1974, 78, 1348), is not enough or even does not apply to others. Instead, large excited-state conformational/electronic changes play a crucial role to the unusual photophysical behaviors of N,N-disubstituted-dihydrodibenzo[a,c]phenazines. Further support is rendered by the relaxation dynamics and simulation of the potential energy surfaces along the structural relaxation, and time-resolved techniques are used to demonstrate the precursor-successor type of kinetic relationship. N,Ndisubstituted-dihydrodibenzo[a,c]phenazines, in their excited states, undergo a sequential three-state emission process: upon photoexcitation, an intramolecular charge transfer reaction (1CTunrelaxed) happens instantly from donor to acceptor units, and subsequently stabilized by the solvent polarity and accompanied/followed by suitably structural changes to a new electronic density distribution (1CTrelaxed), and finally goes for a more planar configuration (1P) with large skeletal motion and charge redistribution to extend the -delocalization for lowering the energy in the excited state. Molecular Dynamics Simulations of Water Coupled Antifreeze Protein Motions in Probing the Water-Ice Recognition Mechanism 分子動力模擬抗凍蛋白水耦合運動探測冰水辨識機理研究 Chi Wei (曲薇), Hsiao-Ching Yang (楊小青)* Department of Chemistry, Fu Jen Catholic University, Hsinchuang, Taipei, Taiwan (天主教輔仁大學化學系) E-mail:[email protected] Antifreeze proteins (AFPs) protect the various organism from freezing injury with protecting the growth of ice crystals, which if uncontrolled may fatal to organism cells. AFPs have been found in fishes, plants, Insects and bacterium. The significant characterization of AFPs is the ability for binding to ice crystal or the inhibition the ice growth, which play an important role in many application and biology. Although many reports had demonstrated the power of AFPs, how the water environment influence the recognition mechanism of AFPs interaction with ice is rare. We develop the smart-jump sampling approach and molecular dynamics simulation to investigate how water interacts with AFPs in different temperature control environments. We are trying to clarify the water coupled AFPs dynamics behavior–characterization of the ice-like structures, exploring the surface induced recognition mechanism. Our results so far reveal the water-protein interface environments for MpAFP (Marinomonas primoryensis), that could be divide into two major factors. One is the grooved recognition effect induced the ice-like structures in the ice binding site interface and the other is the hydrophobic effect of the polypeptide backbone of the AFPs for the control of the ice-like structure size (Fig.1). Figure 1. (a) AFP Anchord clathrate grooved effect with water binding strutre. (b) Characterization of the water binding antifreeze protein to ice-like structures. References [1] Garnham, C. P.; Campbell, R. L.; Davies, P. L. Anchored clathrate waters bind antifreeze proteins to ice. PNAS 2011 , 108, 7363-7367. [2] Jorov, A.; Zhorov, B.; and Yang, D. S. C. Theoretical study of interaction of winter flounder antifreeze protein with ice. Protein Science 2004, 11, 1524-1537 . [3] Lorv, J. S. H.; Rose, D. R.; and Glick, B. R. Bacterial Ice Crystal Controlling Proteins. Scientifica Review 2014. Probing Water Environment of Trp59 in Ribonuclease T1: of the Structure−Water Network Relationship Insight 透過偵測核糖核酸酶中色氨酸周圍的微水環境變化來探討蛋白質結 構與微水結構之交互關聯性 Shih-Hui Weng(翁詩惠), Cheng-Han Yang (楊承翰) ,Yi-Ching Kuo(郭宜靜),Hsiao-Ching Yang (楊 小青)* Department of Chemistry, Fu Jen Catholic University, Hsinchuang, Taipei, Taiwan 24205 (新北市新莊 區,輔仁大學化學系)Tel: 02-2905-2534 ; E-mail: [email protected] In this study, we used the tryptophan analogue, (2,7-aza)Trp, which exhibits water catalyzed proton transfer isomerization among N(1)-H, N(7)-H, and N(2)-H isomers, to probe the water environment of tryptophan-59 (Trp59) near the connecting loop region of ribonuclease Tl (RNase T1) by replacing the tryptophan with (2,7-aza)Trp. The resulting (2,7-aza)Trp59 triple emission bands and their associated relaxation dynamics, together with relevant data of 7-azatryptophan and molecular dynamics (MD) simulation, lead us to propose two Trp59 containing conformers in RNase T1, namely, the loop-close and loop-open forms. Water is rich in the loop-open form around the proximity of (2,7-aza)Trp59, which catalyzes (2,7-aza)Trp59 proton transfer in the excited state, giving both N(1)-H and N(7)-H isomer emissions. Existence of N(2)-H isomer in the loop-open form, supported by the MD simulation, is mainly due to the specific hydrogen bonding between N(2)-H proton and water molecule that bridges N(2)-H and the amide oxygen of Pro60, forming a strong network. The loop-close form is relatively tight in space, which squeezes water molecules out of the interface of α-helix and β2 strand, joined by the connecting loop region; accordingly, the water-scant environment leads to the sole existence of the N(1)-H isomer emission. MD simulation also points out that the Trp-water pairs appear to preferentially participate in a hydrogen bond network incorporating polar amino acid moieties on the protein surface and bulk waters, providing the structural dynamic features of the connecting loop region in RNaseT1. Keywords: ribonuclease Tl, (2,7-aza)Trp, MD simulation, hydrogen bond network Reference : [1] Wei-Chih Chao, Jyh-Feng Lu, Jinn-Shyan Wang, Hsiao-Ching Yang, et.al Chou, Biochemistry, 2013, 52, 1113−1121 [2] Wei-Chih Chao, Hsiao-Ching Yang,* Jinn-Shyan Wang, Jyh-Feng Lu, Pi-Tai Chou,* Nature Commun., 2013, 4, 2611. Probing the Channel Regulation Step to Prostaglandin Isomerization Mechanism by Cytochrome P450 Thromboxane Synthase 探討前列腺素在細胞色素 P450 血栓素合成酶通道調的異構化反應機制 Tai-An Pan(潘泰安) and Hsiao-Ching Yang* (楊小青) Department of Chemistry, Fu Jen Catholic University, Hsinchuang, Taipei, Taiwan (天主教輔仁大學化學系) E-mail:[email protected] Thromboxane synthase (TXAS) belongs to the cytochrome P450 (CYP450) family proteins. TXAS catalytic the isomerization prostaglandin H2 to thromboxane A2(TXA2). We try to confirm that the substrate binding and catalytic activity is associated with the substrate recognition sites (SRSs). The present evidence indicates that there are six water micro-solvation SRS regions, SRS-1 to SRS-6 in sequence assay, in assembly of the protein channel connecting to the buried heme site (Fig. 1a). To explore the minimum energy path (MEP) along the channel regulation coordination and study the substrate dynamics of passage through the dividing surface separating reactants from products. We developed a smart-jump sampling and performed molecular dynamics simulations to investigate the water coupled channel regulation behavior and the relevant amino acids cluster guiding the stereo-selectivity Energy (kcal/mol) of eicosanoid PGH2. Our results reveal the nonbonding interactions, Van der Waals, electrostatics and H-bonding along the MEP (Fig. 1b) and “flip-jump” motion of PGH2 has been demonstrated to be involved in channel regulation of multistate kinetics mechanism. 0 Interaction VdW Electrostatic H-bonds -20 -40 -60 -80 -2 0 2 4 6 8 10 12 14 16 18 20 RMSD Figure 1. (a) TXAS channel with buried heme site. (b) Analysis of eicosanoid PGH2 binding energy along the MEP. References 1. Wei-Chih Chao; Jyh-Feng Lu; Jinn-Shyan Wang; Hsiao-Ching Yang; Hsiao-Hui Chen; Yi-Kang Lan; Ya-Chien Yu; Pi-Tai Chou, and Lee-Ho Wang. , J. Am. Chem. Soc., 2011, 133 (46), pp 18870–18879 2. Wei-Chih Chao; Jyh-Feng Lu; Jinn-Shyan Wang; Hsiao-Ching Yang; Tai-An Pan; Steven Chun-Wei; Chou Lee-Ho Wang; Pi-Tai Chou. , Biochemistry,2013, 52 (6), pp 1113–1121 3. Jiun-Yi Shen; Wei-Chih Chao; Chun Liu; Hsiao-An Pan; Hsiao-Ching Yang; Chi-Lin Chen; Yi-Kang Lan; Li-Ju Lin; Jinn-Shyan Wang. , Nat. Commun, 2013, 4,2611 Vibrational Levels of the C3Ar van der Waals complex Yi-Ren Chen, K. S. Tham, Yi-Jen Wang, and Yen-Chu Hsu Institute of Atomic and Molecular Sciences Academia Sinica P. O. Box 23-166, Taipei 10617, Taiwan, R. O. C. The ground electronic state of the C3 molecule has an extremely low bending vibrational frequency in comparison to other triatomic molecules. As a result the vibrational level structure of C3Ar is expected to be very complicated, with no fewer than four large-amplitude motions. To help assign the wavelength resolved emission spectra of C3Ar1 and to understand the dynamics of its electronic states, we have carried out MCTDH (Multi-channel time dependent Hartree) calculations2 of its lowest vibrational levels. The kinetic operators were derived from Yang and Kühn.3 The ab initio 4D potential energies of C3Ar at the level of CCSD(T)/ccpVQZ were calculated for the following ranges: C-C-C=112-248, vdW bond length=3.4-6.0 Å , and vdW bond angles= 0-180 (azimuth) and 0-90 (colatitude); the C-C bond length was fixed at 1.2981Å , Assuming that only the four low frequency modes of the complex are perturbed by the Ar atom, the 6D potential energy function was obtained by adding the 2D C-C stretching energies of free C3 to the calculated 4D potential energies of the Ar complex. The calculated wavefunction of the zero point level of the complex suggests that the C3 moiety of the complex is bent, rather than linear, as is free C3. The two lowest energy excited vibrational levels are the out-of-plane and in-plane vdW bending fundamentals. The wavefunctions of the first few levels show evidence of strong coupling among the four low frequency modes. Calculations of the higher vibrational levels are in progress. References, 1. G. Zhang, B.-G. Lin, S.-M. Wen, and Y.-C. Hsu, J. Chem. Phys. 120, 3189 (2004). 2. H.-D. Meyer and G. A. Worth, Theor. Chem. Acc. 109, 251(2003). 3. Y. Yang and O. Kühn, Mol. Phys. 106, 2445(2008). Mimicking the Organometallic Complexes for Small Molecule Activation: A Computational Exploration Using Nitrogen doped Carbon Nanotube Yu-Te Chan, Hsiang-Chun Yeh, Yu-Jou Huang, Ming-Kang Tsai* Department of Chemistry, National Taiwan Normal University, Taipei 10677, Taiwan. N doped carbon material have been study as a ORR catalysts where pyridinic-N and graphitic-N are believed to exist in these material.1,2 In the present study, we explored the structure and chemical adsorption properties of 3d and 4d transition metal incorporated into carbon nanotube and graphene (TM-xN-CNT, x = 3, 4)5,6 using density functional theory and showed that the d band splitting is related to ligand field introduced by the curvature of N doped carbon nanotube. The split of the d band of the mounted TM also affects the electronic structure of TM-xN-CNT via the displacement of Fermi level, subsequently transform he pristine CNT into metallic or semiconductor. We would also discuss the adsorption properties for the small molecule activation in terms of CNT curvature for the potential catalytic applications. Unit cell size Higher curvature Higher density Reference 1. Oh, H.-S.; Oh, J.-G.; Roh, B.; Hwang, I.; Kim, H. Electrochem.Commun. 2011, 13, 879. 2. Kiuchi, H.; Niwa, H.; Kobayashi, M.; Harada, Y.; Oshima, M.;Chokai, M.; Nabae, Y.; Kuroki, S.; Kakimoto, M.-a.; Ikeda, T.; Terakura, K.; Miyata, S. Electrochim. Acta 2012, 82, 291. 3. Medard, C.; Lefevre, M.; Dodelet, J.; Jaouen, F.; Lindbergh, G.Electrochim. Acta 2006, 51, 3202–3213. 4. Charreteur, F.; Jaouen, F.; Ruggeri, S.; Dodelet, J.-P. Electrochim.Acta 2008, 53, 2925–2938. DENSITY FUNCTIONAL THEORY STUDY OF THE MECHANISMS OF CLAISEN REARRANGEMENT OF ARYL PROPARGYL ETHER IN THE PRESENCE OF CESIUM FLUORIDE. MENGISTU GEMECH (1), V. SRINIVASADESIKAN (2), SHYI-LONG LEE (1)* (1): Department of Chemistry and Biochemistry, National Chung Cheng University, Taiwan. (2): Department of Chemistry, National Chio Tung University, Taiwan. ABSTRACT Ishii1 and co-workers reported that the Claisen rearrangement of an aryl propargyl ether in the presence of CsF led to exclusive formation of 2-methylarylfuran in excellent yield. The accepted mechanism2 for formation of arylfuran and arylpyran from aryl propargyl ether under the condition of Claisen rearrangement involves a concerted pericyclic [3,3]-sigmatropic rearrangement. A computational study of the mechanisms of the Claisen rearrangement of aryl propargyl ether in the presence of CsF has been performed by using DFT calculations. Our calculations indicated that the [3,3]- sigmatropic rearrangement is the rate limiting step for the formation of arylfuran with G value of 37.1 kcal/mol in N,N-diethylaniline solvent phase. Furthermore, the calculation results reveal that the enolization of -allenyl ketone suffered from a higher energetic barrier and indeed unlikely to allow the rearrangement reaction to proceed towards arylpyran cyclization. However, the abstraction of the -hydrogen atom in the -allenyl ketone has a very low activation barrier and is the favored pathway for the Claisen rearrangement of aryl propargyl ether in the presence of CsF. The effect of substituents, both on phenyl ring and propargyl moiety, on the kinetics of the title reaction was also investigated. Figure 1. Accepted mechanism for formation of arylfuran and arylpyran from aryl propargyl ether under the condition of Claisen rearrangement. References [1] Ishii H., Ishikawa T., Takeda S., Ueki S. and Suzuki M, Chem. Pharm. Bulletin, 40, 5, (1992), 1148-1153. [2] Zsindely J. and Schmid H. Helv. Chim. Acta. 51, (1968), 1510.; Hansen H.-J. and Schmid H. Chem. Brit. 5, (1969), 111. Theoretical Investigations on Electronic Spectra of Protonated Pyrene Chih-Hao Chin and Yu-Jong Wu National Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu Science Park, Hsinchu, Taiwan Keywords:(protonated pyrene, absorption spectrum, Franck-Condon, electronic structure) The electronic absorption spectrum of protonated pyrene has been recorded in the visible spectral region, evidencing that absorptions appear largely red shifted in comparison to that of pyrene. A set of quantum chemistry calculations, the harmonic potential surfaces of the ground state (S0) and the first (S1) excited state of protonated pyrene were investigated, and the electronic structures of the two states were calculated and characterized using the time-dependent density functional theory method in the adiabatic representation. The displaced harmonic oscillator approximation is used to simulate the absorption spectrum of the S1 state along with the Franck–Condon approximation [1]. The assignment of main vibronic transitions was made for S1 state. A tentative plausible assignment has been proposed, which takes into account a weak coupling between these electronic states. Our result shows that the vibronic structures were dominated by the totally symmetric modes with predicted intervals of 539 and 734 cm-1 and this agrees with the experimental observations in solid neon matrices [2]. The present S1 state calculated in the adiabatic representation effectively includes contribution from the adiabatic vibronic coupling through the conical intersection. References: 1. Chin, C.-H.; Mebel, A. M.; Kim, G.-S.; Hayashi, M.; Liang, K. K.; Lin, S. H. Theoretical investigations of spectroscopy and excited state dynamics of adenine. Chem. Phys. Lett. 2007, 445, 361-369. 2. Garkusha, I.; Fulara, J.; Sarre, P. J.; Marier, J. P. Electronic absorption spectra of protonated pyrene and coronene in neon matrixes. J. Phys. Chem. A 2011, 115, 10972-10978. Table of content 報名代號: 分組編號:(主辦單位填寫) Theoretical Study on the Prebiotic Strecker Synthesis of Alanine Catalyzed by Water, Ammonia and the Chiral Molecules Chia-Yu Peng (彭家瑜), Hsiao-Han Chuang (莊曉涵), Wei-Ping Hu (胡維平)* Department of Chemistry and Biochemistry, National Chung Cheng University, Chia-Yi, Taiwan Keywords: alanine, prebiotic, chirality The mechanisms and reaction paths of the prebiotic strecker synthesis of alanine catalyzed by water, ammonia and the chiral molecules have been studied by micro-solvation and PCM model. The calculated results at MP2/6-31+G** level showed the high energy barriers of 31 and 46 kcal/mol in the uncatalyzed reaction for first and second steps, respectively, it also showed that the reaction catalyzed by water and ammonia could significantly reduce the barriers by 20 -30 kcal/mol. When the reaction catalyzed by the chiral molecule CH 3 CHCl(OH); interestingly, not only the barrier could be reduced by 20 -30 kcal/mol, but the different optically active products (D - and L-Alanine) gave the difference between both barriers of 0.6 kcal/mol. This result suggest the chiral catalyst in prebiotic condition might be one of the possible reasons for the majority of the L-amino acids in nature. References: Strecker, A. Liebigs Ann. 1954, 91, 349. 2. Shaw, A. M. Astrochemistry; Wiley, 2006. 3. Shibasaki, M.; Kanai, M.; Mita, K. Org. React. 2008, 70, 1. 1. 報名代號: 分組編號:(主辦單位填寫) Theoretical Study on the isoelectronic OSO and SOO isomers OSO 與 SOO 等電子化合物的理論預測 Ching-Chan Lin(林經展), Wei-Ping Hu*(胡維平) Department of Chemistry and Biochemistry, National Chung Cheng University, Chia-Yi , Taiwan Keywords: isoelectronic ;sulfur compounds; 本篇使用 MP2、DFT 與 CCSD(T) 等方法計算與 O3 等電子化合物 SOO 與 OSO,並以 CASPT2、MRCISD+Q 等方法對 SOO 與 OSO 計算其結構及 能量,並探討 SOO 與 OSO 等電子化合物是否存在 cyclic form,並且比較 open form 和 cyclic form 兩種結構的相對能量。計算結果顯示,以 B3LYP/aug-cc-pVTZ 計算出的結構在 MRCISD+Q 方法下,從 open-OSO 到 open-SOO 的反應能障 138.9 kcal/mol ;open-OSO 到 cyclic-OSO 的反應能障 115.6 kcal/mol 。將 OSO 結構固定在 CS 點群條件,計算 OSO 與 SOO 激發 態能量,得知 open-OSO 到 open-SOO 的 TS 激發態在 Cs 1A' 、1A'' 位能曲面 產生交會,而 open-OSO 到 cyclic-OSO 的 TS 激發態能量,在 Cs 1A' 、1A'' 位 能曲面則沒有交會。從 open-OSO 分解成 SO+O 與 S+O2 ,能量分別為 149.6 kcal/mol 與 145.2 kcal/mol 。於是我們推測 open-OSO 與 open-SOO 可能穩定存 在,cyclic-OSO 則較不穩定存在。 References: Jien-Lian Chen and W.-P. Hu, J.Am. Chem. Soc, 2011 133, 16045 報名代號:LM0704-006 分組編號:(主辦單位填寫) The MC-DFT Approach Including the MC-MP4 Energies using "Calendar" Variations for the Dunning-Type Basis Sets Chia-Yu Peng,Wei-Hong Lin, Wei-Ping Hu* Department of Chemistry and Biochemistry, National Chung Cheng University, ChiaYi , Taiwan Keywords: MC-DFT, SCS-MP2 本篇研究中,我們開發了一種新的 DFT 方法並測試其計算效能。此方法 結合本實驗室先前所開發的 multicoefficient density functional theory (MC-DFT) 及 spin-component-scaled second-order Møller-Plesset perturbation (SCS-MP2) 理 論,並額外增加 E3、E4 能量校正。本方法中我們使用 Dunning-type basis set 以 及經 Truhlar 團隊簡化高角動量 diffuse functions 之 "calendar" 系列 basis sets, 希望能夠提高計算效率。研究結果顯示表現最好的 functional DSD-BLYP 搭配 cc-pVDZ、aug-cc-pVDZ、may- cc-pVTZ 三個基底線性組合,然後將 SCS-MP2 搭配 cc-pVDZ、aug-cc-pVDZ、cc-pVTZ 三個基底線性組合進行計算,其 mean unsigned errors (MUE)為 0.77 kcal/mol,但卻只需約 DSD-BLYP/aug-cc-pVTZ (MUE =1.36 kcal/mol) 44% 的計算時間,若再進一步加入 E3、E4 能量校正, 我們發現 DSD-BLYP 部分改為搭配 cc-pVDZ、aug-cc-pVDZ、cc-pVTZ 效率較 高,其誤差為 0.73 kcal/mol 並只需花費 DSD-BLYP/aug-cc-pVTZ 47% 的計算時 間。 References: 1. Chen, J.-L.; Hong, J.-T.; Wu, K.-J.; Hu, W.-P. Chem. Phys. Lett. 2009, 468, 307. 2. Papajak, E.; Zheng, J.; Leverentz , H. R.;Truhlar,D. G. J. Chem. Theory Comput 2011, 7. 3027. 報名代號:LM0704-008 分組編號:(主辦單位填寫) Theoretical Study on Metal-Containing Noble-Gas Anions 含金屬之惰性氣體陰離子的理論探討 JUN-Yl FU, Ching-Chan Lin,Wei-Ping Hu* Department of Chemistry and Biochemistry, National Chung Cheng University, Chia-Yi , Taiwan Keywords: noble-gas; mental; 本研究以理論預測 MO-NgO 類型的陰離子(M=Li, Na, K, Cu, Ag, Au, Al 等金屬) ,並探討(1) MO-+ Ng + O(S) (2) MO +Ng + O- (3) Ng+ XO2-三種可能 的分解路徑。目前研究發現 MO group 的電子親和力 (EA)對路徑(2) MO +Ng + O- 的反應能影響甚大,是決定陰離子穩定度的關鍵,一般來說 MO group 的 電子親和力越大越利形成穩定的鈍氣陰離子。在 MP2/aug-cc-pVTZ 方法下,以 MO-NgO 為例,可看到隨著 EA 值從 24.2 kcal/mol(M = K) 增加至 78.2 kcal/mol (M = Al),路徑(2) 的反應能量從-25.2 kcal/mol 增加到 27.9 kcal/mol。此外, 本研究也發現 AlO-ArO 在路徑(3)的反應能障為 20.1 kcal/mol,在低溫下應該 可以穩定存在。也預測含硼之 BO-NgO 與 OB-NgO 類型的陰離子,試著探討不 同惰性氣體原子對此類陰離子的穩定性,在 MP2/aug-cc-pVTZ 方法下,以 ONgOB-為例,可看到惰性氣體從 He 至 Kr,其陰離子能量從放熱 24.9 kcal/mol 到吸熱 16.4kcal/mol,可以預測 ONgOB-類型的陰離子會隨著惰性氣體的原子序 變大而趨於穩定。 References: 1. Tsung-Hui Li; C.-H. Mou; H.-R. Chen; W.-P. Hu, J.Am.Chem. Soc. 2005, 127, 9241. 報名代號: 分組編號:(主辦單位填寫) Tunneling Effects in Bimolecular Nucleophilic Substitution (SN2) Reactions Wan-Chen Tsai (蔡婉甄), Chong-En Wang (王崇恩), Wei-Ping Hu* (胡維平) Department of Chemistry and Biochemistry, National Chung Cheng University Chia-Yi 62102, Taiwan Keywords: SN2, VTST/MT, KIE 離子-分子親核取代反應 (SN2) 在有機化學中是很常見的反應,而 SN2 反應 其離子和分子間的作用力會隨著反應在氣態、氣態微水合和水溶液中環境的不 同而有不同的性質,且在動力學上的特性也會有些許的差異。本研究利用雙層 VTST/MT (dual-level dynamics approach with variational transition state theory including multidimensional tunneling) 計算三個 SN2 反應: (1) 氣態 CN+CH3F, (2) 氣態微水合 OH(H2O)+CH3F,(3) 水溶液 OH+CH3F 的反應速率常數與 動力學同位素效應,此三個反應代表不同程度的水合情況。計算結果顯示,此 三個反應能障分別為 11.7、5.1 及 16.2 kcal/mol,而在室溫下穿隧效應對反應速 率就有不可忽略的貢獻,而在低溫下穿隧效應則有更明顯的貢獻。此外,當以 13C 及 14C 取代 alkyl halide 中心原子時,反應在低溫下有呈現明顯的動力學 同位素效應 (kinetic isotope effects, KIEs),以 OH+CH3F 在水溶液中的反應為 例,其 KIE(CD3) 在 35 K 時為 0.608,而 KIE(13C) 和 KIE(14C) 在 35 K 時分 別可高達 7 以及 40,可得知此三個 SN2 反應的穿隧效應主要是由 alkyl halide 的中心碳原子參與,而不是一般認為質量較小的 primary hydrogens。 References: 1. Garver, J. M.; Fang, Y.-R.; Eyet, N.; Villano, S. M.; Bierbaum, V. M.; Westaway, K. C. J. Am. Chem. Soc. 2010, 132, 3808. 2. Liu, Y.-P.; Lu, D.-H.; Gonzalez-lafont, A.; Truhlar, D. G.; Garrett, B. C. J. Am. Chem. Soc. 1993, 115, 2408 報名代號:LM0704-009 非環鹵烷與環鹵烷類之 SN2 和 E2 的動力學分析 0574 Wan-Sam Lu, Wei-Ping Hu* Department of Chemistry and Biochemistry, National Chung Cheng University, Chia-Yi , Taiwan Keywords: KIEs , SN2,E2 有機化學中,雙分子親核基取代(SN2)和消去(E2)反應性質的研究一直是受 到高度重視的課題, SN2 和 E2 的反應物相同,兩反應卻可能互相競爭,而在 分析上,由於產物陰離子相同,實驗上很難分辨主要的反應途徑,但從 deuterium kinetic isotope effects (KIE)常可做正確的判斷,但過去研究都著重在直鏈或有支 鏈的鹵烷類的反應,對於環烷類則少有探討。本研究我們將比較環鹵烷與非環 鹵烷的 SN2 及 E2 的各種反應性質。 此研究探討的反應為 RBr+ SH- (R=CH3CH2、(CH3)2CH、(CH3)3C、C5H9、 C6H11),我們使用 MP2/aug-cc-pVDZ 方法計算這一系列反應 RBr+ SH(R=CH3CH2、(CH3)2CH、(CH3)3C、C5H9)的 SN2 和 E2 路徑下的反應能量和反 應能障,並利用高階理論 CCSD(T)搭配 aug-cc-pVTZ 來計算單點,再將高階理 論所得的反應能障以過渡態理論(TST)來計算反應速率以及動力學同位素效 應。所使用的計算軟體是 Gaussian 09。 References: 1. 2. DePuy, C. H.; Gronert, S.; Mullin, A.; Bierbaum, V. M., Gas-phase SN2 and E2 reactions of alkyl halides. Journal of the American Chemical Society 1990, 112 (24), 8650-8655. Al - Ar: T.H. Dunning, Jr., K. A. Peterson and A. K. Wilson, J. Chem. Phys. 114, 9244 (2001). 報名代號:LM0704-005 分組編號:(主辦單位填寫) Theoretical Prediction of Near-edge X-ray Absorption Fine Structure of Peptides Cheng-Cheng Tsai (蔡承成) Wei-Ping Hu* (胡維平) Department of Chemistry and Biochemistry, National Chung Cheng University Chia-Yi 62102, Taiwan Keywords: NEXAFS, TDDFT, PEPTIDE 近年來由於 X-ray 光譜技術的發展,研究 Near edge X-ray absorption fine structure (NEXAFS)是探討分子內化學環境的一種途徑,NEXAFS 光譜會因為 細微的化學環境不同而有所差異,二者之間的對應關係目前並沒有做很多系統 性的研究,近年來實驗上有意發展此種對應關係成為一種偵測技術,理論上的 模擬方法也因為如此而蓬勃發展。本研究藉由 TDDFT 理論來計算 peptide (Glycine-Tyrosine)的 carbon K-edge NEXAFS 光譜,並且與過去文獻上的實驗光 譜相比較。本研究中分子結構最佳化方法是 B3LYP/6-311+G(d,p),NXAFS 光 譜計算方法是 TD-LB94/6-31+G(d,p)。計算結果顯示,在較低能量區域 明顯的 吸收都是對應到苯環結構上碳的 1s → π*的 core excitation,較高能量區域則是 激發 carboxyl group 的碳到 π*以及一些碳的 1s → Rydberg orbitals,且對於化 學環境不同的原子,激發其 1s 電子所需的 core excitation energy 也會有不小的 差異。分子軌域分析的結果也顯示,如果被激發的 1s 電子所在原子旁邊接電負 度較大的原子,則 core excitation energy 會有變高的趨勢,造成能量不同的吸收 峰主要是因為 (1) 被激發電子從不同的 core orbital 出發 (2) 被激發到不同的 virtual orbital 造成的。光譜上高能量的區域在實驗上可能是對應到電子已經游 離的狀態,與 TDDFT 計算還處在個中性分子 bound state 的假設明顯不同,因 此目前高能量區域的光譜仍不易由理論模擬。整體來說目前 TDDFT 方法預測 Near edge X-ray absorption fine structure 結果在較低能量區域是可行的,可以看 出原子的化學環境與光譜的對應關係。 Besley, N. A.; Asmuruf, F. A. Phys. Chem. Chem. Phys. 2010, 12, 12024. 2. Hirata, S.; Head-Gordon, M. Chem. Phys. Lett. 1999, 314, 291. 3. Boese, J.; Osanna, A.; Jacobsen, C.; Kirz, J. J. Electron. Spectros. Relat. Phenom. 1. 1997, 85, 9. Theoretical Study on the Thiocyanide Formation of 3-mercaptopyruvate Metal and Related Molecular Design Tung-Lin Wua, Jen-Shiang K. Yua,b,* a Department of Biological Science and Technology, National Chiao Tung University b Institute of Bioinformatics and Systems Biology, National Chiao Tung University In this study, computational design of efficient detoxification metal drugs to rescue for the cyanide poisoning by molecular simulation is attempted. Two molecules of 3mercaptopyruvate (3-MP) are expected to chelate one metallic divalent cationic center (MMCu and Zn) to form stable four-coordinate complexes. Geometry optimization of the model complex (1), bis[2-(2-thienyl)-glyoxylato-O,O'] copper(II), is performed using various density functionals including BP86, B3LYP, TPSS, and PBE. The structure of (1) may be further reduced into (2) and optimized at identical levels. The geometries of the ligand coordinations remain almost identical compared to the structure of 3-MP calculated at high level CCSD theory. The energy profile and the mechanism for the detoxification of the bis[3MP]Cu(II) is proposed (Figure 1): in TS, the thiol group (SH) tends to shift to cyanide and produce the thiocyanic acid. Subsequently, the thiocyanic acid approaches the oxygen atom of the carboxyl group and transfers the proton to oxygen, forming thiocyanide, which is less toxic and can be metabolized easily in urea. Energy profile of the dithiolation reaction is also constructed. Figure 1. Energy profile of the detoxification carried at B3LYP and BP86, The relative energy is based on the difference to RC state. [1] Arnaud, C.; Faure, R.; Loiseleur, H. Acta Cryst. (1986). C42, 814-816 Theoretical Study on Relative Stability, Thermal Behaviors and Vibrational Spectra of OH–(H2O)n, n = 4 – 7 Ren-Jie Lin(林仁傑)a, Quoc Chinh Nguyenb, Kaito Takahashi(高橋開人)a, Jer-Lai Kuo(郭哲來)a* a Institute of Atomic and Molecular Science, Academia Sinica, 106 Taipei, Taiwan, ROC. b School of Physical and Mathematical Science, Nanyang Technological University, 637371, Singapore We have demonstrated the relative stability, thermal behaviors and vibrational spectra of deprotonated water cluster OH–(H2O)n, n = 4 – 7 at B3LYP/6-311+G(d,p) level. There are 20, 65, 168 and 695 isomers of OH– (H2O)n, n = 4 – 7, respectively. The isomers of deprotonated water clusters are classified into 5 categories which are multi-ring (MR), double-ring (DR), single-ring (SR), treelike (T) and linear (L). For n = 4 – 7, the categories of most stable isomers with zero-point energy are SR, DR, MR, MR, respectively. The thermal behaviors of OH–(H2O)n, n = 4 – 7 are investigated by calculating the canonical heat capacity in classical harmonic superposition approximation (C-HSA) and quantum harmonic superposition approximation (Q-HSA) regimes. For studying the structural transitions, the canonical probability or population of each topology is calculated as a function of temperature. The vibration spectrum is calculated as the sum of the spectrum of each individual isomer weighted by its corresponding canonical probability. From the results, the free OH-stretching vibration band is shown that a broad peak around 3600 cm-1 of vibrational spectra of OH–(H2O)n, n = 4 – 7.




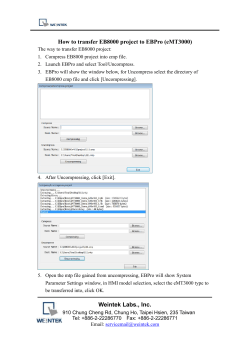





© Copyright 2026