
Allergic contact dermatitis
Review article Pierre SAINT-MEZARD1 Aurore ROSIERES1 Maya KRASTEVA1 Frédéric BERARD1,3 Bertrand DUBOIS2 Dominique KAISERLIAN3 Jean-François NICOLAS1,3 1 INSERM U 503, IFR 128 Bioscience Lyon-Gerland, 21, avenue Tony Garnier 69007 Lyon 2 INSERM U 404, IFR 128 Bioscience Lyon-Gerland, 21, avenue Tony Garnier 69007 Lyon 3 Clinical Immunology and Allergy Unit, CH Lyon-Sud, 69495 Pierre-Benite Cedex, France. Reprints: J.F. Nicolas, Fax: (+33) 478 861 528. E-mail: [email protected] Article accepted on 15/4/2004 Eur J Dermatol 2004; 14: 284-95 Allergic contact dermatitis Contact dermatitis is an inflammatory skin condition induced by exposure to an environmental agent. Eczema and dermatitis are used synonymously to denote a polymorphous pattern of skin inflammation characterized at least in its acute phase by erythema, vesiculation and pruritus. Substances responsible for contact dermatitis after single or multiple exposures are non protein chemicals, i.e. haptens, that induce skin inflammation through activation of innate skin immunity (irritant contact dermatitis) or both innate and acquired specific immunity (allergic contact dermatitis). The present review will focus on allergic contact dermatitis, a delayed-type hypersensitivity reaction, which is mediated by hapten-specific T cells. Recent advances in the pathophysiology of ACD have shown that the occurrence of ACD, as well as its magnitude and duration, is controlled by the opposite functions of CD8 effector T cells and CD4 regulatory T cells. From these studies ACD can be considered as a breakdown of cutaneous immune tolerance to haptens. Key words: haptens, CD8 T cells, CTL, CD4 regulatory T cells, tolerance, contact sensitivity, contact dermatitis C ontact dermatitis is one of the most common skin diseases, with a great socio-economic impact [1]. As the outermost barrier of the human body, the skin is the first to encounter chemical and physical factors from the environment. According to the pathophysiological mechanisms involved, two main types of contact dermatitis may be distinguished. Irritant contact dermatitis is due to the pro-inflammatory and toxic effects of xenobiotics able to activate the skin innate immunity. Allergic contact dermatitis (ACD) requires the activation of antigen specific acquired immunity leading to the development of effector T cells which mediate the skin inflammation. ACD is a T-cell-mediated inflammatory reaction occurring at the site of challenge with a contact allergen in sensitized individuals. It is characterized by redness, papules and vesicles, followed by scaling and dry skin [2]. Knowledge of the pathophysiology of ACD is derived chiefly from animal models in which the skin inflammation induced by hapten painting of the skin is referred to as contact sensitivity (CS) or contact hypersensitivity (CHS). ACD and CS (CHS) are thus considered as synonymous and define a hapten-specific T cell-mediated skin inflammation [3]. The skin and the draining lymph nodes (LN) play a central role in the induction and triggering of a CS reaction. Three elements are necessary for the development of a CS reaction: antigen presenting dendritic cells (DC), haptenspecific T-cells, and the hapten itself. Haptens - contact allergens The origin and nature of the compounds able to induce a CS reaction are very diverse, but they share some common features: contact allergens are low molecular weight chemicals named haptens, that are not immunogenic by themselves and need to bind to epidermal proteins. These then 284 act as carrier proteins to form the hapten-carrier complex that finally acts as the antigen. Most haptens bear lipophilic residues, which enable them to cross through the corneal barrier, and electrophilic residues, which account for covalent bonds to the nucleophilic residues of cutaneous proteins [4-6]. Haptens often derive from unstable chemicals, named prohaptens, which require an additional metabolization step in vivo in the epidermis to be converted into an electrophilic hapten endowed with antigenic properties. This is the case of urushiol (poison ivy) [7] and of photosensitizers, which must be activated by UV-light in order to bind to epidermal proteins. Metal salts do not bind covalently to cutaneous proteins but form complexes with these proteins through weak interactions. Some metal salts also undergo chemical conversions in the skin, such as hexavalent chromium salts, which are turned into trivalent chromium, a highly reactive form binding to cutaneous proteins [8]. Evidence that the conversion of the parent compound into a reactive metabolite is necessary for the development of CS was recently demonstrated for the polyaromatic hydrocarbon (PAH) dimethylbenz(a)anthracene (DMBA). CS to DMBA only occurred in strains of mice that could metabolize the compound and inhibitors of PAH metabolism reduced the magnitude of the reaction. Furthermore, among the PAHs, only those that could induce aryl hydrocarbon hydroxylase, the rate-limiting enzyme in the PAH metabolic pathway, were immunogenic [9]. The implications of these experiments are that at least for some contact allergens, the metabolic status of the host is a key determinant of individual susceptibility to the development of allergic contact dermatitis. Recent studies from several laboratories have shown that T cells recognize haptens as structural entities bound covalently to, or by complexation to, peptides anchored in the grooves of major histocompatibility (MHC) class I and class II molecules. Thus the contact allergen is a chemical EJD, vol. 14, n° 5, September-October 2004 but the antigen able to activate hapten-specific T cells is a haptenated peptide [10]. Pathophysiology of contact sensitivity Knowledge of the mechanisms by which a xenobiotic can induce CS responses comes from the study of strong haptens, also known as “experimental haptens”, since they do not exist in the usual environment of human beings. The pathophysiology of CS consists of two distinct phases (Fig. 1) which are summarized below: Phase 1 - Sensitization phase (also referred to as afferent phase or induction phase of CS) This occurs at the first contact of skin with the hapten and leads to the generation of hapten-specific T-cells in the LN and their migration back to the skin. The ability of a hapten to induce sensitization relies on two distinct properties. Through their pro-inflammatory properties, haptens activate the skin innate immunity and deliver signals able to induce the migration and maturation of cutaneous dendritic cells (DC). Through their binding to amino-acid residues they modify self proteins and allow the expression in the skin of new antigenic determinants. Haptens or haptenated proteins are loaded by cutaneous dendritic cells (DC) and are expressed as haptenated peptides in the groove of MHC class I and class II molecules at Sensitization site the cell surface. Hapten-bearing DC migrate from the skin to the regional LN where specific CD8+ and CD4+ T lymphocytes are primed in the para-cortical area. T cells proliferate and emigrate out of the LNs to the blood where they recirculate between the lymphoid organs and the skin. The sensitization step lasts 10 to 15 days in man, and 5 to 7 days in the mouse. This first step has no clinical consequence. The theory of the first contact of a chemical being immunogenic for the host is true for the strong haptens but cannot be accepted for the vast majority of haptens responsible for ACD. Indeed, ACD to moderate or weak haptens almost never occurs after the first contact but may take years of permanent skin exposure to develop. Phase 2 - Elicitation phase (also known as efferent or challenge phase of CS) Challenge of sensitized individuals with the same hapten leads in 24/72 hours to the apparition of ACD/CS. Haptens diffuse in the skin and are uptaken by skin cells which express MHC I and II/haptenated peptide complexes. Specific T lymphocytes are activated in the dermis and the epidermis, and trigger the inflammatory process responsible for the cutaneous lesions. Recent studies have demonstrated that CD8+ cytotoxic T lymphocytes are the main effector cells of CHS to strong haptens and that they are recruited early after challenge before the massive infiltra- Hapten Elicitation site Hapten 1 Stratum corneum Langerhans cells 6 Epidermis Dermis 7 5 Dermal denritic cells 2 CD4+ Regulatory T cells 4 Efferent lymph Afferent lymph 3 Draining Lymph Node CD8+ Effector T cells Figure 1. Pathophysiology of CHS Sensitization step: Haptens penetrate the stratum corneum. Hapten loading by skin dendritic cells (step 1) parallels activation and migration of DC through the afferent lymphatic vessels to the draining lymph nodes (step 2). Migrating DC are located in the para-cortical area of the draining LN where they can present haptenated peptides on MHC class I and II molecules to CD8+ and CD4+ T cells, respectively (step 3). Specific T cells precursors expand clonally in the draining LN and diffuse to the bloodstream through the efferent lymphatic vessels and the thoracic duct (step 4). During this process they acquire skin-specific homing antigens (CLA and CCR4) and become memory T cells. Primed T cells preferentially diffuse in the skin after transendothelial migration. At the end of the sensitization step everything is ready for the development of a CS reaction upon challenge with the relevant hapten. Elicitation phase: When the hapten is painted for a second (and subsequent) time, it diffuses through the epidermis and could be loaded by LC or other skin cells expressing MHC molecules, such as keratinocytes and dermal dendritic cells, which are then able to activate traffıcking specific T cells (step 5). CD8+ cytotoxic T cell activation initiates the inflammatory process through keratinocyte apoptosis and cytokine/chemokine production (step 6). This is responsible for the recruitment of leukocytes (including regulatory T cells) from the blood to the skin leading to the development of skin lesions (step 7). EJD, vol. 14, n° 5, September-October 2004 285 tion of leucocytes which contain the down-regulatory cells of CHS, found in the CD4+ T cell subset. The efferent phase of CS takes 72 hours in man, and 24 to 48 hours in the mouse. The inflammatory reaction persists only for a few days and rapidly decreases following downregulatory mechanisms. Primary allergic contact sensitivity Although the development of CHS has been postulated to require two spatially and temporally dissociated phases, clinical evidence has demonstrated that ACD could develop after a single skin contact with a strong hapten in previously unsensitized patients. This phenomenon has been referred to as “primary ACD” [11]. We have recently demonstrated, in a murine model, that the pathophysiology of this primary (one step) ACD is identical to the classical (two step) ACD reaction [12]. The afferent and efferent phases of primary ACD can be induced after a single skin contact with haptens due to the persistence of the hapten in the skin for long period of time, allowing the skin recruitment and the activation of T cells which have been primed in the lymphoid organs. The central role of cutaneous dendritic cells There are different subtypes of DC in the skin. Although they are all able to uptake haptens and to present haptenated peptides to T cells, two subsets of cutaneous DC seem crucial in the development of CS. The basic role of epidermal Langerhans cells (LC) has been shown by two sets of experiments. On the one hand, animals painted with a hapten on cutaneous sites naturally or artificially depleted in LC are unable to mount a CS response [13, 14]. On the other hand, sensitization of naive mice can be achieved by injection of in vitro haptenized total epidermal cells, purified LC or FSDC dendritic cell lines, whereas injection of total epidermal cells depleted in LC before haptenization is inefficient in inducing sensitization [15-17]. Dermal DC could also participate in the induction phase of CS, even though their precise contribution and phenotype is not well known [18]. Recent studies by Geissmann et al have brought some insights into the turnover of macrophages and DC in the dermis in normal and inflammatory skin. Two different subsets of monocytes can be defined according to the expression of the chemokine receptor CCR2 [19, 20]. CCR2- monocytes appear to be involved in the physiological turnover of resident macrophages and DC, whereas CCR2+ monocytes are recruited in inflammatory sites where they could differentiate into mature DC able to present exogenous antigens to T cells. Since hapten application is responsible for activation of skin innate immunity, it is possible that the CCR2+ inflammatory monocytes which are recruited in the skin could uptake the hapten and participate in the afferent phase of CHS [21]. Cutaneous DC load haptens in the skin and migrate to the draining LN Activation of naive specific T cell precursors occurs in the regional draining LN upon presentation of haptenated peptides by cutaneous migrating DC. Initial observations 286 showed that induction of a CS reaction requires an intact draining lymphatic system [22], and that after skin painting with the hapten fluorescein isothiocyanate (FITC), dendritic cells bearing the hapten, some of which containing Birbeck granules, accumulate in the draining lymph nodes [23, 24]. Cutaneous DC continuously migrate out of the skin at a low rate which dramatically increases after hapten exposure [25]. This phenomenon is the consequence of numerous factors including the secretion of inflammatory cytokines and chemokines induced by the pro-inflammatory properties of the hapten itself. Fifteen minutes after hapten painting, LC start to synthesize IL-1b mRNA and to release the protein. Then, keratinocytes are activated and release TFN-a and GM-CSF [26]. In the epidermis, LC and keratinocytes are firmly associated by E-cadherine/E-cadherine junctions [27]. Binding of TNF-a and IL-1b on their cognate receptors (TNF-a RII and IL-1 RI and RII) expressed on LC, is followed by a decreased expression of E-cadherine on the LC membrane, allowing their disentanglement from surrounding keratinocytes [28–31]. Furthermore, IL-1b and TFN-a inhibit the expression of chemokine receptors such as CCR1, CCR2, CCR5 and CCR6 on the LC membrane, inducing a loss of sensitivity to their cognate ligands, in particular to MIP-3a (CCL20), a chemokine produced by keratinocytes [32]. In parallel, TNF-a and IL-1b induce the expression of adhesive molecules such as CD54, a6 integrin and different isoforms of CD44, which permit some interactions between LC and the extracellular matrix, allowing the migration of cutaneous DC. In order to reach the lymphatic vessels, LC have to cross the dermo-epidermal junction and the dermis. To this end, LC secrete different types of enzymes, such as metalloproteinase (MMP) 3 and 9 [33], which could cleave macromolecules of the dermo-epidermal junction and of the extracellular matrix. They are also involved in the cleavage of E-cadherin and of pro-TNF-a in its biologically active form [34]. Once in the dermis, DC acquire sensitivity to the chemokines MIP-3b (CCL19) and SLC (CCL21) through the up-regulation of the chemokine receptor CCR7 [32, 35]. CCL21 is expressed on endothelial cells from lymphatic vessels and by stromal cells from the T cell area in LN [36]. The CCL21/CCR7 interaction is crucial during the sensitization phase of CS. Indeed, Endeman and colleagues have described a strong inhibition of CS following injections of SLC-blocking antibodies before and during the sensitization phase [37]. Moreover, TNF-a is able to induce a strong up-regulation of CCL21 on the endothelial cells of skin lymphatic vessels. The over-expression of CCL21 is of critical importance in the migratory properties of peripheral DC and is able to increase, by a factor of 10, the number of cutaneous DC able to migrate from the skin to the LN and subsequently the magnitude of antigenspecific T cell activation [38]. Maturation of cutaneous DC The term “maturation” takes into account a group of morphological, phenotypic and functional modifications which transform skin DC into professional antigen-presenting cells able to prime naïve specific T cell precursors. Although the different steps of DC maturation and differentiation are well described in vitro [18], correlation with the in vivo modifications is not yet totally achieved. Migration EJD, vol. 14, n° 5, September-October 2004 and maturation of cutaneous DC are intimately linked. Indeed, factors known to induce migration of DC are also able to engage DC in a maturation program. Presence of TNF-a and IL-1b in the skin leads to the up-regulation of MHC class II molecules on DC membranes. In the first 3 hours following application of the hapten, expression of MHC II molecules at the DC surface first decreases [39], whereas their intracellular level increases, which probably reflects an endocytosis process triggered by hapten binding [40]. From the sixth hour after hapten painting, synthesis of MHC II mRNA starts to increase to reach a maximum at around 18 hours. This up-regulation of mRNA synthesis, plus an increase in the half life of membrane MHC II molecules [41], is responsible for the strong MHC class II expression observed after 24 hours. In a similar way, the expression and stability of MHC class I molecules on DC increase. DC maturation is also associated to the loss of capability to internalize and process exogenous antigens. Indeed, the expression of DC receptors such as mannose receptors or FcR receptors decreases during DC maturation. Birbeck granules disappear from LC and the intracellular machinery which controls macropinocytosis and phagocytosis is blocked [42]. Although this process was clearly demonstrated in vitro, recent in vivo data suggest, however, that mature migratory skin DC recovered from LN may still be able, at least for a proportion of them, to internalize, process and present some exogenous antigen [43]. In parallel, during the maturation process, DC express costimulatory molecules such as CD80 (B7-1), CD86 (B72), CD40, CD83, adhesive molecules such as CD54 (ICAM-1) and CD58 (LFA-3) and chemokine receptors such as CCR4, CCR7 and CXCR4, which permit them to migrate through lymphatic vessels . In summary, cutaneous DCs uptake haptens in the skin and migrate to draining LNs. This migration is associated with an up-regulation of MHC and costimulatory molecules that confer a high efficiency in the priming of naive T cells to skin DC. Hapten-specific T lymphocytes Hapten specific T cell activation CS reactions are dependant on the priming of effector T cells during the sensitization phase. Adoptive transfer of T cells from sensitized mice into naive recipients results in the transfer of sensitization. Moreover, T cell depletion of sensitized mice totally removes the CS reaction. Finally, patients with thymic aplasia (Di George syndrome) cannot be sensitized [44]. T cell activation requires the combination of two distinct signals. The first signal involves the interaction of TCR and the MHC/peptide complex. The second signal requires costimulatory molecules and/or the secretion of cytokines and chemokines by DC. The absence of this second signal may lead to anergy or the death of the T cell which has already engaged its TCR. Hapten determinants for T cells provide the first signal T lymphocytes usually recognize hapten-modified peptides in the groove of MHC molecules [45]. Most of the results were obtained with the strong hapten TNP (trinitrophenyl) in mouse models. In vitro experiments have shown that for EJD, vol. 14, n° 5, September-October 2004 MHC class I [10, 46] as well as MHC class II restricted determinants [47, 48], T cells react to MHC-associated TNP-peptides and not to covalently TNP-modified MHC molecules, whereas TCR would interact mainly with the hapten TNP and parts of the MHC molecule [45]. However, metal T cell recognition may be different from the general scheme described above for non-metal chemicals. Indeed, Weltzien and co-workers have recently shown that nickel may behave like a superantigen and could directly link TCR and MHC outside the groove of the MHC molecule, in a peptide-independent manner [49]. The special nature of haptens may explain the different routes of processing that they could follow in antigenpresenting cells. Haptens could bind to extracellular and cell surface proteins which are internalized and processed into peptides via the endosomal/lysosomal compartments where they bind to the MHC class II groove. These haptenated peptides can eventually be recognized by class IIrestricted CD4+ T cells. In addition, since most haptens are lipid soluble molecules they may also enter the cell, conjugate with intracytoplasmic proteins, and after processing in the endogenous route may be presented to MHC class I-restricted CD8+ T cells. Alternatively, direct binding of haptens, without processing, to a peptide in the groove of either MHC class I or class II molecules may also contribute to recognition by CD8+ or CD4+ T cells, respectively [50]. These different routes of hapten presentation have been demonstrated for the haptens TNP [51], urushiol [52] and arsonate [53]. These data point to the existence of both hapten-specific class I-restricted CD8+ T cells and class II-restricted CD4+ T cells. The second signal is provided by mature DC During their maturation, DC up-regulate the expression of CD80 and CD86, two ligands of CD28, constitutively expressed on T cells [54, 55]. CD28 ligation is mandatory for the development of CS since mice deficient for the CD28 molecule present a strong reduction of CS [56]. CD86 seems to be the CD28 ligand involved in the second signal [57]. Indeed, injection of anti- CD86 blocking antibodies inhibits the activation of both CD4+ and CD8+ T cells and the development of CS. TCR engagement induces the activation of the nuclear factor NF-AT which regulates the IL-2 gene transcription. The instability of the IL-2 mRNA limits the production of this cytokine, which is essential for the proliferation and activation of T cells. One of the effects of CD28 engagement is to stabilize the IL-2 mRNA, leading to a 20 to 30 fold increase in IL-2 production [58]. Moreover through the activation of AP-1 and NF-kB, CD28 engagement induces an increase in IL-2 and anti apoptotic Bcl-XL gene transcription, which both protect T cells from the apoptotic signals received after TCR engagement [59]. Circulating CD4+ and CD8+ T cells penetrate in the paracortical area of LNs through the post capillary high endothelial venules (HEV) in response to the chemokine DCCK1, secreted by resident DC of this zone [60]. The rare specific T cell precursors are activated by presentation of haptenated peptides by skin derived DC and start a program of clonal expansion and differentiation. This process is initiated by physical contact between T cells and DCs which implicate cell membrane remodeling, allowing the engagement of TCR/MHC-peptide complexes and costimulatory molecules with their cognate ligands. This membrane juxtaposition, associated with transmission of 287 the information, presents some analogy with the neural synapse and is called immune synapse. The plasmic membrane of mature DC expresses a high level of adhesive molecules which are essential for T cell activation. It comprises integrins such as CD54 and CD58 and the lectin DC-SIGN. These molecules interact respectively with LFA-1 (CD11a/CD18), a b2 integrin, CD2 and ICAM-3/2 molecules which are expressed on T cells [61]. In seconds following the T/DC interaction, a small number of TCR engagements leads to cytoskeleton rearrangement by actine polymerisation, and activation of ZAP-70 and the adaptative protein Vav-1 [62]. These modifications generate a central zone, rich in MHC-peptide /TCR complex surrounded by an integrin ring called SMAC (for supramolecular activation cluster). In this zone, CD4+ and CD8+ molecules are progressively replaced by CD28 and CTLA-4 [63]. Other couplings of costimulatory molecules from the TNFTNF family receptors such as CD40/CD40-L or RANK/RANK-L participate in T cell activation. Their interactions lead to the up-regulation of OX40-L on DC membranes. Interaction between OX40-L and OX40, expressed on activated T cells, induces an over expression of CD80 and CD86 and thus a better T cell activation which is of relevance for CS because mice deficient for OX40-L display a dramatic decrease in CS reaction to oxazolone, DNFB, and FITC. This poor CS response was due to deficient T cell priming, as shown by decreased T cell proliferation induced by DC from OX40-L deficient mice [64]. T cell polarization and constitution of CD4+ and CD8+ T cell populations Classically, two distinct roles have been attributed to CD4+ and CD8+ T cell subsets in immunological responses. CD4+ T cells, or T helper (Th) cells, are considered as sources of cytokines and help the establishment of specific B and T specific responses. CD8+ T cells are associated to cytotoxic functions, cleaning of potentially dangerous cells, and produce mainly IFN-c and TNF-a. However, more recent studies on T cells subsets have revealed that CD8+ T cells can synthesize a pattern of cytokines as large as that produced by CD4+ T cells [65]. Moreover, CD4+ T cell functions are not restricted to T cell help or cytokine secretion and may comprise cytotoxicity [66]. Depending on the pattern of cytokine secretion, different functional subpopulations of T lymphocytes serve to determine the qualitative aspects of the adaptative immune response. CD4+ Th1 cells produce IFN-c, IL-2 and TNF-a, and CD4+ Th2 cells produce IL-4, IL-10, IL-13, and IL-5. Similarly, Tc1 cells produce type 1 cytokines (IFN-c, IL-2 and TNF-a) and Tc2 cells type 2 cytokines (IL-4, IL-10, IL-13, and IL-5). However, Tc2 cells could also produce TNF-a in some conditions [65]. A simplification of the nomenclature is the use of “type 1” and “type 2” cytokine pattern for either CD4+ and CD8+ T cells. For CD4+ and CD8+ T cells, orientation through one or the other subtype is dependent on comparable environmental factors [67]. In vitro treatment of APC/T cells mixtures with IL-4 plus anti-IFN-c mAb polarizes T cells to a type 2 phenotype, whereas IL-12 plus anti-IL-4 mAb treatment polarizes T cells to a type 1 phenotype. However, the mechanisms implicated during the T cell-DC dialogue in 288 vivo which are responsible for the polarization of T cells are still poorly understood. Two hypotheses have been recently proposed to explain the basis of the dichotomy of T cell responses. The first hypothesis postulates the existence of distinct DC subpopulations (DC1 versus DC2) involved in the polarization of the immune response (type 1 versus type 2). After CD40 triggering, one subpopulation of DC, derived from monocytes in humans or expressing CD8a in mouse (myeloid), referred to as DC1, would provoke a type 1 response by producing high amounts of IL-12. The other subpopulation, derived from plasmacytoïd DC in human and CD8a neg in mouse (lymphoid) and defined as DC2, produces few IL–12 and might be able to induce a type 2 response [68]. However, CD8a neg cells can prime both type 1 and type 2 responses depending on the activation signal they received [69, 70], demonstrating that the phenotype of a given DC subset cannot be considered as a marker of functionality for the activation of type 1 versus type 2 T cells. The second hypothesis considers that environmental factors will influence the maturation process of a given DC enabling it to prime for type 1 or type 2 T cells [71]. Along this line, Soumelis et al have reported that human epithelial cells produce a cytokine, thymic stromal lymphopoietin (TSLP), that binds to specific receptors on CD11c+DCs [72]. This induces the production of Th2-attracting chemokines and primes naïve T cells for a Th2 phenotype. Polarization of type 1 T cells is crucial to the development of specific effector T cell populations and optimal CS reaction. CS to DNFB is due to CD8+ effector type 1 T cells and is regulated by CD4+ type 2 T cells [73, 74]. Type 1 polarization by injection of IL-12 at the time of sensitization favors CD8+ T cell differentiation and increases the CS reaction [75]. On the contrary, type 2 polarization using IL-4 (or anti-IFNc mAbs) leads to a diminished CS response associated with an altered CD8+ T cell priming [75]. Need for CD4+ help in hapten-specific CD8+ responses? CS to strong haptens is mediated by CD8+ T cells and regulated by CD4+ T cells. More importantly, CD8+ effectors can develop in the absence of CD4+ T cells. This has been demonstrated by different sets of experiments: i) mice depleted in CD8+ T cells or deficient in CD8+ T cells (MHC class I-KO mice) cannot develop CS responses; ii) mice depleted in CD4+ T cells or genetically deficient in CD4+ T cells (MHC class II-KO mice) develop an enhanced CS response; iii) DC recovered from MHC class I-deficient mice cannot sensitize for CS in transfer experiments whereas DC recovered from MHC class II-deficient mice are able to induce a normal CS reaction [73, 76–82]. That CD4+ T cell help is not necessary for development of CHS is in keeping with recent studies showing that antigenspecific cytotoxic lymphocyte (CTL) responses can be induced in the absence of help provided that i) the immunogen has intrinsic proinflammatory properties (e.g. endotoxins and pathogenic microorganisms) able to generate a danger signal to the DC [83]; ii) the affinity of TCR for MHC/peptide complexes is high; iii) the frequency of specific CD8+ T cell precursors is high [85]. Contact sensitizing haptens have two important properties that may explain their immunogenicity in the absence of CD4+ help. First, they are proinflammatory xenobiotics through induction of chemokine and cytokine production by skin cells [3]. SecEJD, vol. 14, n° 5, September-October 2004 ond, covalent binding of haptens, such as DNFB or TNCB, on amino acid residues of proteins generates a high number of haptenated peptides allowing activation of high numbers of CTL precursors [85]. Migration of specific T cells in the blood and in the skin Once activated, T cells emigrate from the LN through the efferent lymphatic vessels and then circulate in the blood. Emigration of T cells outside the LNs is associated with modifications in the expression of chemokine and adressine receptors. Different subsets of T cells are generated during an immune response. A subset of activated specific T cells down-regulates the expression of CCR7 and then loses the ability to re-circulate into the LNs. The CCR7- T cells constitute the peripheral memory T cell subsets able to enter peripheral tissues and especially the skin. CCR7+ T cells constitute the other memory subset called central memory T cells, which has kept the ability to re-circulate from the blood to LN, but which cannot be recruited in peripheral tissues [86]. Upon a subsequent antigen challenge, peripheral memory T cells may act as innate cells in respect to their quick and strong release of IFN-c and RANTES which confer an increased efficiency in the T cell response. The central memory T cells have a role in the preservation of relative high frequency of hapten-specific T cells. CD4+ and CD8+ T cells found in the skin of sensitized mice, express CCR4, a4b1 integrin and cutaneous lymphocyte antigen (CLA) [87]. Skin-selective homing of primed T cells depends on tissue microenvironment and more specifically on skin dendritic cells [88]. Migration of T cells from the blood to the skin occurs at the site of post capillary HEV through interactions of CLA and CCR4 with their respective ligands, E-, P-selectine and TARC (CCL17), constitutively expressed on endothelial cells [89–92]. The passage of T cells in the dermis requires the sequential interaction of VLA-4 and LFA-1 receptors on T cells with VCAM-1 and ICAM-1 on endothelial cells [93]. Thus, at the end of the afferent phase of CHS, specific T cells which have been activated by hapten-bearing DC are found in the LNs (central memory cells), in the blood and in the skin (peripheral memory cells). The skin has a normal looking appearance. Specific T cells will be activated directly in the skin and massively recruited upon a subsequent skin contact with the same hapten. Figure 2. Clinical aspect of ACD. followed by the production of cytokines and chemokines. This first signal induces the recruitment of hapten specific T cells from the blood to the dermis and the epidermis. One of characteristic features of CS consists in the early recruitment of type 1 CD8+ effector T cells followed by a late arrival of the CD4+ T cell population which contains the regulatory T cells responsible for the resolution of the inflammation [96]. That CD8+ and CD4+ T cells are recruited at different times after hapten painting may be explained by the differential expression of chemokine receptors by T cell subsets as well as by the sequential production of chemokines during the development of the skin inflammation. Even if it is still unclear which chemokines drive the initial influx of T cells, the recruitment of activated CD8+ T cells seems to be under the control of the IP-10/CXCR3 chemokine/ chemokine receptor pathway [97–99]. The contribution of IP-10 (CXCL10) has been recently suggested by a study showing that IP-10 deficient mice present a deficient skin recruitment in IFN-c producing T cells and a diminished CS reaction [100]. The recruitment of CD4+ T cells is possibly under the control of the MDC (CCL22) and TARC (CCL17) chemokines and their receptor CCR4 expressed on activated CD4+ T cells [92]. These two chemokines are up regulated in the skin around 12 hours after hapten exposure concomitantly with the infiltration of mononuclear Epidermal spongiosis and edema Expression of contact sensitivity reaction Hapten skin painting in sensitized individuals induces the skin inflammatory reaction which occurs in three steps. First, activation of the skin innate immunity recruits hapten-specific T cells. Second, T cells are activated, produce IFN-c and cytotoxicity which results in the activation of skin resident cells and in the production of new mediators of the inflammatory reaction. Third, leucocytes (polymorphonuclears, monocytes, T cells) are recruited and progressively induce the morphological changes typical of contact dermatitis (Figs. 2 and 3). T cell recruitment Skin contact with the hapten induces, as during the sensitization phase, the release of TNF-a and IL-1b [94, 95], EJD, vol. 14, n° 5, September-October 2004 Epidermotropism of mononuclear cells Dermal inflammatory cellular infiltrate Figure 3. Histopathologic features of ACD. 289 cells. Another recently described chemokine, CTACK (CCL27) and its receptor CCR10, is also important in the traffic of activated T cells in the skin [101, 102]. This chemokine is constitutively expressed by keratinocytes and its synthesis is up-regulated following IL-1b and TNF-a exposure. In parallel to chemokines and chemokine receptors, adhesion molecules (CLA, VLA-4 and LFA-1) are involved in the infiltration of T cells in the skin. Under the influence of TNF-a, endothelial cells up-regulate their respective ligands (E-, P-selectine, VCAM-1 and ICAM-1) allowing a direct interaction with T cells and their extravasation in the dermis. Characterization of effector T cells During the 1980s, studies from Cher and colleagues clearly established that classical delayed-type hypersensitivity (DTH) to nominal protein antigens was mediated by CD4+ T cells [103]. The over-representation of CD4+ T cells in established ACD lesions and the presence of haptenspecific CD4+ T cells in the blood of sensitized patients [104, 105], have led to the wrong conclusion that CS was mediated by CD4+ T cells. Animals models have contributed to elucidate the nature of the effector T cell population in CS. Mice deficient in CD8+ T cells, following the invalidation of the MHC class I b2 microglobulin gene, or mice depleted in CD8+ T cells, are unable to develop a CS reaction to experimental haptens [73]. However, lack of CD8+ T cells in these mice does not affect the classical DTH to proteins. Alternatively, mice deficient in CD4+ T cells, following the invalidation of the MHC class II Ab gene, or mice depleted in CD4+ T cells, develop a stronger and sustained CS reaction, suggesting a regulatory function for the CD4+ T cell compartment [73]. Thus CS appears as very different from classical DTH reactions in term of T cell subset involved in the effector pro-inflammatory functions. Recent studies on nickel ACD have confirmed that the pathophysiology of ACD in humans was similar to that of CS in mice and involved CD8+ effector T cells and CD4+ regulatory T cells [106]. IFN-c production by CD8+ T cells Once in the skin, type 1 CD8+ T cells recognize haptenmodified peptides presented by cutaneous cells. In part due to their chemical properties, haptens are able to cross through plasmic cell membranes, to bind to intracellular proteins and are then presented in an MHC class I context by resident skin cells. It is certainly by this mechanism, or by a direct binding to external MHC I/peptide complexes, that haptenated peptides are presented to CD8+ T cells [10, 51]. TCR engagement induces the release of type 1 cytokines such as IFN-c, which in turn is responsible for the increased production in the skin of IP-10, IP-9 and Mig, of IL-1, IL-6, TFN-a, GM-CSF and MIP-2 (CXCL8). This complex cytokine and chemokine production amplifies the inflammatory response initiated by the hapten (IL-1, TNF-a) and is responsible for the massive infiltration of leukocytes. Recruited cells comprise polynuclear neutrophils, T cells and inflammatory monocytes able to differentiate into macrophages and dendritic cells. Resident mast cells have been recently proposed to be key regulators of the amplification phase of the skin inflammation [107]. Indeed, following CD8+ T cell activation, mast 290 cells produce TNF-a and MIP-2 which are both needed for the recruitment of neutrophils constitutively expressing CXCR1 and 2. Cytotoxic activity of CD8+ T cells IFN-c release by CD8+ T cells is not sufficient for the full development of the CS reaction, since CS is only moderately impaired in mice deficient for IFNc receptors [108]. Recent studies have shown that CD8+ T cell cytotoxicity was mandatory for the development of CS responses since mice deficient in the two cytotoxic pathways, i.e. Fas/Fas-L and perforin, were unable to develop a CS reaction although IFN-c producing CD8+ T cells were detected at the site of hapten challenge [109]. Moreover, the two CD8+ CTL pathways are redundant since abrogation of CS occurs only when the 2 pathways are inactivated in the same animal. Thus, the CD8+ T cells are effectors of CHS through cytotoxicity. Keratinocytes are the main targets of the cytotoxic effect of hapten specific CD8+ T cells [110]. Keratinocyte apoptosis coincides with CD8+ T cell arrival in the epidermis and increases proportionally with the number of infiltrating CD8+ T cells [96]. These epidermal damages facilitate the penetration of haptens present on the skin which may increase the inflammation [111]. Finally, perforin release by CD8+ T cells is associated to the production of RANTES, MIP-1a and MIP-1b, which mediate the recruitment of CCR1+ and CCR5+ monocytes and granulocytes [112]. In summary, hapten-specific type 1 T cell activation leads to the production of a cascade of cytokines and chemokines by skin cells which induce a massive recruitment of leukocytes responsible for the cutaneous changes typical of CS. After a peak obtained at 24/48 hours, the inflammation starts to resolve slowly by active down-regulatory mechanisms which limit the tissular damages and maintain the skin integrity. Regulation of contact sensitivity Down-regulation of CS was initially attributed to clearance of the hapten from the skin in the few days following hapten painting. However, recent studies have shown that a hapten could stay in the epidermis for as long as two weeks after a single skin contact [12]. In parallel, several groups have reported that the down-regulation of CS was an active immune phenomenon mediated by a subset of suppressor/regulatory CD4+ T cells [73, 81, 113]. Several CD4+ T cell subsets with down-regulatory activities have been described in murine models and in human diseases, among which are Th2 cells, Th3 cells, Tr1 cells, CD4+25+ cells, which could be involved in the resolution of the CS reaction [114]. The regulation of CS could be divided into two phases, a central and peripheral phase [115, 116]. The central regulatory phase controls the expansion and differentiation of CD8+ effector T cells in the LNs while the peripheral phase limits the inflammatory process generated in the skin. CD4+CD25+ natural regulatory T cells seem to be involved in the first phase. Indeed, a recent study has shown that CD4+CD25+ T cells are necessary in the oral tolerance phenomenon to haptens through the total inhibition of the clonal expansion of hapten-specific CD8+ T cells [114, EJD, vol. 14, n° 5, September-October 2004 117]. Moreover, mice treated with an IL-2-IgG2b fusion protein, showed a decreased CS reaction associated with an increase in the CD4+CD25+ T cell subset [118]. Finally, depletion of CD4+25+ T cells by in vivo treatment of mice with an anti-CD25 mAb at the time of sensitization led to an enhanced CS reaction and an increased CD8+ T cell priming (Dubois B and Kaiserlian D, unpublished data). The mechanisms by which CD4+ T cells (CD4+25+ and/or CD4+25- T cells) limit the skin inflammatory reaction are still not understood and may involve IL-10 and other immunosuppressive cytokines [106]. The immunosuppressive effect occurs through the inhibition of production of IFN-c, IL-6, IL-1, GM-CSF and TNF-a. IL-10 plays a crucial role in the down-regulation of CS reactions inasmuch as IL-10deficient mice mount an exaggerated CS reaction to oxazolone, increased in both magnitude and duration as compared to wild type mice. Moreover, IL-10 injection before challenge totally abrogated the CS response [119]. Finally, resolution of CS is associated with the recruitment in the skin of a regulatory CCR8+ T cell population which produces a large amount of IL-10 around 24 hours postchallenge [120]. The production of IL-10 is not restricted to T cells and can be supplied locally by other cells such as keratinocytes which synthesize the cytokine by 48/72 hours after hapten painting [121]. Other regulatory mechanisms may be involved in the resolution of CS. As an example, in the presence of a large quantity of IFN-c, endothelial cells down-regulate the expression of E and P selectins, thereby limiting the arrival of new infiltrating leucocytes in the dermis [122]. Clinical hallmarks In a sensitized individual, ACD appears 24 to 96 hours after contact with the causative allergen. Its initial localization is at the site of contact [2]. The edges of the lesions may be well demarcated, but unlike irritant contact dermatitis it may propagate in the immediate vicinity or to distant unrelated sites. In its acute phase, ACD is characterized by erythema and edema, followed by the appearance of papules, closely set vesicles, oozing and crusting. In the chronic stages, the involved skin becomes lichenified, fissured and pigmented, but new episodes of oozing and crusting may occur, usually as a consequence of a new exposure to the causative allergen. ACD is usually accompanied by intense pruritus. Systemically induced eczema or hematogenous contact dermatitis is induced by oral or parenteral application of certain contact allergens in previously sensitized individuals. The best known example is the “flare-up” phenomenon at sites of previous eczematous skin changes following an experimental challenge by oral or parenteral application. Substances most often implicated in inducing hematogenous contact eczema are metal salts and drugs. Histopathology of allergic contact dermatitis The histopathologic findings are different in acute and chronic contact dermatitis and are dependent on the severEJD, vol. 14, n° 5, September-October 2004 ity of the inflammatory reaction. The most common histologic feature is spongiosis, which results from intercellular edema. It is often limited to the lower epidermis but, if the reaction is severe, it may affect the upper layers. The clinical expression of intense fluid accumulation in the acute stage is the formation of vesicles that may rupture at the epidermal surface. The papillary vessels are dilated, with perivascular lymphohistiocytic infiltrate, and the upper dermis is edematous. The lymphohistiocytic infiltrate extends in the epidermis (exocytosis) and accumulates in the spongiotic vesicles. In subacute and chronic ACD the spongiotic pattern gradually fades out, the epidermis becomes hyperplastic, and parakeratosis develops. Diagnosis The site and clinical appearance of the lesions frequently suggest the etiologic factor when the patient is first seen. Thus sharply delineated geometric lesions are evocative of sensitivity to rosin in adhesive tape [123]. Dermatitis at the site of contact with jewelry, blue jeans buttons, wrist watches, and other metallic objects are seen in nickel dermatitis. It is important to know the location of the initial skin changes and to try to establish a list of possible contactants that may have caused them. If the dermatitis has taken a chronic course, the patient’s observations about factors causing relapses may be helpful. A search for possible sources should concentrate on occupation, hobbies, clothing and personal objects, home environment, and past and previous treatment. Inhalents, dust exposure and ingestion have to be considered. A family history or a past history of atopy and psoriasis may be decisive particularly when a diagnosis of hand eczema is discussed. Patch testing is the universally accepted method for the detection of the causative contact allergens. The positive patch test reproduces an experimental contact dermatitis on a limited area of the skin. A good patch test indicates contact sensitization of past or present relevance and produces no false-positive reaction. Based on the principles of evidence-based medicine, patch testing is cost-effective only if patients are selected on the basis of a clear-cut clinical suspicion of contact allergy and only if patients are tested with chemicals relevant to the problem [124]. Finn chambers and several other tape methods are currently in use [125]. Most allergens used in patch testing are welldefined chemical substances. To save place and time, mixes of chemically related chemicals may be used. The most frequently encountered contact allergens have been selected by various international contact dermatitis groups and included in standard patch test series [126]. There are additional series aimed towards specific occupations and other spheres of activities. Most commercially available allergens supplied in syringes are incorporated in petrolatum. Considerable efforts have been made to standardize the concentration of the allergens to ensure comparable results worldwide. Great care must be taken in testing with non standardized chemicals not found in commercially available kits because testing with irritant concentrations may result in false positive reactions [127]. Patch tests are usually applied for 48 hours on the upper half of the back. Patches are read at least 20 minutes after their removal. 291 The method of recording recommended by the European and North American contact dermatitis groups is as follows [128]: + weak positive reaction: erythema, infiltration, possibly papules ++ strong positive reaction: erythema, infiltration, papules, vesicles +++ extreme positive reaction: intense erythema and infiltration, coalescing vesicles, bullous reaction. ? doubtful reaction (weak erythema only) IR irritant reaction of different types NR negative reaction NT not tested It is recommended to perform a second reading 24 or 48 hours after patch test removal. In doing only a single reading, a large number of delayed reactions will be missed, while others due to early irritant effects will be considered as allergic [127]. The type of positive reaction that can safely be interpreted as indicating allergic contact sensitivity exhibits erythema, edema, and small vesicles extending slightly beyond the patch border. Pruritus and reactivation of previous eczematous skin lesions at the time of testing indicate allergy. When a positive patch test is considered to reveal a genuine contact sensitivity, a decision has to be taken as to its relevance. Current relevance is related to “current clinical symptoms, occurring in the last few days or weeks”; past relevance refers to older clinical events [129]. The major prerequisites for a contact allergy to be clinically relevant are: i) exposure to the sensitizer; ii) presence of a dermatitis which is understandable and explainable with regard to the exposure, on the one hand, and type, localization and course of the dermatitis, on the other hand [130]. Common causes of allergic contact dermatitis Metals Nickel is the most common cause of ACD in women in almost all countries. The greater exposure of women to high-nickel content jewelry is a predisposing factor. Ear piercing is considered to be the principal inducer of nickel contact dermatitis. Hand eczema in nickel sensitive patients is often of the dyshidrotic type and may be aggravated by nickel ingestion. A threshold of 0.5 microgram of nickel/cm2/week has been established to which only a small number of nickel-sensitive patients will react [131]. The Danish nickel exposure regulation and the nickel directive (European union) regulating nickel content in objects which are in direct and prolonged contact with the skin have resulted in a significant decrease in nickel sensitization in young patients [132, 133]. Chromate is the most common contact allergen in men and sensitization to it is usually occupational. Occupational exposure is most frequent in construction workers who handle cement. Other common sources are chrome-tanned leather, bleaching agents, paints, and printing solutions. Cosmetics and skin care products Compulsory ingredient labeling of cosmetic products (excluding perfumes) has greatly facilitated the diagnosis and treatment of cosmetic contact dermatitis. Positive patch 292 tests are found most frequently to preservatives, perfumes, active or category-specific ingredients, excipients/ emulsifiers and sunscreens [134]. The relevance of the positive patch tests is confirmed if the contact dermatitis disappears upon discontinuation of the use of the product. Most allergic reactions are caused by cosmetics that remain on the skin: “stay-on” or “leave-on” products [135]. Dermatitis from clothes and shoes Contact dermatitis to clothes is usually located in the axillae, which is due to the release of allergens from the textile under the action of sweat and friction. Clothing dermatitis from formaldehyde is rare nowadays. Textile dye dermatitis is usually related to disperse dyes [136]. Leather articles contain several substances that may cause ACD: chrome, adhesives (paratertiary butyl phenol formaldehyde resin), and dyes. A number of accelerators and antioxidants used in the production of synthetic rubber may also cause contact dermatitis. Drug dermatitis Drug dermatitis may be elicited by the active ingredient of a topical drug, by the vehicle or by a preservative. Contact sensitization to antibiotics, antiseptics, and anesthetics is relatively frequent, especially in leg ulcer patients. ACD from topical corticosteroids has been reported with increasing frequency [137, 138]. Systemic application of a drug to which an individual has been sensitized by a previous cutaneous exposure may cause systemic contact dermatitis. Plant dermatitis Plant dermatitis can manifest itself in a variety of ways, depending upon the plant and the means of exposure. Airborne contact dermatitis mimicking photodermatitis may be caused by sesquiterpene lactones found in the Compositae family, while contact dermatitis to plants from the Liliaceae and Alstroemeriaceae families may present as a dry painful dermatitis of the fingers in bulb growers, called “tulip fingers”. Urushiol, present in poison ivy and poison oak is the most common cause of ACD in the United States, with 50% of the adult population clinically sensitive to it. Treatment The only available etiologic treatment of ACD is elimination of the contact allergen. The patients should be informed about the identity of the offending agent and the possible sources of the sensitizer. Cross-reacting substances should be listed. Topical steroids are used in the acute stage and are gradually replaced by ointments and cold creams as the skin lesions withdraw. If ACD is widespread and severe, systemic corticosteroids may be indicated for a short period of time. Reducing the total body load of nickel has been attempted in nickel eczema by means of a nickel-restricted diet and by treatment with disulfiram. Trials have yielded conflicting results as regards the clinical effect of the treatment and the application of the metal-chelator disulfiram was limited by serious side effects [139]. Oral hyposensitization to urushiol and nickel has been attempted but is not performed in practice. EJD, vol. 14, n° 5, September-October 2004 Conventional immunosuppressive therapy is not appropriate in the management of ACD. New immunomodulating macrolactams have been successfully tested in clinical trials [140–142]. Perspectives in pharmacological intervention include new classes of immunosuppressors, inhibitors of cellular metabolic activity, inhibitors of cell adhesion molecules, targetted skin application of regulatory cytokines and neutralization of pro-inflammatory cytokines (antisense oligonucleoides, anticytokine antibodies, soluble cytokine receptors). Conclusion Recent advances in knowledge of the mechanisms by which haptens can generate a specific T cell activation leading to ACD have reinforced the importance of hapten presentation by Langerhans cells to specific T cells. The induction of ACD depends on the production by epidermal cells, within minutes or hours following hapten application, of a rather specific pattern of cytokines. This cytokine milieu seems necessary for efficient hapten handling by LC and for T cell priming in the regional draining lymph nodes. More recently, it was demonstrated that LC have a dual function in the pathophysiology of ACD. On the one hand, LC activate effector cells which mediate the inflammatory reaction aimed at eliminating the potentially harmful haptens. On the other hand, LC are able to activate regulatory cells which limit the skin inflammation. These regulatory cells are extremely important for the outcome of ACD, since their absence will lead to a chronic skin inflammation with major tissue damage. Although CD4+ T cells have been shown to comprise regulatory cells in ACD, the molecular mechanisms by which they exert their regulatory properties are presently unknown. Ongoing studies will undoubtedly provide more information on how CD4+ regulatory T cells could be specifically activated and thus provide new ways of treating ACD. j Acknowledgments . This work was supported by the Region Rhône Alpes Grant 8HC07H and by Institutional Grants from INSERM. Pierre Saint-Mezard is supported by Laboratoire BIODERMA, 75 Cours Albert Thomas, 69003 Lyon, France. References 1. Uter W, Schnuch A, Geier J, Frosch PJ. Epidemiology of contact dermatitis. The information network of departments of dermatology (IVDK) in Germany. Eur J Dermatol 1998; 8: 36-40. 2. Krasteva M,et al. Contact dermatitis II. Clinical aspects and diagnosis. Eur J Dermatol 1999; 9: 144-59. 3. Krasteva M, et al. Contact dermatitis I. Pathophysiology of contact sensitivity. Eur J Dermatol 1999; 9: 65-77. 4. Lepoittevin J, Leblond I. Hapten-peptide T cell receptor interactions: molecular basis for the recognition of haptens by T lymphocytes. Eur J Dermatol 1997; 7: 151-4. 5. Dupuis GB. Nature of hapten-protein interactions. Chemically reactive function in haptens and proteins, in allergic contact dermatitis to simple chemicals. A molecular approach. New-York: Ed Marcel Dekker, Inc.: 1982. 6. Berard F, Marty JP, Nicolas JF. Allergen penetration through the skin. Eur J Dermatol 2003; 13: 324-30. EJD, vol. 14, n° 5, September-October 2004 7. Dupuis G. Studies on poison ivy. In vitro lymphocyte transformation by urushiol-protein conjugates. Br J Dermatol 1979; 101: 617-24. 8. Saloga J, Knop J, Kolde G. Ultrastructural cytochemical visualization of chromium in the skin of sensitized guinea pigs. Arch Dermatol Res 1988; 280: 214-9. 9. Anderson C, et al. Metabolic requirements for induction of contact hypersensitivity to immunotoxic polyaromatic hydrocarbons. J Immunol 1995; 155: 3530-7. 10. Martin S, Ortmann B, Pflugfelder U, Birsner U, Weltzien HU. Role of hapten-anchoring peptides in defining hapten-epitopes for MHC-restricted cytotoxic T cells. Cross-reactive TNP-determinants on different peptides. J Immunol 1992; 149: 2569-75. 11. Vigan M, Girardin P, Adessi B, Laurent R. Late reading of patch tests. Eur J Dermatol 1997; 7: 574-6. 12. Saint-Mezard P,et al. Afferent and efferent phases of allergic contact dermatitis (ACD) can be induced after a single skin contact with haptens: evidence using a mouse model of primary ACD. J Invest Dermatol 2003; 120: 641-7. 13. Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol 1980; 124: 445-53. 14. Lynch DH, Gurish MF, Daynes RA. Relationship between epidermal Langerhans cell density ATPase activity and the induction of contact hypersensitivity. J Immunol 1981; 126: 1892-7. 15. Ptak W, Rozycka D, Askenase PW, Gershon RK. Role of antigen-presenting cells in the development and persistence of contact hypersensitivity. J Exp Med 1980; 151: 362-75. 16. Tamaki K, Fujiwara H, Levy RB, Shearer GM, Katz SI. Hapten specific TNP-reactive cytotoxic effector cells using epidermal cells as targets. J Invest Dermatol 1981; 77: 225-9. 17. Girolomoni G, et al. Establishment of a cell line with features of early dendritic cell precursors from fetal mouse skin. Eur J Immunol 1995; 25: 2163-9. 18. Caux C. Pathways of development of human dendritic cells. Eur J Dermatol 1998; 8: 375-84. 19. Kurimoto I, Grammer SF, Shimizu T, Nakamura T, Streilein JW. Role of F4/80+ cells during induction of hapten-specific contact hypersensitivity. Immunology 1995; 85: 621-9. 20. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003; 19: 71-82. 21. Taylor PR, Gordon S. Monocyte heterogeneity and innate immunity. Immunity 2003; 19: 2-4. 22. Frey JR, Wenk P. Experimental studies on the pathogenesis of contact eczema in the guinea-pig. Int Arch Allergy Appl Immunol 1957; 11: 81-100. 23. Macatonia SE, Edwards AJ, Knight SC. Dendritic cells and the initiation of contact sensitivity to fluorescein isothiocyanate. Immunology 1986; 59: 509-14. 24. Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J Exp Med 1987; 166: 1654-67. 25. Kamath AT, Henri S, Battye F, Tough DF, Shortman K. Developmental kinetics and lifespan of dendritic cells in mouse lymphoid organs. Blood 2002; 100: 1734-41. 26. Enk AH, Angeloni VL, Udey MC, Katz SI. An essential role for Langerhans cell-derived IL-1 beta in the initiation of primary immune responses in skin. J Immunol 1993; 150: 3698-704. 27. Borkowski TA, et al. Expression of E-cadherin by murine dendritic cells: E-cadherin as a dendritic cell differentiation antigen characteristic of epidermal Langerhans cells and related cells. Eur J Immunol 1994; 24: 2767-74. 28. Schwarzenberger K, Udey MC. Contact allergens and epidermal proinflammatory cytokines modulate Langerhans cell E-cadherin expression in situ. J Invest Dermatol 1996; 106: 553-8. 29. Wang B, et al. Tumour necrosis factor receptor II (p75) signalling is required for the migration of Langerhans’ cells. Immunology 1996; 88: 284-8. 30. Wang B, et al. Depressed Langerhans cell migration and reduced contact hypersensitivity response in mice lacking TNF receptor p75. J Immunol 1997; 159: 6148-55. 31. Tang A, Amagai M, Granger LG, Stanley JR, Udey MC. Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature 1993; 361: 82-5. 32. Dieu MC, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med 1998; 188: 373-86. 33. Kobayashi Y, Matsumoto M, Kotani M, Makino T. Possible involvement of matrix metalloproteinase-9 in Langerhans cell migration and maturation. J Immunol 1999; 163: 5989-93. 293 34. Gearing AJ, et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature 1994; 370: 555-7. 35. Cyster JG. Chemokines and the homing of dendritic cells to the T cell areas of lymphoid organs. J Exp Med 1999; 189: 447-50. 36. Ngo VN, et al. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med 1999; 189: 403-12. 37. Engeman TM, Gorbachev AV, Gladue RP, Heeger PS, Fairchild RL. Inhibition of functional T cell priming and contact hypersensitivity responses by treatment with anti-secondary lymphoid chemokine antibody during hapten sensitization. J Immunol 2000; 164: 5207-14. 38. Martin-Fontecha A, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med 2003; 198: 615-21. 39. Becker D, Neiss U, Neis S, Reske K, Knop J. Contact allergens modulate the expression of MHC class II molecules on murine epidermal Langerhans cells by endocytotic mechanisms. J Invest Dermatol 1992; 98: 700-5. 40. Becker D, Mohamadzadeh M, Reske K, Knop J. Increased level of intracellular MHC class II molecules in murine Langerhans cells following in vivo and in vitro administration of contact allergens. J Invest Dermatol 1992; 99: 545-9. 41. Pierre P, Mellman I. Developmental regulation of invariant chain proteolysis controls MHC class II trafficking in mouse dendritic cells. Cell 1998; 93: 1135-45. 42. Garrett WS,et al. Developmental control of endocytosis in dendritic cells by Cdc42. Cell 2000; 102: 325-34. 43. Ruedl C, Koebel P, Karjalainen K. In vivo-matured Langerhans cells continue to take up and process native proteins unlike in vitro-matured counterparts. J Immunol 2001; 166: 7178-82. 44. Waksman BH. Cellular hypersensitivity immunity : inflammation and cytotoxicity. Philadelphia: Saunders, 1978. p. 173-218. 45. Weltzien HU, et al. T cell immune responses to haptens. Structural models for allergic and autoimmune reactions. Toxicology 1996; 107: 141-51. 46. von Bonin A, Ortmann B, Martin S, Weltzien HU. Peptideconjugated hapten groups are the major antigenic determinants for trinitrophenyl-specific cytotoxic T cells. Int Immunol 1992; 4: 869-74. 47. Kohler J, Hartmann U, Grimm R, Pflugfelder U, Weltzien HU. Carrier-independent hapten recognition and promiscuous MHC restriction by CD4+ T cells induced by trinitrophenylated peptides. J Immunol 1997; 158: 591-7. 48. Cavani A, Hackett CJ, Wilson KJ, Rothbard JB, Katz SI. Characterization of epitopes recognized by hapten-specific CD4+ T cells. J Immunol 1995; 154: 1232-8. 49. Gamerdinger K, et al. A new type of metal recognition by human T cells: contact residues for peptide-independent bridging of T cell receptor and major histocompatibility complex by nickel. J Exp Med 2003; 197: 1345-53. 50. Sinigaglia F. The molecular basis of metal recognition by T cells. J Invest Dermatol 1994; 102: 398-401. 51. Martin S, von Bonin A, Fessler C, Pflugfelder U, Weltzien HU. Structural complexity of antigenic determinants for class I MHCrestricted, hapten-specific T cells. Two qualitatively differing types of H-2Kb-restricted TNP epitopes. J Immunol 1993; 151: 678-87. 52. Kalish RS, Wood JA, LaPorte A. Processing of urushiol (poison ivy) hapten by both endogenous and exogenous pathways for presentation to T cells in vitro. J Clin Invest 1994; 93: 2039-47. 53. Nalefski EA, Rao A. Nature of the ligand recognized by a hapten- and carrier-specific, MHC-restricted T cell receptor. J Immunol 1993; 150: 3806-16. 54. Reiser H, Schneeberger EE. Expression and function of B7-1 and B7-2 in hapten-induced contact sensitivity. Eur J Immunol 1996; 26: 880-5. 55. Symington FW, Brady W, Linsley PS. Expression and function of B7 on human epidermal Langerhans cells. J Immunol 1993; 150: 1286-95. 56. Kondo S, Kooshesh F, Wang B, Fujisawa H, Sauder DN. Contribution of the CD28 molecule to allergic and irritant-induced skin reactions in CD28 -/- mice. J Immunol 1996; 157: 4822-9. 57. Xu H, Heeger PS, Fairchild RL. Distinct roles for B7-1 and B7-2 determinants during priming of effector CD8+ Tc1 and regulatory CD4+ Th2 cells for contact hypersensitivity. J Immunol 1997; 159: 4217-26. 58. Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science 1991; 251: 313-6. 59. Boise LH, et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 1995; 3: 87-98. 60. Adema GJ, et al. A dendritic-cell-derived C-C chemokine that preferentially attracts naive T cells. Nature 1997; 387: 713-7. 294 61. Geijtenbeek TB,et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 2000; 100: 575-85. 62. Krawczyk C,et al. Vav1 controls integrin clustering and MHC/peptide-specific cell adhesion to antigen-presenting cells. Immunity 2002; 16: 331-43. 63. Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Threedimensional segregation of supramolecular activation clusters in T cells. Nature 1998; 395: 82-6. 64. Chen AI, et al. Ox40-ligand has a critical costimulatory role in dendritic cell: T cell interactions. Immunity 1999; 11: 689-98. 65. Sad S, Marcotte R, Mosmann TR. Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokines. Immunity 1995; 2: 271-9. 66. Hahn S, et al. Down-modulation of CD4+ T helper type 2 and type 0 cells by T helper type 1 cells via Fas/Fas-ligand interaction. Eur J Immunol 1995; 25: 2679-85. 67. Li L, Sad S, Kagi D, Mosmann TR. CD8+Tc1 and Tc2 cells secrete distinct cytokine patterns in vitro and in vivo but induce similar inflammatory reactions. J Immunol 1997; 158: 4152-61. 68. Maldonado-Lopez R, Maliszewski C, Urbain J, Moser M. Cytokines regulate the capacity of CD8+alpha(+) and CD8+alpha(-) dendritic cells to prime Th1/Th2 cells in vivo. J Immunol 2001; 167: 4345-50. 69. Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD4+0L drive a potent TH1 polarization. Nat Immunol 2000; 1: 305-10. 70. MacDonald AS, Straw AD, Bauman B, Pearce EJ. CD8+- dendritic cell activation status plays an integral role in influencing Th2 response development. J Immunol 2001; 167: 1982-8. 71. Ardavin C, et al. Origin and differentiation of dendritic cells. Trends Immunol 2001; 22: 691-700. 72. Soumelis V, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol 2002; 3: 673-80. 73. Bour H, et al. Major histocompatibility complex class I-restricted CD8+ T cells and class II-restricted CD4+ T cells, respectively, mediate and regulate contact sensitivity to dinitrofluorobenzene. Eur J Immunol 1995; 25: 3006-10. 74. Xu H, DiIulio NA, Fairchild RL. T cell populations primed by hapten sensitization in contact sensitivity are distinguished by polarized patterns of cytokine production: interferon gamma-producing (Tc1) effector CD8+ T cells and interleukin (Il) 4/Il-10-producing (Th2) negative regulatory CD4+ T cells. J Exp Med 1996; 183: 1001-12. 75. Gorbachev AV, DiIulio NA, Fairchild RL. IL-12 augments CD8+ T cell development for contact hypersensitivity responses and circumvents anti-CD154 antibody-mediated inhibition. J Immunol 2001; 167: 156-62. 76. Gocinski BL, Tigelaar RE. Roles of CD4+ and CD8+ T cells in murine contact sensitivity revealed by in vivo monoclonal antibody depletion. J Immunol 1990; 144: 4121-8. 77. Bouloc A, Cavani A, Katz SI. Contact hypersensitivity in MHC class II-deficient mice depends on CD8+ T lymphocytes primed by immunostimulating Langerhans cells. J Invest Dermatol 1998; 111: 44-9. 78. Kolesaric A, Stingl G, Elbe-Burger A. MHC class I+/II- dendritic cells induce hapten-specific immune responses in vitro and in vivo. J Invest Dermatol 1997; 109: 580-5. 79. Krasteva M,et al. Dual role of dendritic cells in the induction and down-regulation of antigen-specific cutaneous inflammation. J Immunol 1998; 160: 1181-90. 80. Martin S,et al. Peptide immunization indicates that CD8+ T cells are the dominant effector cells in trinitrophenyl-specific contact hypersensitivity. J Invest Dermatol 2000; 115: 260-6. 81. Xu H, DiIulio NA, Fairchild RL. T cell populations primed by hapten sensitization in contact sensitivity are distinguished by polarized patterns of cytokine production: interferon gamma-producing (Tc1) effector CD8+ T cells and interleukin (Il) 4/Il-10-producing (Th2) negative regulatory CD4+ T cells. J Exp Med 1996; 183: 1001-12. 82. Xu H, Banerjee A, Dilulio NA, Fairchild RL. Development of effector CD8+ T cells in contact hypersensitivity occurs independently of CD4+ T cells. J Immunol 1997; 158: 4721-8. 83. Matzinger P. The danger model: a renewed sense of self. Science 2002; 296: 301-5. 84. Wang B,et al. Multiple paths for activation of naive CD8+ T cells: CD4+-independent help. J Immunol 2001; 167: 1283-9. 85. Martin S,et al. A high frequency of allergen-specific CD8+ Tc1 cells is associated with the murine immune response to the contact sensitizer trinitrophenyl. Exp Dermatol 2003; 12: 78-85. 86. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999; 401: 708-12. 87. Santamaria-Babi LF. CLA+ T cells in cutaneous diseases. Eur J Dermatol 2004; 14: 13-8. EJD, vol. 14, n° 5, September-October 2004 88. Dudda JC, Simon JC, Martin S. Dendritic cell immunization route determines CD8+ T cell trafficking to inflamed skin: role for tissue microenvironment and dendritic cells in establishment of T cell-homing subsets. J Immunol 2004; 172: 857-63. 89. Berg EL,et al. The cutaneous lymphocyte antigen is a skin lymphocyte homing receptor for the vascular lectin endothelial cellleukocyte adhesion molecule 1. J Exp Med 1991; 174: 1461-6. 90. Campbell JJ,et al. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature 1999; 400: 776-80. 91. Erdmann I,et al. Fucosyltransferase VII-deficient mice with defective E-, P-, and L-selectin ligands show impaired CD4+ and CD8+ T cell migration into the skin, but normal extravasation into visceral organs. J Immunol 2002; 168: 2139-46. 92. Reiss Y, Proudfoot AE, Power CA, Campbell JJ, Butcher EC. CC chemokine receptor (CCR)4 and the CCR10 ligand cutaneous T cell-attracting chemokine (CTACK) in lymphocyte trafficking to inflamed skin. J Exp Med 2001; 194: 1541-7. 93. Santamaria LF, Perez Soler MT, Hauser C, Blaser K. Allergen specificity and endothelial transmigration of T cells in allergic contact dermatitis and atopic dermatitis are associated with the cutaneous lymphocyte antigen. Int Arch Allergy Immunol 1995; 107: 359-62. 94. Enk AH, Katz SI. Early molecular events in the induction phase of contact sensitivity. Proc Natl Acad Sci U S A 1992; 89: 1398402. 95. Heufler C, et al. Cytokine gene expression in murine epidermal cell suspensions: interleukin 1 beta and macrophage inflammatory protein 1 alpha are selectively expressed in Langerhans cells but are differentially regulated in culture. J Exp Med 1992; 176: 1221-6. 96. Akiba H, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol 2002; 168: 3079-87. 97. Albanesi C,et al. A cytokine-to-chemokine axis between T lymphocytes and keratinocytes can favor Th1 cell accumulation in chronic inflammatory skin diseases. J Leukoc Biol 2001; 70: 617-23. 98. Bonecchi R, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 1998; 187: 129-34. 99. Flier J, et al. The CXCR3 activating chemokines IP-10, Mig, and IP-9 are expressed in allergic but not in irritant patch test reactions. J Invest Dermatol 1999; 113: 574-8. 100. Dufour JH,et al. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol 2002; 168: 3195-204. 101. Homey B, et al. Cutting edge: the orphan chemokine receptor G protein-coupled receptor-2 (GPR-2, CCR10) binds the skinassociated chemokine CCL27 (CTACK/ALP/ILC). J Immunol 2000; 164: 3465-70. 102. Homey B,et al. CCL27-CCR10 interactions regulate T cellmediated skin inflammation. Nat Med 2002; 8: 157-65. 103. Cher DJ, Mosmann TR. Two types of murine helper T cell clone. II. Delayed-type hypersensitivity is mediated by TH1 clones. J Immunol 1987; 138: 3688-94. 104. Gawkrodger DJ, Vestey JP, Wong WK, Buxton PK. Contact clinic survey of nickel-sensitive subjects. Contact Dermatitis 1986; 14: 165-9. 105. Silvennoinen-Kassinen S, Ikaheimo I, Karvonen J, Kauppinen M, Kallioinen M. Mononuclear cell subsets in the nickelallergic reaction in vitro and in vivo. J Allergy Clin Immunol 1992; 89: 794-800. 106. Cavani A, Albanesi C, Traidl C, Sebastiani S, Girolomoni G. Effector and regulatory T cells in allergic contact dermatitis. Trends Immunol 2001; 22: 118-20. 107. Biedermann T,et al. Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J Exp Med 2000; 192: 1441-52. 108. Saulnier M, Huang S, Aguet M, Ryffel B. Role of interferongamma in contact hypersensitivity assessed in interferon-gamma receptor-deficient mice. Toxicology 1995; 102: 301-12. 109. Kehren J,et al. Cytotoxicity is mandatory for CD8+(+) T cellmediated contact hypersensitivity. J Exp Med 1999; 189: 779-86. 110. Traidl C,et al. Disparate cytotoxic activity of nickel-specific CD8+ and CD4+ T cell subsets against keratinocytes. J Immunol 2000; 165: 3058-64. 111. Trautmann A, Akdis M, Brocker EB, Blaser K, Akdis CA. New insights into the role of T cells in atopic dermatitis and allergic contact dermatitis. Trends Immunol 2001; 22: 530-2. 112. Wagner L,et al. Beta-chemokines are released from HIV-1specific cytolytic T-cell granules complexed to proteoglycans. Nature 1998; 391: 908-11. EJD, vol. 14, n° 5, September-October 2004 113. Gorbachev AV, Fairchild RL. CD4+(+) T Cells Regulate CD8+(+) T Cell-Mediated Cutaneous Immune Responses by Restricting Effector T Cell Development through a Fas Ligand-Dependent Mechanism. J Immunol 2004; 172: 2286-95. 114. Dubois B, Chapat L, Goubier A, Kaiserlian D. CD4+CD25+ T cells as key regulators of immune responses. Eur J Dermatol 2003; 13: 111-6. 115. Gorbachev AV, Fairchild RL. Regulatory role of CD4+ T cells during the development of contact hypersensitivity responses. Immunol Res 2001; 24: 69-77. 116. Gorbachev AV, Fairchild RL. Induction and regulation of T-cell priming for contact hypersensitivity. Crit Rev Immunol 2001; 21: 451-72. 117. Dubois B,et al. Innate CD4+CD25+ regulatory T cells are required for oral tolerance and control CD8+ T cells mediating skin inflammation. Blood 2003; 102:3295-330. 118. Ruckert R, Brandt K, Hofmann U, Bulfone-Paus S, Paus R. IL-2IgG2b fusion protein suppresses murine contact hypersensitivity in vivo. J Invest Dermatol 2002; 119: 370-6. 119. Ferguson TA, Dube P, Griffith TS. Regulation of contact hypersensitivity by interleukin 10. J Exp Med 1994; 179: 1597-604. 120. Sebastiani S,et al. Chemokine receptor expression and function in CD4+ T lymphocytes with regulatory activity. J Immunol 2001; 166: 996-1002. 121. Enk AH, Katz SI. Identification and induction of keratinocytederived IL-10. J Immunol 1992; 149: 92-5. 122. Melrose J, Tsurushita N, Liu G, Berg EL. IFN-gamma inhibits activation-induced expression of E- and P-selectin on endothelial cells. J Immunol 1998; 161: 457-64. 123. Kanerva L, Alanko K. Allergic contact dermatitis from 2-hydroxyethyl methacrylate in an adhesive on an electrosurgical earthing plate. Eur J Dermatol 1998; 8: 521-4. 124. van der Valk PG, Devos SA, Coenraads PJ. Evidence-based diagnosis in patch testing. Contact Dermatitis 2003; 48: 121-5. 125. Suneja T, Belsito DV. Comparative study of Finn Chambers and T.R.U.E. test methodologies in detecting the relevant allergens inducing contact dermatitis. J Am Acad Dermatol 2001; 45: 836-9. 126. Lachapelle JM,et al. Proposal for a revised international standard series of patch tests. Contact Dermatitis 1997; 36: 121-3. 127. Loffler H, et al. Evaluation of skin susceptibility to irritancy by routine patch testing with sodium lauryl sulfate. Eur J Dermatol 2001; 11: 416-9. 128. Fregert S. Manual of Contact Dermatitis. ed. 2. Copenhagen: Munksgaard; 1981. 129. Lachapelle JM. A proposed relevance scoring system for positive allergic patch test reactions: practical implications and limitations. Contact Dermatitis 1997; 36: 39-43. 130. Bruze M. What is a relevant contact allergy? Contact Dermatitis 1990; 23: 224-5. 131. Menne T. Prevention of nickel allergy by regulation of specific exposures. Ann Clin Lab Sci 1996; 26: 133-8. 132. Jensen CS, Lisby S, Baadsgaard O, Volund A, Menne T. Decrease in nickel sensitization in a Danish schoolgirl population with ears pierced after implementation of a nickel-exposure regulation. Br J Dermatol 2002; 146: 636-42. 133. Schnuch A, Geier J, Lessmann H, Uter W. Decrease in nickel sensitization in young patients--successful intervention through nickel exposure regulation? Results of IVDK, 1992-2001. Hautarzt 2003; 54: 626-32. 134. Goossens A, et al. Adverse cutaneous reactions to cosmetic allergens. Contact Dermatitis 1999; 40: 112-3. 135. De Groot AC. Fatal attractiveness: the shady side of cosmetics. Clin Dermatol 1998; 16: 167-79. 136. Hatch KL, Maibach HI. Textile dye allergic contact dermatitis prevalence. Contact Dermatitis 2000; 42: 187-95. 137. Rocha N, Silva E, Horta M, Massa A. Contact allergy to topical corticosteroids 1995-2001. Contact Dermatitis 2002; 47: 362-3. 138. Corazza M, Mantovani L, Maranini C, Bacilieri S, Virgili A. Contact sensitization to corticosteroids: increased risk in long term dermatoses. Eur J Dermatol 2000; 10: 533-5. 139. Veien NK, Hattel T, Laurberg G. Low nickel diet: an open, prospective trial. J Am Acad Dermatol 1993; 29: 1002-7. 140. Saripalli YV, Gadzia JE, Belsito DV. Tacrolimus ointment 0.1% in the treatment of nickel-induced allergic contact dermatitis. J Am Acad Dermatol 2003; 49: 477-82. 141. Ruzicka T, Assmann T, Lebwohl M. Potential future dermatological indications for tacrolimus ointment. Eur J Dermatol 2003; 13: 331-42. 142. Marsland AM, Griffiths CE. The macrolide immunosuppressants in dermatology: mechanisms of action. Eur J Dermatol 2002; 12: 618-22. 295





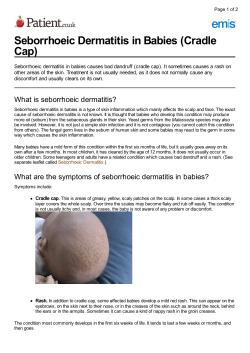






© Copyright 2026