
The Management of Ascites and Hyponatremia in Cirrhosis Pere Gine`s, M.D.,
The Management of Ascites and Hyponatremia in Cirrhosis Pere Gine`s, M.D.,1 and Andre´s Ca´rdenas, M.D., M.M.Sc.2 ABSTRACT Ascites is the most common complication of cirrhosis and is associated with an increased risk for the development of infections, dilutional hyponatremia, renal failure, and mortality. Cirrhotic patients who develop ascites and associated complications have a low probability of long-term survival without liver transplantation, and therefore should be referred for evaluation of liver transplantation. While the initial management of uncomplicated ascites with low-sodium diet and diuretic treatment is straightforward in the majority of patients, there is a group of patients who fail to respond to diuretics and develop refractory ascites. The development of specific associated complications such as dilutional hyponatremia may further challenge the management of patients with ascites. New pharmacological agents such as the V2 receptor antagonists, drugs that directly antagonize the effects of elevated plasma antidiuretic hormone levels, induce solute-free water diuresis and seem to be promising in the management of patients with cirrhosis, ascites, and dilutional hyponatremia. This article focuses on the pathophysiology, clinical consequences, current management, and new treatment modalities for ascites and dilutional hyponatremia in cirrhosis. KEYWORDS: Ascites, hyponatremia, vaptans C irrhosis is the twelfth-leading cause of mortality in the United States, accounting for over 26,000 deaths.1 Ascites is the most common complication of cirrhosis resulting in poor quality of life, high risk of development of complications of cirrhosis, increased morbidity and mortality associated with surgical interventions, high risk of renal failure, and poor long-term outcome.2–4 Cirrhotic patients who develop ascites have a probability of survival of 85% at 1 year and 56% at 5 years without liver transplantation.3 Hyponatremia is a frequent complication of patients with cirrhosis and ascites that is also associated with increased morbidity and mortality, the importance of which is increasingly being recognized.5 This article reviews current concepts of the pathophysiology and management of ascites and dilutional hyponatremia (DH) in patients with cirrhosis. Moreover, we review the significance of hyponatremia in the setting of cirrhosis and the possible efficacy of the arginine vasopressin (AVP) V2 receptor antagonists that are currently under investigation.6 1 M.D., Liver Unit, Hospital Clı´nic, Villarroel 170, 08036 Barcelona, Spain (e-mail: [email protected]). Complications of Cirrhosis; Guest Editor, Pere Gine`s, M.D. Semin Liver Dis 2008;28:43–58. Copyright # 2008 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York, NY 10001, USA. Tel: +1(212) 584-4662. DOI 10.1055/s-2008-1040320. ISSN 0272-8087. Liver Unit, Hospital Clı´nic and University of Barcelona, Institut d’Investigacions Biome`diques August Pi-Sunyer (IDIBAPS), Ciber de Enfermedades Hepaticas y Digestivas (CIBERHED), Barcelona, Spain; 2GI Unit, Hospital Clı´nic and University of Barcelona, Institut d’Investigacions Biome`diques August Pi-Sunyer (IDIBAPS), Ciber de Enfermedades Hepaticas y Digestivas (CIBERHED), Barcelona, Spain. Address for correspondence and reprint requests: Pere Gine`s, PATHOPHYSIOLOGY OF ASCITES The major factor contributing to ascites formation is a splanchnic vasodilation resulting in a decreased effective arterial blood volume.7 Increased hepatic resistance to portal flow due to cirrhosis causes gradual development of portal hypertension and collateral vein formation with 43 44 SEMINARS IN LIVER DISEASE/VOLUME 28, NUMBER 1 2008 Figure 1 Pathogenesis of ascites formation in cirrhosis and current therapeutic options for ascites and dilutional hyponatremia. Portal hypertension is the main factor responsible for the development of splanchnic vasodilation and a decrease in effective arterial blood volume. The transjugular intrahepatic portosystemic shunt (TIPS), by reducing portal pressure, is an effective treatment for reducing ascites, particularly in patients with refractory ascites. The compensatory homeostatic response occurs secondary to a reduced effective arterial blood volume and leads to the activation of antinatriuretic factors (mainly the renin angiotensin-aldosterone system) with subsequent sodium retention. The use of diuretics (spironolactone) that promote natriuresis is the preferred therapy for patients with sodium retention and fluid accumulation. Patients with largevolume ascites should be treated with therapeutic paracentesis and albumin administration (8 g per liter tapped) followed by diuretics as required. In the later stages of the disease solute-free water retention may occur due to high levels of arginine vasopressin; this contributes to hyponatremia and ascites accumulation. The new V2 receptor antagonists that block the action of arginine vasopressin in the distal tubule of the kidney are the most promising therapy for dilutional hyponatremia. shunting of blood to the systemic circulation. As portal hypertension develops, splanchnic arterial vasodilation occurs due to the enhanced local production of nitric oxide and other vasodilators (including calcitonin generelated peptide, substance P, carbon monoxide, and endogenous cannabinoids) as a consequence of endothelial stretching and possibly bacterial translocation.8,9 Recent studies have provided evidence indicating that bacterial translocation to mesenteric lymph nodes with increased production of bacterial products and subsequent stimulation of cytokine synthesis play a major role in the pathogenesis of arterial vasodilation and associated circulatory abnormalities that occur in cirrhosis.10–12 In early stages of cirrhosis, splanchnic arterial vasodilation is moderate and has no major effect on effective arterial blood volume, which is maintained within normal limits due to an increase in plasma volume and cardiac output.7 In advanced stages, splanchnic arterial vasodilation is so intense that effective arterial blood volume becomes markedly reduced and arterial pressure falls. A reduction in cardiac output, possibly related to the presence of cirrhotic cardiomyopathy, occurs in the late stages of cirrhosis and may also contribute to the impairment in effective arterial blood volume.13–15 As a consequence of the impairment in effective arterial blood volume, there is homeostatic activation of vasoconstrictor and antinatriuretic factors to maintain arterial pressure, resulting in renal sodium and fluid retention. The combination of portal hypertension and splanchnic arterial vasodilation alters intestinal capillary pressure and permeability, which facilitates the accumulation of the retained fluid in the abdominal cavity. With disease progression, there is a marked impairment in renal solute-free water excretion and renal vasoconstriction, leading to DH and hepatorenal syndrome (HRS), respectively. The pathophysiological rationale for the treatment of patients with cirrhosis and hyponatremia is described in Fig. 1. EVALUATION OF PATIENTS WITH ASCITES The evaluation of patients with ascites should include standard hematology, electrolyte, renal (serum creatinine and blood urea nitrogen), coagulation (prothombin time MANAGEMENT OF ASCITES AND HYPONATREMIA/GINE`S, CA´RDENAS or international normalized ratio), and liver tests (aminotransferases, bilirubin, albumin, total protein, alkaline phosphatase).16,17 An abdominal ultrasonography to rule out hepatocellular carcinoma and evaluate the patency of the portal venous system should be performed. In addition, an upper gastrointestinal endoscopy to assess the presence and characteristics of esophageal and gastric varices is recommended. In patients with renal failure (serum creatinine greater than 1.5 mg/dL), urine sediment and 24-hour urine protein should be assessed and the kidneys examined by ultrasonography. Evaluation of circulatory function should include measurement of arterial pressure and heart rate. A diagnostic paracentesis (30 mL of fluid) is required in all patients presenting with their first episode of ascites and in all patients with any evidence of clinical deterioration such as fever, abdominal pain, gastrointestinal bleeding, hepatic encephalopathy, hypotension, or renal failure. The ascitic fluid in cirrhotics is mostly transparent and yellow/amber in color. Tests in the ascitic fluid should include cell count, albumin, total protein, and cultures in blood culture bottles (10 mL of fluid injected at the bedside).17–20 The cell count is the most helpful test in determining bacterial infection. In most cases the ascitic fluid white blood cell count is less than 100/mm3 with a predominance of mononuclear cells (> 75%) and a low number of neutrophils. An increased number of white blood cells with predominance of neutrophils indicates peritoneal infection. The diagnosis of spontaneous bacterial peritonitis (SBP) is made when the fluid sample has > 250/mm3 neutrophils and there are no signs of peritonitis due to perforation or inflammation of intrabdominal organs.19–21 The difference between serum albumin concentration and ascites albumin concentration (serum-ascites albumin gradient) in patients with cirrhosis and ascites is usually greater than 11 g/L; values lower than 11 g/L suggest a cause of ascites other than cirrhosis.20 A low total protein concentration in ascitic fluid (< 15 g/L) is associated with an increased risk of SBP and in selected patients may indicate a need for antibiotic prophylaxis with oral quinolones to reduce the risk of SBP and HRS (see below). Finally, cultures in blood culture bottles increase the probability of isolating an organism to 50% of cases if the fluid is inoculated into the bottles at the bedside.20,22 and elimination of ascites in only 10% of patients. Fluid restriction is not necessary unless patients have DH, a condition defined as serum sodium less than 130 mEq/L together with ascites and/or edema5,23 (see below). In DH, fluid restriction of 1000 to 1500 mL/day is recommended.6,23 Patients with serum sodium concentration between 130 and 135 mEq/L should be cautious about fluid intake to prevent fluid retention and development of DH. An evaluation by a nutritionist is of utmost importance for appropriate education regarding suitable caloric and salt intake. Improvement of the nutritional status is essential because patients with advanced liver disease have decreased intake and absorption of nutrients, increased energy expenditure, and altered fuel metabolism with an accelerated starvation metabolism.24,25 Nutritional therapy in cirrhotic patients can improve nutritional status, reduce infection rates, and decrease perioperative morbidity.24,25 It is also recommended that nutritional therapy be instituted for long periods of time or until patients reach liver transplantation. The goal is for nutritional supplementation to correct the underlying protein energy malnutrition. In advanced cases of malnutrition, supplemental enteral nutrition in cirrhotic patients with ascites may improve liver function and hepatic encephalopathy.24–27 MANAGEMENT OF PATIENTS WITH ASCITES MODERATE-VOLUME ASCITES General Measures A low-sodium diet of 2000 mg per day (90 mmol per day) may facilitate the elimination of ascites and delay the reaccumulation of fluid.16,17 However, a reduction in sodium intake alone achieves a negative sodium balance Specific Measures The current classification of ascites defined by the International Ascites Club divides patients into three groups.19 Patients with grade 1 ascites are those in whom ascites is detected only by ultrasonography; these patients do not require any specific treatment, but they should be cautioned about avoiding foods with large amounts of salt. Patients with grade 2 ascites are those in whom ascites cause moderate distension of the abdomen associated with mild/moderate discomfort. Patients with grade 3 ascites have large amounts of ascitic fluid causing marked abdominal distension and associated with significant discomfort. Patients with refractory ascites are those who do not respond to high doses of diuretics or develop side effects that preclude their use. The discussion of the management of ascites may therefore be divided into three parts: moderate-volume ascites (grade 2 ascites), large-volume ascites (grade 3), and refractory ascites. A summary of recommendations for the management of ascites is included in Table 1. Patients with moderate ascites usually accumulate fluid slowly, such that they typically do not develop large ascites unless their sodium intake is very high or they do not seek medical attention for long periods of time. Renal solute-free water excretion and glomerular filtration rate are normal in most cases; therefore, serum sodium and creatinine concentration are usually within 45 46 SEMINARS IN LIVER DISEASE/VOLUME 28, NUMBER 1 2008 Table 1 Management Practice Points in Patients with Ascites and Cirrhosis Treatment strategy for patients with cirrhosis and moderate volume (grade 2) ascites Start with a low-sodium diet (90 mmol/day) and spironolactone (50–100 mg/day) to reach goal of weight loss: 300–500 g/day. If needed, doses to be increased every 7 days up to with diuretic therapy, may require a reduction in diuretic dosage. Isolated reports indicate that quinidine (quinidine sulfate, 400 mg/day)31 and intravenous albumin administration (25 g/week)32 reduce the frequency and intensity of muscle cramps in cirrhotic patients with ascites treated with diuretics. 400 mg/day of spironolactone. Furosemide can be added LARGE-VOLUME ASCITES at a starting dose of 20–40 mg/day and subsequently increased Patients with large-volume ascites can be treated as outpatients unless they have associated complications. The majority of these patients have intense sodium retention (urine sodium frequently lower than 10 mEq/ L), so that ascites accumulate rapidly even under sodium restriction. Some of these patients have a mild impairment in solute-free water excretion and may develop DH if they have increased fluid intake. Serum creatinine levels are normal or only moderately increased, indicating normal or moderately reduced glomerular filtration rate. Patients can be treated with diuretics alone or with large-volume paracentesis followed by diuretics. Diuretics alone are given at increasing doses (maximum of 400 mg/day of spironolactone and 160 mg/day of furosemide) following the same schedule as indicated above. Nonetheless, complete removal of ascites with one session of large-volume paracentesis with intravenous albumin (8 g per liter tapped) is a faster and more effective measure in controlling tense ascites and is associated with a lower number of complications than conventional diuretic therapy.33,34 Therefore, current guidelines support paracentesis followed by diuretics as the method of choice to treat large-volume ascites.19,35 Although there is no difference between the two strategies in regard to long-term mortality, large-volume paracentesis is faster, more effective, and associated with fewer adverse events compared with diuretics. Large-volume paracentesis without plasma expanders is associated with a derangement in circulatory function, characterized by reduction of effective arterial blood volume with subsequent activation of vasoconstrictor and antinatriuretic factors.36–38 Circulatory dysfunction after large-volume paracentesis is associated with high ascites recurrence rate, development of HRS and/or DH in 20% of cases, and shortened survival.36–38 This disorder can be effectively prevented with the administration of plasma expanders.33–38 When less than 5 L of ascites are removed, artificial plasma expanders, saline, and albumin are equally effective.38–40 However, if more than 5 L are removed, albumin is recommended.36–38 Since ascites will invariably recur, patients need to be started or continued on diuretics to prevent a positive sodium balance.41 Although the use of albumin after paracentesis is controversial due to the lack of data proving a survival benefit and high cost in some countries, the protective effect of albumin on the circulatory system compared with other expanders favors its use in this setting. to 160 mg/day if needed. Treatment strategy for patients with cirrhosis and large-volume (grade 3) ascites Total paracentesis plus intravenous albumin (8 g per liter of ascites removed) followed by a low-sodium diet (90 mmol/day) and diuretics if patient tolerated them beforehand. Treatment strategy for refractory ascites Total paracentesis plus intravenous albumin can be performed as needed. Consider use of TIPS in patients with very frequent recurrent ascites without severely impaired hepatic function, aged < 70 years, and no hepatic encephalopathy. TIPS, transjugular intrahepatic portosystemic shunts. normal limits. These patients typically can be managed as outpatients and do not require hospitalization unless other complications of cirrhosis are present. A negative sodium balance with loss of ascites is quickly and easily obtained in most cases with low doses of diuretics.17,19,20,28,29 Diuretics of choice are either spironolactone (starting dose: 50 to 100 mg/day) or amiloride (5 to 10 mg/day). Low doses of furosemide (20 to 40 mg/ day) may be useful during the first few days to increase natriuresis, especially when marked peripheral edema is present; however, it should be used with caution because it can cause overdiuresis leading to hypokalemia and prerenal renal failure. The goal of treatment is to produce an average weight loss of 300 to 500 g/day in patients without peripheral edema and 800 to 1000 g/ day in those with peripheral edema until ascites have decreased markedly, following which diuretic dosage can be reduced.17,19 In the uncommon cases of no response to low doses of diuretics, spironolactone may be increased up to a dose of 400 mg/day and furosemide up to 160 mg/day in progressively increasing doses.29 In patients who do not respond, compliance with a lowsodium diet and diuretics should always be assessed. In these cases measurement of urine sodium, which provides an accurate assessment of the response to diuretics, may help in deciding whether there is a need to increase dose. Spironolactone may cause tender gynecomastia in some patients and in most instances the dose needs to be reduced or the medication needs to be stopped. Tamoxifen (20 mg by mouth twice a day) may be useful in reducing pain in some cases; however, information is very limited.30 Muscle cramps, which also may appear MANAGEMENT OF ASCITES AND HYPONATREMIA/GINE`S, CA´RDENAS Complications related to paracentesis, such as bleeding, infection, or intestinal perforation, are exceedingly rare if paracentesis is performed under sterile conditions and with an appropriate technique and needle.20,33,35–38,42–45 The recommended location for performing paracentesis is the left lower quadrant, 2 finger breadths cephalad and 2 finger breadths medial to the anterior superior iliac spine. In this area, the skin is thin and there is a larger pool of fluid than at the midline.46 The risk of bleeding in cirrhotic patients when performing a paracentesis is extremely low; the frequency of severe hemorrhage after a tap is 0.20% and a lethal outcome occurs in less than 0.01% of cases.43 In some cases, severe hemorrhage due to a paracentesis may occur due to trauma of the inferior hypogastric artery. In such cases, the patient requires urgent arteriography with embolization to arrest bleeding (P. Gine`s, unpublished observations). Therefore, when performing paracentesis, the area of the inferior hypogastric arteries (midway between the pubis and anterior superior iliac spines) should be avoided. Most clinical trials in patients with cirrhosis and ascites have excluded patients with an elevated prothrombin time greater than 21 seconds or international normalized ratio greater than 1.6 or platelet count below 50,000 per mL. Therefore, the risk of bleeding complications in patients with more severe coagulopathy is unknown and deserves investigation. Nonetheless, consensus meetings, guidelines, and expert opinion consider that the abnormal coagulation profile of the cirrhotic patient (mild prolonged prothrombin time and low platelets with a value > 50,000 per mL) is not an absolute contraindication for paracentesis and the routine administration of platelets or fresh frozen plasma as prophylaxis for bleeding is not recommended.20,35,44 In patients with severe thrombocytopenia with a platelet count < 50,000 per mL, transfusion of platelets should be considered prior to the procedure.35 Patients with suspected disseminated intravascular coagulopathy should not undergo a paracentesis.20 REFRACTORY ASCITES Refractory ascites occur in nearly 10% of patients with cirrhosis and ascites.19,47 In patients with nonrefractory ascites, sodium excretion may be increased with the use of diuretics, but in patients with refractory ascites, sodium excretion cannot be increased with diuretics, either because patients do not respond to high doses of diuretics (spironolactone 400 mg/day plus furosemide 160 mg/day) or they develop side effects with lower doses that preclude their use19,47 (Table 2). These patients in general have features of advanced liver disease, a high recurrence rate of ascites after large-volume paracentesis, an increased risk of Type-1 HRS, and a poor prognosis.48 Patients can be treated with either repeated large-volume paracentesis with plasmaexpansion or transjugular intrahepatic portosystemic Table 2 Definition and Diagnostic Criteria for Refractory Ascites in Cirrhosis Diuretic-resistant ascites Ascites that cannot be mobilized or the early recurrence of which cannot be prevented because of a lack of response to sodium restriction and diuretic treatment. Diuretic-intractable ascites Ascites that cannot be mobilized or the early recurrence of which cannot be prevented because of the development of diuretic-induced complications that preclude the use of an effective diuretic dosage. Requisites (1) Treatment duration: Patients must be on intensive diuretic therapy (spironolactone 400 mg/day and furosemide 160 mg/day) for at least 1 week and on a salt-restricted diet of less than 90 mmol/day. (2) Lack of response: Mean weight loss of < 0.8 kg over 4 days and urinary sodium output less than the sodium intake. (3) Early ascites recurrence: Reappearance of grade 2 or 3 ascites within 4 weeks of initial mobilization. (4) Diuretic-induced complications: Diuretic-induced hepatic encephalopathy is the development of encephalopathy in the absence of any other precipitating factor. Diuretic-induced renal impairment is an increase of serum creatinine by > 100% to a value > 2 mg/dL in patients with ascites responding to treatment. Diuretic-induced hyponatremia is defined as a decrease of serum sodium by > 10 mEq/L to a serum sodium of < 125 mEq/L. Diuretic induced hypo- or hyperkalemia is defined as a change in serum potassium to < 3 mEq/L or > 6 mEq/L despite appropriate measures. Modified with permission from Moore KP, Wong F, Gine`s P, et al. The management of ascites in cirrhosis: report on the consensus conference of the International Ascites Club. Hepatology 2003;38:258–266. shunts (TIPS). Repeated large-volume paracentesis plus albumin is the most accepted initial therapy for refractory ascites. Patients generally require paracentesis every 2 to 4 weeks, which can be done in the outpatient setting. This approach is therefore easy to perform and relatively inexpensive49.The main drawback is early recurrence of ascites, because paracentesis does not modify the mechanisms responsible for ascites formation. In contrast to paracentesis, TIPS, a nonsurgical method of portal decompression that consists of the insertion of an intrahepatic stent between one hepatic vein and the portal vein using a transjugular approach, is effective in preventing ascites recurrence in refractory ascites because reduction in portal pressure is accompanied by a resolution of ascites in most patients. Uncovered TIPS are very effective in relieving ascites, but are frequently complicated by stenosis of the prosthesis (18 to 78%).50 Polytetrafluoroethylene-covered prostheses seem to improve TIPS patency and decrease the number of clinical relapses and reinterventions without increasing the risk of encephalopathy.51 A randomized trial comparing large-volume paracentesis plus albumin versus covered 47 48 SEMINARS IN LIVER DISEASE/VOLUME 28, NUMBER 1 2008 TIPS is being conducted in patients with refractory ascites (www.clinicaltrials.gov). Randomized clinical trials comparing uncovered TIPS versus repeated paracentesis demonstrate that TIPS controls ascites effectively and is associated with a lower rate of ascites recurrence.49,52–55 In addition, patients with ascites who undergo TIPS improve their nutritional status as measured by resting energy expenditure, total body nitrogen, body fat, and food intake.56 Hepatic encephalopathy occurs in 30 to 50% of patients.49,50,52,53 Two studies showed a survival benefit with TIPS,53,55 but two other studies demonstrated no difference in survival.49,54 Three meta-analyses of these randomized controlled studies conclude that TIPS is better at controlling ascites but does not improve survival compared with paracentesis.57–59 Another recent meta-analysis that reviewed individual patient data of three trials53–55 indicates that TIPS significantly improves transplant-free survival of cirrhotic patients with refractory ascites.60 Although meta-analysis using individual data is in theory a more reliable tool compared with standard meta-analysis, it cannot overcome deficiencies of individual studies, such as inclusion of patients who did not have refractory ascites in two of the studies,53,55 use of large-volume paracentesis without albumin in one study,53 and use of TIPS as rescue therapy in a large proportion of patients treated with large-volume paracentesis in another study.55 Until more information becomes available, the higher frequency of hepatic encephalopathy as well as the higher cost of TIPS compared with large-volume paracentesis plus albumin, in the context of a lack of survival benefit, leaves TIPS as a second-line therapy in the management of refractory ascites. The first line of treatment for refractory ascites is repeated large-volume paracentesis associated with albumin.49,54,57–59 TIPS placement may an option for patients with very rapid recurrence of ascites and preserved liver function (bilirubin < 3 mg/dL, serum sodium level > 130 mEq/ L, Child-Pugh score < 12, model for end-stage liver disease (MELD) score < 18), aged < 70, without hepatic encephalopathy, central hepatocellular carcinoma, or cardiopulmonary disease.58–61 EVALUATION FOR LIVER TRANSPLANTATION Patients with cirrhosis developing their first episode of ascites have a relatively good mid-term prognosis (probability of survival of 85% at 1 year and 76% at 3 years).3 This prognosis is similar to the outcome of liver transplant recipients (probability of survival of 87% at 1 year and 78% at 3 years) (www.unos.org). Several factors associated with poor prognosis have been described in patients with cirrhosis and ascites. These factors, which are usually not present at the time of development of first ascites and occur later in the course of the disease, include low serum sodium concentration, low arterial pressure, high serum creatinine, intense sodium retention (urine sodium less than 10 mEq/day), and high Child-Pugh scores.62,63 Thus, patients with cirrhosis and the first episode of ascites without associated poor prognostic factors may not benefit from liver transplantation, particularly if they have a MELD score < 15 (A. Ca´rdenas, P. Gine`s, unpublished data). Nevertheless, whenever some of these factors are present patients should be promptly referred for evaluation of liver transplantation. The allocation of organs for patients awaiting liver transplantation is currently based on MELD score in many countries.64 Although MELD score predicts survival in patients with cirrhosis and ascites, some studies have suggested that it may not be accurate enough for all patients with ascites, particularly for those with persistent or refractory ascites, who may have a poor prognosis despite low MELD scores. In fact, in one study, half of a cohort of patients awaiting liver transplantation with a MELD score < 21 died within 180 days, suggesting that these patients did not get transplanted soon enough.65 Therefore, there is a need for scores with higher accuracy in the prediction of survival in patients with cirrhosis and ascites. In this regard, several recent studies have shown that serum sodium concentration is a very sensitive prognostic factor, independent of that of the MELD score, in patients with cirrhosis and ascites awaiting liver transplantation.66–71 Moreover, some studies, but not all, have shown that serum sodium improves the prognostic accuracy of the MELD score in the prediction of survival in these patients. Therefore, the suggestion has been made that a new score, MELD-sodium, could be more useful than MELD score alone.68 Nevertheless, the inclusion of serum sodium in a new score for survival prediction in cirrhosis has in our opinion two major limitations. The first and more important one is that serum sodium is a very modifiable parameter. While the parameters currently included in the MELD score (bilirubin, creatinine, and international normalized ratio) are relatively steady despite common therapeutic measures, serum sodium concentration can fluctuate markedly. In fact, the administration of diuretics or moderate amounts of hypotonic fluids may induce significant reductions in serum sodium concentration which may not reflect changes in the prognostic status of the patients. Second, studies are being conducted to investigate the effects of V2 receptor antagonists, drugs that increase serum sodium concentration by improving solute-free water excretion, in the management of DH in cirrhosis.6 Should these drugs have beneficial effects in the management of patients with cirrhosis, their clinical use may be hampered by the inclusion of serum sodium in a prognostic score. In this regard, it is important to remark that hyponatremia before liver transplantation is associated with an increased risk of neurological as well as other complications which result in a poor outcome after transplantation.72,73 Therefore, the use of drugs that can MANAGEMENT OF ASCITES AND HYPONATREMIA/GINE`S, CA´RDENAS increase serum sodium before transplantation could result in reduction of complications and improved posttransplantation outcome. Taken together, all these data suggest that a greater understanding of hyponatremia and its clinical consequences as well as management in patients with cirrhosis should be obtained before serum sodium is included as a new parameter in an improved MELD score to be used for organ allocation in liver transplantation. Future Therapies New treatments for ascites are being evaluated based on two approaches. The first stems from what is known regarding the underlying pathophysiological abnormalities that cause sodium and water retention. In this approach the use of new agents (V2 vasopressin receptor antagonists) that act on the kidney and promote solutefree water diuresis can be combined with diuretics to improve mobilization of ascites and edema. Animal and human studies suggest that these drugs, apart from causing solute-free water excretion, enhance sodium excretion.74,75 The second approach relates to the use of vasoconstrictor drugs that are able to improve the underlying hemodynamic derangements that lead to ascites formation in cirrhosis, thereby reducing the activity of antinatriuretic factors. These drugs may theoretically improve the action of diuretics to maximize ascites mobilization. The use of specific antagonists of the vasopressin receptor (V2 receptor antagonists) on the distal tubule of the kidney by means of counteracting AVP and increasing the excretion of solute-free water is being investigated in the management of ascites in cirrhosis. These drugs are not yet approved for patients with cirrhosis and ascites; however, an intravenous V1-V2 receptor antagonist (conivaptan) is approved by the U.S. Food and Drug Administration for the treatment of euvolemic hyponatremia in hospitalized patients (www.fda.gov). Two large, multicenter, randomized controlled studies evaluated the effects of satavaptan (an oral V2 receptor antagonist) in patients with cirrhosis and ascites.76,77 In one study, 184 cirrhotic patients with ascites were randomized to receive three fixed doses of satavaptan (5 mg, 12.5 mg, and 25 mg daily) or placebo for 14 days.76 Patients were given spironolactone (100 mg/ day) and furosemide (20 to 25 mg/day) during the study period. In the study, 24-hour urine volume of those receiving satavaptan significantly increased in a dosedependent fashion compared with placebo. In addition there was a significant reduction in body weight in the satavaptan group, without any significant side effects compared with placebo. In the other study, 151 cirrhotic patients with recurrent ascites requiring large-volume paracentesis were randomized to receive either three fixed doses of satavaptan (5 mg, 12.5 mg, and 25 mg daily) or placebo for 12 weeks.77 All patients were given spironolactone (100 mg/day) during the study period. The frequency of recurrent ascites was decreased in all groups receiving satavaptan compared with placebo. The clinical utility of these agents in cirrhosis has been assessed only in short-term studies; therefore further controlled studies during longer periods of time are needed to evaluate the safety, efficacy, and applicability of these agents in patients with cirrhosis and ascites. Other potential therapies for patients with ascites stem from data on the use of vasoconstrictors with plasma expansion in patients with cirrhosis and HRS.78,79 These agents improve renal function, urinary sodium excretion, and serum sodium levels, and decrease the levels of vasoctive/antinatriuretic systems in patients with advanced cirrhosis and HRS. Therefore the use of an oral vasoconstrictor is an attractive approach in the treatment of patients with ascites without HRS who do not respond to diuretics. In one study of 39 patients with cirrhosis, oral midodrine (an a-1 adrenergic agonist) 10 mg, three times a day, was given to 11 patients without ascites and 12 with ascites and compared with placebo in 8 patients without ascites and 8 with ascites for 7 days.80 In the midodrine group there was a moderate increase in urine sodium excretion in patients with ascites (urinary sodium from 30 15 to 49 16 mEq/L) as well as glomerular filtration rate (from 80 23 mL/min to 92 27 mL/min) and urine volume (from 0.98 0.26 L to 1.15 0.34 L) and significant decreases in plasma renin activity and aldosterone. Nonetheless, in this study neither body weight or abdominal girth as measures of an effect on ascites were recorded. Finally, clonidine, a centrally acting a2-agonist and sympatholitic agent, was evaluated as an adjunct treatment in patients with cirrhosis and ascites. In a randomized study, oral clonidine (0.075 mg twice a day) led to a more rapid mobilization of ascites with fewer complications than placebo in patients with cirrhosis, ascites, and a plasma norepinephrine level of > 300 pg/mL.81 The use of these drugs in cirrhosis and ascites is limited to a small number of reports; therefore, more controlled studies are needed to evaluate their efficacy and safety in treating patients with ascites. HYPONATREMIA IN CIRRHOSIS In a subset of patients with cirrhosis, hyponatremia may occur due to excessive losses of sodium and extracellular fluid, either from the kidney (as a result of high doses of diuretics) or from the gastrointestinal tract (due to diarrhea or excessive vomiting). In this type of hyponatremia (also termed hypovolemic hyponatremia), there is significant volume depletion and serum sodium levels improve after correcting the precipitating cause; that is, stopping diuretics and replacing sodium losses. Nevertheless, in the majority of patients with advanced 49 50 SEMINARS IN LIVER DISEASE/VOLUME 28, NUMBER 1 2008 cirrhosis, hyponatremia develops in the setting of ascites where there is an expanded extracellular fluid volume along with increased renal sodium retention. This type of hyponatremia is known as hypervolemic hyponatremia or DH. In this condition, renal retention of solute-free water is disproportionate to that of sodium retained and serum sodium levels decrease despite the existence of increased total body sodium. DH in cirrhosis is defined as a reduction in serum sodium concentration below 130 mEq/L in the setting of ascites and/or edema.5,23 The cut-off level of 130 mEq/L was defined arbitrarily in a consensus meeting; however, there are many patients with cirrhosis and ascites with serum sodium levels between 130 and 135 mEq/L that also have an impaired ability to eliminate solute-free water. In patients with refractory ascites or HRS, this proportion may increase up to 50%.49 Prospective data on 997 patients in a multicenter trial showed that the prevalence of hyponatremia in cirrhosis as defined by a serum sodium level 135 mEq/L was 49%; with levels 130 mEq/L, 125 mEq/L, and 120 mEq/L the prevalence was 21.6%, 5.7%, and 1.2%, respectively.5 When compared with patients without hyponatremia, those with hyponatremia had more severe liver disease, worse control of their ascites, a higher rate of hepatic encephalopathy, SBP, and HRS.5 Pathogenesis Sodium concentration in blood is maintained by a complex interaction between baroreceptors, osmoreceptors, and central neurohormonal systems, which include the thirst drive and the antidiuretic hormone or AVP. A decrease in serum sodium concentration may lead to a range of symptoms from mild cognitive and motor dysfunction to seizures and/or coma. Abnormalities in AVP arise from either an increased or decreased production or a deranged action. Diseases such as cirrhosis may cause hyponatremia by nature of an increased production of AVP. In cirrhosis, splanchnic vasodilation leads to arterial underfilling which unloads high-pressure baroreceptors that stimulate a nonosmotic hypersecretion of AVP, leading to solute-free water retention and DH (Fig. 1).7,82 The biological effects of AVP are mediated through three types of G protein-coupled receptors known as V1a, V1b, and V2 receptors. V1a and V1b are associated to the phosphoinositol-signaling pathway with intracellular calcium as second messenger. V1a is responsible for vascular smooth muscle cell contraction, platelet aggregation, and hepatic glycogenolysis, and V1b is expressed in the anterior pituitary where it mediates adrenocorticotropin release.83,84 The V2 receptors are located on the basolateral (capillary) membrane of the principal cells of the collecting ducts and are responsible for the AVP-induced water reabsorption. The effect of AVP on V2-mediated water reabsorption is due to selective water channels called aquaporins (AQP). The most important one is AQP2. This water channel has been characterized in human and rat kidneys and is expressed almost exclusively in the principal cells of the collecting ducts.85,86 The binding of AVP to the V2 receptor stimulates adenyl cyclase via the stimulatory G protein and promotes the formation of cyclic AMP (cAMP). This cAMP binds to a regulatory subunit of protein kinase A, which in turn phosphorylates AQP2, which is then translocated from vesicular bodies present in the cytosol to the luminal (apical) plasma membrane of the collecting duct cells, and acts as a water channel thereby increasing water permeability.83 The water entering the cell by the luminal plasma membrane leaves the cell through the basolateral membrane and enters the capillaries that are in close contact with tubular cells. Recent data indicate that in patients with cirrhosis and ascites the excretion of AQP2 is reduced possibly as a protective mechanism that would prevent ongoing and continuous solute-free water absorption from the collecting duct.87 Data from large clinical studies using specific V2 receptor antagonists of AVP indicate that hypersecretion of AVP plays a major role in the development of DH because the administration of these drugs is associated with an increase in serum sodium concentration in the large proportion of patients with DH.88–92 Cerebral Adaptation to Hyponatremia In health, brain osmolality and extracellular fluid osmolality are in balance with similar amounts of electrolytes and water. When serum sodium falls, water moves into brain cells in response to osmotic gradients to attain osmotic equilibrium, thereby producing brain edema. This cell swelling leads to extrusion of intracellular solutes (mainly potassium) and organic osmolytes within 24 to 48 hours to decrease brain cell osmolality and to therefore try to match that of plasma93,94 (Fig. 2). Organic osmolytes (glutamine, glutamate, taurine, and myo-inositol) are intracellular compounds involved in the long-term adaptation of osmotic changes. Once the solute removal is successfully achieved, osmolytes are extruded afterward and osmotic equilibrium will be maintained between brain and plasma, and no symptoms arise from hyponatremia. In acute hyponatremia, adaptation to this new environment is difficult to achieve and therefore patients may develop symptoms. In chronic hyponatremia there is a slow coordinated loss of solutes and osmolytes that allows effective management of brain volume and explains why in chronic hyponatremia patients often do not have symptoms.95–97 CEREBRAL CHANGES IN CIRRHOSIS Although it has been proposed that patients with cirrhosis and hepatic encephalopathy have some degree of MANAGEMENT OF ASCITES AND HYPONATREMIA/GINE`S, CA´RDENAS Figure 2 Brain volume adaptation to hyponatremia. (A) Under normal conditions brain osmolality and the extracellular fluid osmolality are in balance (the intracellular solutes are potassium and osmolytes and the extracellular solute is sodium). (B) With hyposmolality, water moves into the brain due to changes in osmotic gradients causing edema depicted by the dotted line (middle A). In response to this swelling, the brain loses both intra- and extracellular solutes (B). Water losses accompany losses of brain solute and the expanded volume returns to normal (C). If hyposmolality is sustained, brain volume will eventually normalize and the brain will become adapted to the hyponatremic extracellular fluid. (Adapted from Verbalis JG. The syndrome of inappropriate secretion of antidiuretic hormone secretion and other hyposmolar disorders. In: Schrier R, ed. Diseases of the Kidney and Urinary Tract. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:2214.) cerebral edema, they do not develop signs of significant brain edema or increased intracranial pressure. Studies with 1H-MRS spectroscopy in patients with hepatic encephalopathy reveal a reduction of myo-inositol along with an increased glutamine/glutamate signal that likely contributes to the development of a low-grade brain edema.98 It has therefore been suggested that in cirrhosis, hepatic encephalopathy may be a manifestation of a low-grade cerebral edema, which may deteriorate with other precipitant factors and thereby alter astrocyte function.99,100 Magnetic resonance imaging (MRI) findings in patients with minimal hepatic encephalopathy show changes consistent with a low-grade cerebral edema and these changes become more evident with advanced stages of hepatic encephalopathy or with the insertion of TIPS.98–101 Interestingly, patients with portal hypertension and minimal hepatic encephalopathy but without cirrhosis also have MRI findings consistent with increased brain water content.102 Other factors including drugs, hyponatremia, and inflammatory cytokines can induce astrocyte swelling that in turn leads to a complex series of steps that are implicated in the pathogenesis of hepatic encephalopathy.100 In cirrhosis there is evidence of an increase in brain glutamine and reduction in other organic osmolytes, particularly myo-inositol.103,104 The high levels of glutamine lead to a compensatory loss of myo-inositol and other organic osmolytes but do not appear to completely prevent some degree of brain swelling.98,105,106 The relationship between hyponatremia and hyperammonemia in animals and humans has shed light into the complex interactions between these two states which commonly occur in cirrhosis. In a study of patients with cirrhosis and hyponatremia, levels of organic osmolytes, as measured by MR spectroscopy, correlated directly with serum sodium and osmolality; brain glutamine levels were higher in patients with cirrhosis when compared with those of healthy subjects and correlated with ammonia levels but not with serum sodium. The high levels of glutamine, which in noncirrhotic patients with hyponatremia were low, may be the consequence of a prolonged exposure of the brain to 51 52 SEMINARS IN LIVER DISEASE/VOLUME 28, NUMBER 1 2008 high ammonia levels.106 These findings suggest that organic osmolytes play an important role in cerebral fluid homeostasis in cirrhosis. Hyponatremia in cirrhosis is accompanied by significant changes in osmolytes that possibly prevent significant neurological damage and occur as high levels of glutamine trigger a compensatory response against cell swelling. Several studies indicate that a low serum sodium concentration increases the risk of hepatic encephalopathy: (1) A study in an animal model of acute liver failure and hepatic encephalopathy induced by ammonia infusion indicated that the presence of hyponatremia increases brain edema compared with animals with normal serum sodium levels107; (2) Patients with low brain myo-inositol levels have a marked impairment in neuropsychological tests after oral administration of an amino acid solution (mimicking a protein content that can trigger hepatic encephalopathy), compared with patients with normal brain myo-inositol content108; (3) In patients with acute liver failure, hyponatremia, and high ammonia levels, the administration of hypertonic saline reduces intracerebral pressure109; (4) Hyponatremia by means of producing metabolic changes in brain cells (either the direct effects of low sodium on the brain or the adaptation of the brain to hyponatremia) may act as predisposing factor of hepatic encephalopathy110; (5) Hyponatremia is an independent predictive factor for the development of hepatic encephalopathy in patients with refractory ascites111; (6) Prospective data from a large multicenter trial in patients with hyponatremia indicate there is an inverse relationship between serum sodium levels and the frequency of hepatic encephalopathy, SBP, and HRS. Patients with a serum sodium concentration 130 mEq/L have a higher rate of these complications compared with subjects with normal serum sodium concentration5 (Fig. 3). Clinical Features There is limited information on the clinical consequences of DH in the setting of cirrhosis. Most of the clinical symptoms associated with hyponatremia in patients without liver disease are related to the central nervous system. The typical manifestations of acute hyponatremic encephalopathy in patients without liver disease, which range from confusion and disorientation to more severe symptoms associated with brain herniation, such as coma and seizures, rarely occur in cirrhosis. Hyponatremia in cirrhosis usually develops slowly over several days or weeks, giving the brain time to adapt to a hypotonic state, although occasionally some patients may present with an acute onset of hyponatremia. As discussed before, recent data suggest that hyponatremia is a risk factor for the development of hepatic encephalopathy.111 Moreover, a recent analysis of health-related Figure 3 Clinical data from a large multicenter trial in patients with hyponatremia. Patients with hyponatremia (serum sodium 130 mEq/L) had a higher frequency of spontaneous bacterial peritonitis (SBP), hepatic encephalopathy, and hepatorenal syndrome (HRS) compared with patients without hyponatremia. (Adapted from Angeli P et al. Hyponatremia in cirrhosis: results of a patient population survey. Hepatology 2006;44:1535–1542.) quality of life data by using the SF-36 questionnaire collected on 523 patients with cirrhosis with and without hyponatremia enrolled in multicenter double-blind studies of treatment with the V2-receptor antagonist satavaptan showed that the serum sodium concentration was an independent predictor of six out of the eight domains analyzed.112 Its predictive value was independent of liver function, strongly suggesting that serum sodium concentration is a major determinant of quality of life in patients with cirrhosis. Hyponatremia before liver transplantation has important clinical implications. Since liver transplantation is associated with a rapid correction of low serum sodium levels in hyponatemic patients, this sudden normonatremic state may put patients at risk for deleterious effects on the brain. In fact, there are reports of severe neurological complications, particularly osmotic demyelination syndrome, in patients with cirrhosis with hyponatremia submitted to liver transplantation.113,114 Also, as mentioned above, patients that undergo liver transplantation with low serum sodium levels are prone to more neurological complications, renal failure, and bacterial infections during the first 30 days after transplant and increased 3-month mortality with respect to patients without hyponatremia.73 Prevention There is limited information regarding specific measures that prevent DH. Patients with ascites who undergo large-volume paracentesis without plasma expansion may develop DH due to a postparacentesis circulatory dysfunction.37 This complication may be effectively prevented with the administration of albumin (see MANAGEMENT OF ASCITES AND HYPONATREMIA/GINE`S, CA´RDENAS above). Patients with ascites who receive large amounts of intravenous hypotonic fluids (i.e., 5% dextrose) may develop worsening DH as the renal capacity to excrete a water load is limited in most patients with advanced cirrhosis. Therefore, care should be taken when administering hypotonic fluids to patients with cirrhosis and ascites to prevent the development of DH. hyponatremia, although probably impractical due to the need for daily intravenous administration, could be further investigated in a subset of patients that would benefit from a short-term therapy (i.e., patients with very advanced liver disease awaiting liver transplantation). PHARMACOLOGICAL THERAPY Pharmacological approaches to the management of DH in cirrhosis have focused on inhibiting the actions of AVP. Although some drugs used for DH in cirrhosis, such as demeclocycline, urea, and kappa-opioids, have been reported to be effective, their use has been abandoned due to important side effects.116–119 The aim of therapy in DH is to increase solute-free water excretion, which has been done by antagonizing the action of AVP by means of blocking the V2 receptor for AVP with specific antagonists. These drugs have been evaluated in the treatment of DH in hypervolemic states such as congestive heart failure, the syndrome of inappropriate antidiuretic hormone secretion (SIADH), and cirrhosis. Several nonpeptide V2 receptor antagonists including mozavaptan (OPC-31260), lixivaptan (VPA-985), satavaptan (SR-121463), tolvaptan (OPC-41061), and RWJ-351647 have been tested in patients with cirrhosis and ascites.88–92,120–122 Published clinical studies using the V2 receptor antagonist satavaptan in patients with cirrhosis and hyponatremia are included in Table 3. Management The most commonly accepted method for the management of DH in cirrhosis and other water-retaining states is fluid restriction of 1 to 1.5 L/day 23. However, fluid restriction in patients with cirrhosis and DH has very limited efficacy in improving serum sodium levels.88,89 The routine administration of hypertonic saline solution is not recommended because additional expansion of the extracellular fluid worsens edema and ascites and its effect in increasing serum sodium is modest. Plasma expansion with albumin seems to be beneficial in transiently correcting DH in patients with cirrhosis and ascites. In a preliminary report, 24 patients with refractory ascites and DH (serum sodium 130 mEq/L) were randomized to fluid restriction (1.5 L/day) versus intravenous albumin (40 gm/day) for 7 days. In patients who received albumin, serum sodium levels improved in 8/12 patients. In addition, there was a reduced incidence of infections, hepatic encephalopathy, and in-hospital mortality in the albumin group.115 Although these results are encouraging, the study period was only 1 week and the effects therefore short-lived. The use of albumin for V2 RECEPTOR ANTAGONISTS IN CIRRHOSIS AND DH Two multicenter, randomized, placebo-controlled trials evaluated the use of lixivaptan in cirrhotic patients with Table 3 Published Clinical Studies Using V2 Receptor Antagonists in Patients with Cirrhosis and Ascites and Hyponatremia Author (ref) Compound Dose Phase Patients Comments Wong et al (2003)88 Lixivaptan* 50–500 mg/day p.o. II 44y with Increased urine output, CH2O, for 7 days hyponatremia S osm, serum Na. Dehydration with doses of 500 mg. Drop-out rate was 27% Gerbes et al (2003)89 Lixivaptan* 100–200 mg/day p.o. II for 7 days 60 with hyponatremia Increased serum Na, decreased U osm and body weight. Thirst appeared in patients at the 200-mg dose. Gine`s et al (2006)91 Satavaptan þ 5 mg, 12.5 mg, and II 25 mg daily for 110 with hyponatremia Concomitant spironolactone 100 mg/day. Serum Na increased to 135 mEq/L or > 5 mEq/L in 2 weeks 54–64% of cases, depending on dose. Gine`s et al (2007)92 Satavaptan þ 5 mg, 12.5 mg, and 25 mg daily for 1 year II 73 with hyponatremia Concomitant spironolactone 100 mg/day. Median serum Na increased from 131 mEq/L up to 136 mEq/L by week 40. *Randomized, double-blind, placebo-controlled trial. y Included 5 patients with cardiac disease and 5 with SIADH. p.o., by mouth; U osm, urinary osmolality; S osm, serum osmolality; CH2O, solute-free water clearance; Serum Na, serum sodium; U vol, urine volume. 53 54 SEMINARS IN LIVER DISEASE/VOLUME 28, NUMBER 1 2008 DH.88,89 In these two studies this agent caused a doserelated increase in serum sodium levels and free water clearance. Unfortunately, the drug caused significant dehydration in some patients at the effective doses in one study,88 and in the other study, which evaluated two doses of the drug versus placebo, a small number of patients in each group was included (20 per arm) and the effects of lixivaptan were only evaluated until serum sodium normalized without information on long-term response.89 Recent data from a large multicenter randomized controlled study evaluating the use of tolvaptan in patients with euvolemic or hypervolemic hyponatremia (SIADH, congestive heart failure, and cirrhosis) indicate that this agent is effective in raising serum sodium concentration when used for 30 days.90 In this study, a subgroup of 63 patients with cirrhosis and DH were assigned tolvaptan and 57 placebo starting at 15 mg in ascending doses according to response.122 There were no significant side effects compared with placebo. Tolvaptan rapidly improved serum sodium and fluid balance and caused significant weight loss in patients with cirrhosis and hyponatremia compared with placebo, and markedly increased solute-free water clearance without adversely affecting renal function. In patients with cirrhosis and DH, normalization of sodium levels (> 135 mEq/L) occurred in 30% and 22% of subjects at days 4 and 30, respectively.122 This agent therefore seems to be safe, effective, and likely to be promising in the treatment of DH. Finally, data from a multicenter study indicates that the short- and long-term use of satavaptan in patients with cirrhosis and DH improves serum sodium levels.91,92 The short-term use (14 days) of satavaptan at doses of 5 mg, 12.5 mg, 25 mg once daily in patients with cirrhosis and hyponatremia receiving 100 mg of spironolactone was effective in correcting serum sodium levels. After 5 days, patients with a dose of 5 mg, 12.5 mg, and 25 mg increased serum sodium level to 135 mEq/L or > 5 mEq/L in 61%, 54%, and 64% of cases, respectively. Seventy-three patients where followed for a year. Forty-seven were randomized to satavaptan and 26 to placebo.92 Satavaptan was used at 5 mg/day and titrated up as needed to 50 mg/day. The majority of patients in the satavaptan group were maintained on 5 mg or 12.5 mg daily. Median serum sodium concentration at baseline in patients treated with satavaptan was 131 mEq/L and increased up to136 mEq/L by week 40. By contrast, in the placebo group serum sodium values did not change. In a subset of patients, serum sodium levels fell back to their baseline after discontinuation of satavaptan. There were no significant differences between the two groups with respect to the incidence of serious adverse events. These results demonstrate that the improvement of hyponatremia by satavaptan in patients with cirrhosis is maintained with long-term treatment and that discontinuation of treatment leads to recurrence of hyponatremia.92 The results of these studies demonstrate that V2 receptor antagonists are beneficial in correcting DH in advanced cirrhosis. The clinical utility of these agents in cirrhosis has been assessed in short-term studies and one long-term study; however, more controlled studies are needed to evaluate the safety, efficacy, and applicability of these agents in patients with cirrhosis and ascites. ACKNOWLEDGMENTS Grant support: Some of the studies reported in this manuscript and performed in the authors’ Unit have been supported by grants from the Fondo de Investigacio´n Sanitaria (FIS 05/0246), Ministerio de Educacio´n y Ciencia (SAF 2005/01917), and Fundacio´ Marato´ TV3 (U-2000-TV2710). ABBREVIATIONS AQP aquaporins AVP arginine vasopressin cAMP cyclic AMP DH dilutional hyponatremia HRS hepatorenal syndrome MELD model for end-stage liver disease MRI magnetic resonance imaging SBP spontaneous bacterial peritonitis SIADH syndrome of inappropriate antidiuretic hormone secretion TIPS transjugular intrahepatic portosystemic shunt REFERENCES 1. Minino AM, Heron MP, Smith BL. Deaths: preliminary data for 2004. Natl Vital Stat Rep 2006;54:1–49 2. Gine`s P, Quintero E, Arroyo V, et al. Compensated cirrhosis: natural history and prognostic factors. Hepatology 1987;7:122–128 3. Planas R, Montoliu S, Balleste´ B, et al. Natural history of patients hospitalized for management of cirrhotic ascites. Clin Gastroenterol Hepatol 2006;4:1385–1394 4. Gine`s A, Escorsell A, Gine`s P, et al. Incidence, predictive factors, and prognosis of hepatorenal syndrome in cirrhosis. Gastroenterology 1993;105:229–236 5. Angeli P, Wong F, Watson H, Gine`s PCAPPS Investigators. Hyponatremia in cirrhosis: results of a patient population survey. Hepatology 2006;44:1535–1542 6. Gine`s P. Vaptans: a promising therapy in the management of advanced cirrhosis. J Hepatol 2007;46:1150–1152 7. Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rode´s J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology 1988;8:1151–1157 8. Iwakiri Y, Groszmann R. The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule. Hepatology 2006;43:S121–S131 MANAGEMENT OF ASCITES AND HYPONATREMIA/GINE`S, CA´RDENAS 9. Iwakiri Y, Groszmann R. Vascular endothelial dysfunction in cirrhosis. J Hepatol 2007;46:927–934 10. Wiest R, Das S, Cadelina G, et al. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. J Clin Invest 1999; 104:1223–1233 11. Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology 2005;41:422–433 12. Riordan SM, Williams R. The intestinal flora and bacterial infection in cirrhosis. J Hepatol 2006;45:744–757 13. Ruiz-del-Arbol L, Urman J, Ferna´ndez J, et al. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology 2003;38:1210–1218 14. Ruiz-del-Arbol L, Monescillo A, Arocena C, et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology 2005;42:439–447 15. Lee SS, Liu H. Cardiovascular determinants of survival in cirrhosis. Gut 2007;56:746–748 16. Sherlock S, Dooley J. Ascites. In: Sherlock S, Dooley J, eds. Diseases of the Liver and Biliary System. 11th ed. Oxford, England: Blackwell Science; 2002:127–146 17. Gine`s P, Ca´rdenas A, Arroyo V, Rode´s J. Management of cirrhosis and ascites. N Engl J Med 2004;350:1646–1654 18. Ca´rdenas A, Arroyo V. Ascites. In: Al-Knawy B, Sniffman M, Wiesner R, eds. Hepatology: A Practical Approach. Philadelphia: Elsevier Science; 2005:347–360 19. Moore KP, Wong F, Gine`s P, et al. The management of ascites in cirrhosis: report on the consensus conference of the International Ascites Club. Hepatology 2003;38:258–266 20. Runyon BA. Management of adult patients with ascites due to cirrhosis. Hepatology 2004;39:841–856 21. Ghassemi S, Garcia-Tsao G. Prevention and treatment of infections in patients with cirrhosis. Best Pract Res Clin Gastroenterol 2007;21:77–93 22. Runyon BA, Antillon MR, Akriviadis EA, McHutchison JG. Bedside inoculation of blood culture bottles is superior to delayed inoculation in the detection of spontaneous bacterial peritonitis. J Clin Microbiol 1990;28:2811–2812 23. Gine`s P, Berl T, Bernardi M, et al. Hyponatremia in cirrhosis: from pathogenesis to treatment. Hepatology 1998;28:851–864 24. Henkel AS, Buchman AL. Nutritional support in patients with chronic liver disease. Nat Clin Pract Gastroenterol Hepatol. 2006;3:202–209 25. McCullough AJ. Nutrition and malnutrition in liver disease. In: Wolfe MM, Davis GL, Farraye F, Giannella RA, Malagelada JR, Steer ML, eds. Therapy of Digestive Disorders. 2nd ed. Philadelphia: Saunders; 2006:67–83 26. Kearns PJ, Young H, Garcia G, et al. Accelerated improvement of alcoholic liver disease with enteral nutrition. Gastroenterology 1992;102:200–205 27. Cabre` E, Gonzalez-Hux F, Abad-Lacruz A, et al. Effect of total enteral nutrition on the short-term outcome of severely malnourished cirrhotics: a randomized controlled trial. Gastroenterology 1990;98:715–720 28. Perez-Ayuso RM, Arroyo V, Planas R, et al. Randomized comparative study of efficacy of furosemide versus spironolactone in nonazotemic cirrhosis with ascites: relationship between the diuretic response and the activity of the reninaldosterone system. Gastroenterology 1983;84:961–968 29. Santos J, Planas R, Pardo A, et al. Spironolactone alone or in combination with furosemide in the treatment of 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. moderate ascites in nonazotemic cirrhosis: a randomized comparative study of efficacy and safety. J Hepatol 2003;39: 187–192 Li CP, Lee FY, Hwang SJ, et al. Treatment of mastalgia with tamoxifen in male patients with liver cirrhosis: a randomized crossover study. Am J Gastroenterol 2000;95: 1051–1055 Lee FY, Lee SD, Tsai YT, et al. A randomized controlled trial of quinidine in the treatment of cirrhotic patients with muscle cramps. J Hepatol 1991;12:236–240 Angeli P, Albino G, Carraro P, et al. Cirrhosis and muscle cramps: evidence of a causal relationship. Hepatology 1996; 23:264–273 Gine`s P, Arroyo V, Quintero E, et al. Comparison of paracentesis and diuretics in the treatment of cirrhotics with tense ascites: results of a randomized study. Gastroenterology 1987;93:234–241 Salerno F, Badalamenti S, Incerti P, et al. Repeated paracentesis and i.v. albumin infusion to treat ‘‘tense’’ ascites in cirrhotic patients. A safe alternative therapy. J Hepatol 1987;5:102–108 Moore KP, Aithal GP. Guidelines on the management of ascites in cirrhosis. Gut 2006;55(suppl 6):vi1–vi12 Gine`s P, Tito L, Arroyo V, et al. Randomized comparative study of therapeutic paracentesis with and without intravenous albumin in cirrhosis. Gastroenterology 1988;94: 1493–1502 Ruiz-del-Arbol L, Monescillo A, Jime´nez W, et al. Paracentesis-induced circulatory dysfunction: mechanism and effect on hepatic hemodynamics in cirrhosis. Gastroenterology 1997;113:579–586 Gine`s A, Ferna´ndez-Esparrach G, Monescillo A, et al. Randomized trial comparing albumin, dextran 70, and polygeline in cirrhotic patients with ascites treated by paracentesis. Gastroenterology 1996;111:1002–1010 Moreau R, Valla DC, Durand-Zaleski I, et al. Comparison of outcome in patients with cirrhosis and ascites following treatment with albumin or a synthetic colloid: a randomised controlled pilot trial. Liver Int 2006;26:46–54 Sola-Vera J, Minana J, Ricart E, et al. Randomized trial comparing albumin and saline in the prevention of paracentesis-induced circulatory dysfunction in cirrhotic patients with ascites. Hepatology 2003;37:1147–1153 Ferna´ndez-Esparrach G, Guevara M, Sort P, et al. Diuretic requirements after therapeutic paracentesis in non-azotemic patients with cirrhosis: a randomized double-blind trial of spironolactone versus placebo. J Hepatol 1997;26:614–620 Thomsen TW, Shaffer RW, White B, Setnik GS. Videos in clinical medicine: paracentesis. N Engl J Med 2006;355:e21 Pache I, Bilodeau M. Severe haemorrhage following abdominal paracentesis for ascites in patients with liver disease. Aliment Pharmacol Ther 2005;21:525–529 Caldwell SH, Hoffman M, Lisman T, et al. Coagulation disorders and hemostasis in liver disease: pathophysiology and critical assessment of current management. Hepatology 2006;44:1039–1046 Ca´rdenas A, Guevara M, Gine`s P. Paracentesis. In: Weinstein W, Hawkey CJ, Bosch J, eds. Clinical Gastroenterology and Hepatology. Philadelphia: Elsevier Science; 2005:1097–1100 Sakai H, Sheer TA, Mendler MH, Runyon BA. Choosing the location for non-image guided abdominal paracentesis. Liver Int 2005;25:984–986 55 56 SEMINARS IN LIVER DISEASE/VOLUME 28, NUMBER 1 2008 47. Arroyo V, Gine`s P, Gerbes A, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. Hepatology 1996;23:164–176 48. Guardiola J, Baliellas C, Xiol X, et al. External validation of a prognostic model for predicting survival of cirrhotic patients with refractory ascites. Am J Gastroenterol 2002;97: 2374–2378 49. Gine`s P, Uriz J, Calahorra B, et al. Transjugular intrahepatic portosystemic shunting versus paracentesis plus albumin for refractory ascites in cirrhosis. Gastroenterology 2002;123:1839–1847 50. Boyer TD, Haskal Z. The role of transjugular intrahepatic portosystemic shunt in the management of portal hypertension. Hepatology 2005;41:386–400 51. Bureau C, Garcı´a-Pagan JC, Otal P, et al. Improved clinical outcome using polytetrafluoroethylene-coated stents for TIPS: results of a randomized study. Gastroenterology 2004;126:469–475 52. Lebrec D, Giuily N, Hadengue A, et al. Transjugular intrahepatic portosystemic shunts—comparison with paracentesis in patients with cirrhosis and refractory ascites: a randomized trial. J Hepatol 1996;25:135–144 53. Rossle M, Ochs A, Gulberg V, et al. A comparison of paracentesis and transjugular intrahepatic portosystemic shunting in patients with ascites. N Engl J Med 2000; 342:1701–1707 54. Sanyal AJ, Genning C, Reddy RK, et al. The North American Study for Treatment of Refractory Ascites. Gastroenterology 2003;124:634–641 55. Salerno F, Merli M, Riggio O, et al. Randomized controlled study of TIPS versus paracentesis plus albumin in cirrhosis with severe ascites. Hepatology 2004;40:629–635 56. Allard JP, Chau J, Sandokji K, Blendis LM, Wong F. Effects of ascites resolution after successful TIPS on nutrition in cirrhotic patients with refractory ascites. Am J Gastroenterol 2001;96:2442–2447 57. Deltenre P, Mathurin P, Dharancy S, et al. Transjugular intrahepatic portosystemic shunt in refractory ascites: a meta-analysis. Liver Int 2005;25:349–356 58. Albillos A, Banares R, Gonzales M, et al. A meta-analysis of transjugular intrahepatic portosystemic shunt versus paracentesis for refractory ascites. J Hepatol 2005;43:990–996 59. D’Amico G, Luca A, Morabito A, et al. Uncovered transjugular intrahepatic portosystemic shunt for refractory ascites: a meta-analysis. Gastroenterology 2005;129:1282–1293 60. Salerno F, Camma C, Enea M, et al. Transjugular intrahepatic portosystemic shunt for refractory ascites: a meta-analysis of individual patient data. Gastroenterology 2007;133:825–834 61. Ca´rdenas A, Gine`s P. Management of refractory ascites. Clin Gastroenterol Hepatol 2005;3:1187–1191 62. Llach J, Gine`s P, Arroyo V, et al. Prognostic value of arterial pressure, endogenous vasoactive systems, and renal function in cirrhotic patients admitted to the hospital for the treatment of ascites. Gastroenterology 1988;94:482–487 63. Guevara M, Ca´rdenas A, Uriz J, Gine`s P. Prognosis of patients with ascites and cirrhosis. In: Gine`s P, Arroyo V, Rode´s J, Schrier R, eds. Ascites and Renal Dysfunction in Liver Disease. 2nd ed. Oxford: Blackwell Publishing; 2005:260–270 64. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology 2001;33:464–470 65. Heuman DM, Abou-Assi SG, Habib A, et al. Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death. Hepatology 2004;40:802–810 66. Biggins SW, Rodriguez HJ, Bacchetti P, Bass NM, Robert JP, Terrault NA. Serum sodium predicts mortality in patients listed for liver transplantation. Hepatology 2005; 41:32–39 67. Ruf AE, Kremers WK, Chavez LL, Descalzi VI, Podesta LG, Villamil FG. Addition of serum sodium into the MELD score predicts waiting list mortality better than MELD alone. Liver Transpl 2005;11:336–343 68. Biggins SW, Kim WR, Terrault NA, et al. Evidence-based incorporation of serum sodium concentration into MELD. Gastroenterology 2006;130:1652–1660 69. Luca A, Angermayr B, Bertolini G, et al. An integrated MELD model including serum sodium and age improves the prediction of early mortality in patients with cirrhosis. Liver Transpl 2007;13:1174–1180 70. Mathur S, Gane EJ, McCall JL, Plank LD. Serum sodium and hydration status predict transplant-free survival independent of MELD score in patients with cirrhosis. J Gastroenterol Hepatol 2007; April 19 (Epub ahead of print) 71. London˜o MC, Cardenas A, Guevara M, et al. MELD score and serum sodium in the prediction of survival of patients with cirrhosis awaiting liver transplantation. Gut 2007;56: 1283–1290 72. Dawwas MF, Lewsey JD, Neuberger JM, et al. The impact of serum sodium concentration on mortality after liver transplantation: a cohort multicenter study. Liver Transpl 2007;13:1115–1124 73. London˜o MC, Guevara M, Rimola A, et al. Hyponatremia impairs early posttransplantation outcome in patients with cirrhosis undergoing liver transplantation. Gastroenterology 2006;130:1135–1143 74. Ferna´ndez-Varo G, Ros J, Cejudo-Martin P, et al. Effect of the V1a/V2-AVP receptor antagonist, conivaptan, on renal water metabolism and systemic hemodynamics in rats with cirrhosis and ascites. J Hepatol 2003;38:755–761 75. Decaux G. Difference in solute excretion during correction of hyponatremic patients with cirrhosis or syndrome of inappropriate secretion of antidiuretic hormone by oral vasopressin V2 receptor antagonist VPA-985. J Lab Clin Med 2001;138:18–21 76. Gine`s P, Wong F, Watson HR, et al. Effects of a selective vasopressin V2 receptor antagonist, satavaptan (SR121463B), in patients with cirrhosis and ascites without hyponatremia. Hepatology 2006;44:691A 77. Wong F, Gine`s P, Watson HR, et al. Effects of a selective vasopressin V2 receptor antagonist, satavaptan (SR121463B), on recurrence of ascites after large volume paracentesis. Hepatology 2006;44:180A 78. Angeli P, Volpin R, Gerunda G, et al. Reversal of type 1 hepatorenal syndrome with the administration of midodrine and octreotide. Hepatology 1999;29:1690–1697 79. Ortega R, Gine`s P, Uriz J, et al. Terlipressin therapy with and without albumin for patients with hepatorenal syndrome: results of a prospective, non-randomized study. Hepatology 2002;36:941–948 80. Kalambokis G, Fotopoulos A, Economou M, et al. Effects of a 7-day treatment with midodrine in non-azotemic cirrhotic patients with and without ascites. J Hepatol 2007; 46:213–221 MANAGEMENT OF ASCITES AND HYPONATREMIA/GINE`S, CA´RDENAS 81. Lenaerts A, Codden T, Meunier JC, et al. Effects of clonidine on diuretic response in ascitic patients with cirrhosis and activation of sympathetic nervous system. Hepatology 2006;44:844–849 82. Schrier RW. Water and sodium retention in edematous disorders: role of vasopressin and aldosterone. Am J Med 2006;119(suppl 1):S47–S53 83. Ishikawa SE, Schrier RW. Pathogenesis of hyponatremia: the role of arginine vasopressin in cirrhosis. In: Gine`s P, Arroyo V, Rode´s J, Schrier RW, eds. Ascites and Renal Dysfunction in Liver Disease: Pathogenesis, Diagnosis and Treatment. 2nd ed. Oxford: Blackwell Science; 2005:305–314 84. Thibonnier M, Conarty DM, Preston JA, Wilkins PL, Berti-Mattera LN, Mattera R. Molecular pharmacology of human vasopressin receptors. Adv Exp Med Biol 1998; 449:251–276 85. Kwon TH, Hager H, Nejsum LN, Andersen ML, Frokiaer J, Nielsen S. Physiology and pathophysiology of renal aquaporins. Semin Nephrol 2001;21:231–238 86. Nielsen S, Frokiaer J, Marples D, Kwon TH, Agre P, Knepper MA. Aquaporins in the kidney: from molecules to medicine. Physiol Rev 2002;82:205–244 87. Esteva-Font C, Baccaro ME, Ferna´ndez-Llama P, et al. Aquaporin-1 and aquaporin-2 urinary excretion in cirrhosis: relationship with ascites and hepatorenal syndrome. Hepatology 2006;44:1555–1556 88. Wong F, Blei AT, Blendis LM, Thuluvath PJ. A vasopressin receptor antagonist (VPA-985) improves serum sodium concentration in patients with hyponatremia: a multicenter, randomized, placebo-controlled trial. Hepatology 2003;37:182–191 89. Gerbes AL, Gu¨lberg V, Gine`s P, et al. The VPA Study Group. Therapy of hyponatremia in cirrhosis with a vasopressin receptor antagonist: a randomized double-blind multicenter trial. Gastroenterology 2003;124:933–939 90. Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med 2006;355:2099–2112 91. Gine`s P, Wong F, Milutinovic D, et al. Effects of satavaptan (SR121463B), a selective vasopressin V2 receptor antagonist on serum sodium concentration and ascites in patients with cirrhosis hyponatremia. J Hepatol 2006;44:732A 92. Gine`s P, Wong F, Watson H. Long-term improvement of serum sodium by the V2-receptor antagonist satavaptan in patients with cirrhosis and hyponatremia. J Hepatol 2007; 46:90A 93. Fraser CL, Arieff AI. Epidemiology, pathophysiology, and management of hyponatremic encephalopathy. Am J Med 1997;102:67–77 94. Berl T, Verbalis J. Pathophysiology of water metabolism. In: Brenner & Rector’s The Kidney. 7th ed. Philadelphia: Elsevier; 2004:857–920 95. Verbalis JG, Gullans SR. Hyponatremia causes large sustained reductions in brain content of multiple organic osmolytes in rats. Brain Res 1991;567:274–282 96. Verbalis JG. The syndrome of inappropriate secretion of antidiuretic hormone secretion and other hyposmolar disorders. In: Schrier R, ed. Diseases of the Kidney and Urinary Tract. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:2214 97. Halperin ML, Kamel KS. A new look at an old problem: therapy of chronic hyponatremia. Nat Clin Pract Nephrol. 2007;3:2–3 98. Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994;107:1475–1480 99. Ha¨ussinger D, Kircheis G, Fischer R, Schliess F, vom Dahl S. Hepatic encephalopathy in chronic liver disease: a clinical manifestation of astrocyte swelling and low-grade cerebral edema. J Hepatol 2000;32:1035–1038 100. Haussinger D. Low-grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology 2006;43:1187–1190 101. Laubenberger J, Haussinger D, Bayer S, et al. Proton magnetic resonance spectroscopy of the brain in symptomatic and asymptomatic patients with liver cirrhosis. Gastroenterology 1997;112:1610–1616 102. Minguez B, Garcia-Pagan JC, Bosch J, et al. Noncirrhotic portal vein thrombosis exhibits neuropsychological and MR changes consistent with minimal hepatic encephalopathy. Hepatology 2006;43:707–714 103. Seery JP, Taylor-Robinson SD. The application of magnetic resonance spectroscopy to the study of hepatic encephalopathy. J Hepatol 1996;25:988–998 104. Butterworth RF. Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab Brain Dis 2002;17: 221–227 105. Butterworth RF. Pathogenesis of hepatic encephalopathy: new insights from neuroimaging and molecular studies. J Hepatol 2003;39:278–285 106. Restuccia T, Go´mez-Anso´n B, Guevara M, et al. Effects of dilutional hyponatremia on brain organic osmolytes and water content in patients with cirrhosis. Hepatology 2004; 39:1613–1622 107. Co´rdoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after porto-caval anastomosis. J Hepatol 1998;29:589–594 108. Shawcross DL, Balata S, Olde Damink SW, et al. Low myo-inositol and high glutamine levels in brain are associated with neuropsychological deterioration after induced hyperammonemia. Am J Physiol Gastrointest Liver Physiol 2004;287:G503–G509 109. Murphy N, Auzinger G, Bernel W, Wendon J. The effect of hypertonic sodium chloride in intracranial pressure in patients with acute liver failure. Hepatology 2004;39:464– 470 110. Baccaro ME, Guevara M, Torre A, et al. Hyponatremia predisposes to hepatic encephalopathy in patients with cirrhosis: results of a prospective study with time-dependent analysis. Hepatology 2006;44:118A 111. Guevara M, Uriz J, Arcos E, et al. Hyponatremia is a risk factor of hepatic encephalopathy in cirrhosis: a post hoc time-dependent analysis of the international study group on refractory ascites trial. Hepatology 2006;44:677A 112. Gine`s P, Smajda Lew E, Diamand F. Hyponatremia is a major determinant of impaired health-related quality of life in cirrhosis with ascites. Hepatology 2007;46:744A 113. Abbasoglu O, Goldstein RM, Vodapally MS, et al. Liver transplantation in hyponatremic patients with emphasis on central pontine myelinolysis. Clin Transplant 1998;12:263– 269 114. Wszolek ZK, McComb RD, Pfeiffer RF, et al. Pontine and extrapontine myelinolysis following liver transplantation: relationship to serum sodium. Transplantation 1989;48: 1006–1012 57 58 SEMINARS IN LIVER DISEASE/VOLUME 28, NUMBER 1 2008 115. Jalan R, Mookerjee R, Cheshire L, et al. Albumin infusion for severe hyponatremia in patients with refractory ascites: a randomized clinical trial. J Hepatol 2007;46:232A 116. De Troyer A, Pilloy W, Broeckaert I, Demanet JC. Demeclocycline treatment of water retention in cirrhosis. Ann Intern Med 1976;85:336–337 117. Carrilho F, Bosch J, Arroyo V, et al. Renal failure associated with demeclocycline in cirrhosis. Ann Intern Med 1977;87: 195–197 118. Decaux G, Mols P, Cauchie P, et al. Treatment of hyponatremic cirrhosis with ascites resistant to urea. Nephron 1986;44:337–343 119. Gadano A, Moreau R, Pessione F, et al. Aquaretic effects of niravoline, a kappa-opioid agonist, in patients with cirrhosis. J Hepatol 2000;32:38–42 120. Inoue T, Ohnishi A, Matsuo A, et al. Therapeutic and diagnostic potential of a vasopressin-2 antagonist for impaired water handling in cirrhosis. Clin Pharmacol Ther 1998;63:561–570 121. Thuluvath PJ, Maheshwari A, Wong F, et al. Oral V2 receptor antagonist (RWJ-351647) in patients with cirrhosis and ascites: a randomized, double-blind, placebo-controlled, single ascending dose study. Aliment Pharmacol Ther 2006;24:973–982 122. Afdhal N, Cardenas A, Gine`s P, et al. Randomized, placebo-controlled trial of tolvaptan, a novel V2-receptor antagonist, in hyponatremia: results of the SALT 2 trial with emphasis on efficacy and safety in cirrhosis. Hepatology 2005;42:LB19A


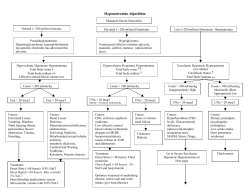









© Copyright 2026