
CO2 Capture by Aqueous Absorption Summary of 4th Quarterly
CO2 Capture by Aqueous Absorption Summary of 4th Quarterly Progress Reports 2008 Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption by Gary T. Rochelle Department of Chemical Engineering The University of Texas at Austin February 3, 2009 Introduction This research program is focused on the technical obstacles to the deployment of CO2 capture and sequestration from flue gas by alkanolamine absorption/stripping and on integrating the design of the capture process with the aquifer storage/enhanced oil recovery process. The objective is to develop and demonstrate evolutionary improvements to monoethanolamine (MEA) absorption/stripping for CO2 capture from coal-fired flue gas. The Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption support 14 graduate students. These students have prepared detailed quarterly progress reports for the period October 1, 2008 to December 31, 2008. Conclusions The addition of Inhibitor A to 8 m PZ with 1 mM Fe++ eliminated foaming when this solution was oxidized for 160 hrs at low gas flow. There also appeared to be no measurable change in PZ concentration over this period. A pilot plant campaign with 8 m and 5 m PZ including 100 mM inhibitor encountered no serious problems with solids precipitation or foaming. No significant degradation was observed after about 2 weeks of operation. In the pilot campaign with 8 m PZ the optimum solvent rate appears to be lower than we had predicted or operated. The minimum L/G was 18 gpm/350 cfm. The observed energy use to get 80% CO2 removal was 22 kJ equivalent work to get 1 gmol of CO2 at 60 psia. The equivalent work was practically the same with a simple stripper at 20 or 50 psia. Even though the pseudo-first order theory for the liquid film mass transfer coefficient, kg’ contains both temperature and concentration dependent terms, the measured kg’ values are generally not affected by T from 40 to 100 °C or amine concentration from 3 to 13 m in MEA, PZ, or MEA/PZ solvents. Some deviation from this generalization is evident in rich PZ at 80 and 100°C. Addition of formaldehyde to loaded solutions of PZ causes foaming and results in the formation of piperazine oligimers. However, these oligimers were not detected by NMR in recently oxidized PZ solutions that foam. 1 Structured packing P500 exhibited slightly higher area (~10%) than M500Y. On a fractional area basis, M2Y yielded the same results as M250Y, despite being a coarser packing. The mass transfer area database (updated to include P500 and M2Y) was still represented well by the correlation that was regressed as a function of (WeL)(FrL)-1/3. The contribution of pre-wetting to mass transfer performance was found to become negligible after a period of about 20 minutes. Oxidation of 7 m MDEA/2 m PZ with 1 mM Fe++ with or without Inhibitor A produces about 0.01 mM formate/hr, more than an order of magnitude less formate than oxidation of 7 m MEA. However with 1 mM Fe+ and 100 mM Inhibitor A, oxidation produces a foaminess factor greater than we can measure. The predictions of the Hilliard model for PZ/CO2 heat capacity at stripper conditions are not reliable. This thermodynamic model will be fixed as new data become available. 70% recovery of PZ was achieved in a preliminary experiment of batch reclaiming by evaporation at 1 atm. The partial heat capacity of total CO2 in concentrated loaded PZ appears to be practically zero, as we have observed in measurements with MEA and in PZ at lower amine concentration. Dynamic modeling of a stripper integrated with a CO2 compressor with proportional step change in solvent rate and reboiler duty shows that the reboiler pressure and temperature approach the new steady state in 10 and 30 min. However, the lean loading remains almost constant. A 10% reduction in stripper load resulted in 4% decrease in stripper pressure with constant speed compressor. 1. Oxidative Degradation of Amines by Andrew Sexton A Ph.D. dissertation was completed this quarter. It is available as a separate document on our website. Here is the abstract. Aqueous amine solutions were batch loaded into 500 mL glass jacketed reactors and subjected to oxidative degradation at both low and high gas rates. Solutions at low gas were degraded with 100 mL/min of 98%O2/2%CO2 with mass transfer achieved by vortexing. Samples were drawn from the reactor during the course of the experiment and analyzed for degradation using ion chromatography and HPLC with evaporative light scattering detecion. In a parallel apparatus 7.5 L/min of 15%O2/2%CO2 was sparged through 350 mL of solution; additional mass transfer was achieved by vortexing. A Fourier Transform Infrared Analyzer collected continuous gas-phase data on amine volatility and volatile degradation products. Hydroxyethyl-formamide (HEF), hydroxyethylimidazole (HEI) and formate are the major carbon containing monoethanolamine (MEA) oxidation products; HEF, HEI and ammonia are the major nitrogen containing products. In terms of catalyst oxidation potential, Cu > Cr/Ni (combined) > Fe > V. The oxygen stoichiometry (ν) ranges from 1.5 mol MEA degraded/mol O2 consumed for Cu and Fe catalyzed systems to 1.0 for V catalyzed systems. Estimation of rates from an industrial absorber show degradation costs to range from $1.17/metric ton (MT) CO2 captured for a system controlled by the solubility of O2 to $2.22/MT CO2 for a mass transfer controlled system. 2 Inhibitors A and B (reaction mechanism inhibitors) and EDTA (a chelating agent) were established as effective MEA oxidation inhibitors. EDTA and Inhibitor A were successful inhibitors at 100 mM, while 7.5 mM Inhibitor B successfully inhibited degradation. Sodium sulfite and reaction intermediates formaldehyde and formate (expected oxygen scavengers) were unsuccessful at inhibiting MEA oxidation. Cu catalyzes concentrated PZ oxidation, while Fe has no effect on PZ oxidation even at high catalyst concentration. MEA/PZ blends were more susceptible to oxidation than any other amine system investigated. It is believed that free radicals formed in the MEA oxidation process serve to accelerate the degradation of the PZ structure. All MEA analogs (glycine, ethylenediamine and ethylene glycol) and secondary/hindered amines (diethanolamine, diglycolamine and 2amino-2-methyl-1-propanol) were resistant to oxidation in the presence of Fe or Cu, except for diethanolamine. 2. Solvent Management of Concentrated Piperazine p. 9 by Stephanie Freeman Oxidative degradation of concentrated PZ was studied this quarter. Six conditions were studied in the presence of 1.0 mM of iron. PZ showed little production of degradation products in studies with just iron, iron with 100 mM Inhibitor A, and iron with 30 mM Inhibitor B. In the case of iron and 30 mM EDTA, the most EDA and formate was produced as compared with the other oxidative inhibitors. All degradation observed was lower than that of MEA in similar conditions although the reduction of PZ observed in all cases was not wholly matched to the amount of degradation products detected. A pilot plant campaign was performed this quarter on 8 m PZ at Pickle Research Campus at the University of Texas. The goal of the campaign was to tackle any operational difficulties with using concentrated PZ and to demonstrate the improved mass transfer performance of the solvent. Fourteen runs were performed with sampling at five points throughout the system, with a few samples missing due to empty sample bombs. The final five runs were performed at 5 m PZ. Initial analysis shows enhanced mass transfer performance of the 8 m PZ over similar 7 m MEA systems. The equivalent work was minimized in systems with higher liquid flow rates and higher lean loadings. The optimum liquid flow rate was not found during the course of the campaign and appears to be at a value higher than 18 gallons per minute. Further analysis is ongoing, including a comparison of these pilot plant results to that of an MEA system. 3. Rate Measurements for MEA and PZ p. 34 by Ross E. Dugas The remaining measurements of CO2 partial pressure and CO2 absorption/desorption rate at 80 and 100˚C for monoethanolamine (MEA) and piperazine (PZ) solutions were completed. 7 m MEA/2 m PZ solutions were also tested at 40, 60, 80 and 100˚C with four CO2 loadings. The measured values of CO2 partial pressure match very well with available literature sources. PZ absorbs CO2 2–3 times faster than MEA. 7 m MEA/2 m PZ solution absorbs CO2 faster than MEA and slightly slower than PZ. Even though the pseudo-first order theory for the liquid film mass transfer coefficient, kg’, contains both temperature and concentration dependent terms, the measured kg’ values are generally not affected by changes in temperature or amine concentration. Varying temperature 3 and MEA concentration did not affect kg’ in the wetted wall column. Piperazine experiments in the wetted wall column show lower kg’ values at higher temperature and higher CO2 loading. These conditions require greater CO2 fluxes in the wetted wall column experiments and may increase the significance of the liquid film physical mass transfer coefficient, klo. Under these conditions, diffusion limitations of reactants and products near the interface can restrict CO2 mass transfer giving reduced kg’ values. 4. Analysis of Degraded Piperazine Solutions that Result in Foaming p. 42 by Xi Chen NMR and mass spectroscopy (MS) analysis were conducted on PZ solutions with and without formaldehyde added and an oxidized PZ solution to identify contaminants that account for increased foaming tendency of degraded PZ solutions. Most peaks in the NMR spectrum were tentatively interpreted and correlated to different molecules. There were no additional peaks found in foaming solution that had been oxidized for two weeks. For another degraded PZ sample, NMR peaks with same position as in formaldehyde-added PZ were found, which may indicate that formaldehyde is the cause of increased foaming tendency. MS analysis seems not to render useful information on contaminants. 5. Influence of Liquid Properties on Effective Mass Transfer Area of Structured Packing p. 72 by Robert Tsai (also supported by the Separations Research Program} Two additional structured packings were evaluated: a prototype 500-series packing (P500) (ap = 500 m2/m3) and Sulzer Mellapak 2Y (M2Y) (ap = 205 m2/m3). P500 exhibited higher pressure drops (15%) than Mellapak 500Y (M500Y), a geometrically similar packing, under dry conditions and for liquid loads up to 10 gpm/ft2. At higher loads, the pressure drop behavior of the two packings became less distinct. In contrast, hold-ups for P500 were consistently lower for all investigated liquid loads (up to 20 gpm/ft2). Base case and low surface tension (σ ~ 30 dynes/cm) mass transfer tests with P500 yielded results comparable to those obtained with M500Y. The reduction in surface tension increased the effective mass transfer area, although the effect was not as pronounced as with M500Y. M2Y was compared to Mellapak 250Y (M250Y), which had a comparable albeit slightly higher geometric area (ap = 250 m2/m3). M2Y displayed lower pressure drops by about 20–25% under both dry and irrigated (up to 30 gpm/ft2) conditions. Hold-up values were comparable to M250Y. M2Y also appeared to have similar fractional areas as M250Y, despite being a coarser packing. The mass transfer area database was updated to incorporate P500 and M2Y. The current global (ae/ap) correlation, able to represent the entire database within limits of ±15%, is as follows: 6. Modeling Stripper Performance for CO2 Removal by David Van Wagener 4 p. 88 Previous simulations using the Hilliard (2008) piperazine thermodynamic model produced unexpected results in the optimization process for 8 m PZ, and further investigation revealed that the heat capacity predictions are wildly inaccurate at high temperatures. Heat capacity data for 8 m PZ was not yet available, so values were extrapolated from 2.0 m PZ and 3.6 m PZ data sets. Additionally, the relationship between heat capacity and temperature is strongly linear, so heat capacities at high temperatures were determined by extending the linear trend. Selected parameters in the Hilliard PZ model were regressed using two high concentration data sets extrapolated by different methods. The regressions did not improve the accuracy of heat capacity predictions by Aspen Plus®, and the VLE predictions weakened. The important parameters to be varied during regressions need to be determined in future work to generate an accurate model. In addition, high concentration heat capacity data is expected, so the questionable extrapolation can be eliminated in future model improvement. 7. CO2 Absorption Modeling Using Aqueous Amines p. 96 by Jorge M. Plaza The MEA model presented at GHGT-9 was used by AspenTech in a rate-based process modeling study of CO2 capture with MEA. Pilot plant data from the Master’s Thesis by Dugas were used to assess the advantages of rate-based modeling over the traditional equilibrium-stage models. Results show excellent match of rate-based model predictions against the comprehensive pilot plant data sets. During this period collaboration with AspenTech led to the submission of this work for publication in Industrial & Engineering Chemistry Research. (A copy is attached at the end of this report.) Work also focused on absorber intercooling and the critical L/G. Intercooling was evaluated as a process option in CO2 absorption by piperazine (PZ) promoted potassium carbonate. The system performance with 4.5 m K+/4.5 m PZ was simulated by a model in Aspen Plus® RateSepTM. The absorber was evaluated for use with a double matrix stripper by optimizing the position of the semilean feed and intercooling stages to maximize CO2 removal. Additionally, a simple absorber system was modeled to observe the effect of intercooling on systems with variable CO2 lean loading. Intercooling increases CO2 removal by as much as 10% with the double matrix configuration. With a simple absorber, the effectiveness of intercooling depends on solvent rate. Near a critical liquid/gas ratio (L/G) there is a large improvement with intercooling. This is related to the position of the temperature bulge. An approximation was proposed to estimate the critical L/G where intercooling may maximize removal. This work has also led to an article that is being finalized for publication. The final draft of this article is attached to this report. 8. Thermal Degradation by Jason Davis This quarter we purchased and installed a dionex ion chromatograph to operate upstream an existing mass spectrometer managed by our Civil Engineering Department. This system will allow us to identify by molecular weight the unknown peaks eluted in both cation and anion chromatography. No results were developed with this system in this quarter. 9. Reclaiming by Crystallization of Potassium Sulfate by Qing Xu 5 p. 121 In previous work sulfate was removed from CO2 capture solvents by crystallization (Solvent reclaiming by crystallization of potassium sulfate, GHGT-9 paper by Q. Xu and G. Rochelle, 2008; M.S. thesis by Q. Xu 2008). The thesis is now available on our website. But other impurities like formate cannot be removed through that process so a thermal reclaimer, probably with a smaller size, is still needed. In this period a thermal reclaiming apparatus under 1 atm was developed for 8 m PZ solution. The main impurities considered here include sulfate and formate. Apparatus was developed and modified through a series of experiments. The results show that a sharp separation of anion impurities was possible: formate levels were reduced by a factor of 200 and sulfate levels were reduced by a factor of 20. The experiments indicated that no CO2 was left in the liquid after 80 minutes. A recovery of 70.6% of PZ was accomplished in Run 3. One issue is the precipitation of PZ in the vapor line when its concentration became too high. The apparatus needs to be modified to solve this problem. 10. Experimental Heat Capacity of Concentrated Piperazine p. 138 by Bich-Thu Nguyen This report discusses the heat capacity, Cp, of concentrated piperazine solution (8–12 m PZ). Heat capacity is analyzed as a function of CO2 loading (nominal loading of 0.3 and 0.4 mol CO2/equivalent PZ) and temperature (40 ºC–120 ºC). The range of heat capacity values, expressed in J/g*K, at different concentrations and loadings are: 8 m (α = 0.21): 3.19–3.58; 8 m (α = 0.29): 3.11–3.56; 8 m (α = 0.4): 3.02–3.59; 10 m (α = 0.31): 2.89–3.33; 12 m (α = 0.29): 2.74–3.19. While Cp increases with temperature, it decreases with increasing loading and amine concentration. It is also determined that CO2 has a finite, though negligible, heat capacity. Finally, it must be noted that Cp measurements at high temperatures (90 ºC or greater) ought to be used with care as the magnitudes of these values also reflect CO2 and H2O vaporization in addition to the true heat of the solution. 11. Dynamic Operation of CO2 Capture p. 144 by Sepideh Ziaii The work this quarter focused on dynamic simulation of flexible CO2 capture in response to variation in electricity load. The rate-based dynamic model of the stripper, which was created in ACM® for 30 wt % MEA, was combined with the steady state performance model of a multistage compressor. Simple ratio-control strategy was implemented to control the rich solvent rate proportional to the reboiler heat rate. When decreasing the heat rate suddenly by 10%, the reboiler pressure and temperature change and approach the new steady state in 10 and 30 minutes respectively, while the lean loading remains almost constant. With a constant speed compressor, the stripper pressure varies in dynamic operation. In order to avoid the maximum discharge temperature with a constant speed compressor, the compressor should be designed based on minimum. 12. Electric Grid Level Implications of Flexible CO2 Capture Operation p. 152 by Stuart Cohen Cost analysis of coal-based power plants with flexible CO2 capture finds that the operating point with the lowest marginal cost is highly dependent on CO2 price. There is a $5/MWh incremental cost of flexibility that is primarily the result of pessimistically assuming that maintenance costs 6 for a flexible CO2 capture system are more than double the maintenance costs of inflexible systems. To investigate the long-term economics of flexible CO2 capture systems in the Electric Reliability Council of Texas (ERCOT) electric grid, this study uses projected fuel and CO2 prices from analysis of the Lieberman-Warner Climate Bill, ERCOT expected future electricity demand, and the current and planned ERCOT power plant fleet as inputs into a dynamic model of electricity dispatch that calculates annual generation, profits, and CO2 emissions in the years 2012–31. A scenario without CO2 capture is compared to scenarios with flexible and inflexible CO2 capture. Over the 20 years, changes in the contribution of each plant type to meeting electricity demand and in their operating profits are primarily associated with the relative marginal costs of electricity production for each plant type. Utilization of coal-based plants remains high despite rising CO2 costs even without CO2 capture because coal remains much less expensive than natural gas. Coal-fired plants become less expensive to operate with CO2 capture than without in the late 2010s to early 2020s. Flexible CO2 capture systems become more economical to operate at maximum CO2 removal by the mid to late 2010s, but annual operating profits are greater for inflexible systems due to the incremental cost of flexibility. After maximum CO2 removal is most economical, coal-fired plants earn much greater annual operating profits if CO2 capture is available. When capital cost considerations are included in a discounted cash flow analysis over the 20year study period, no scenarios with CO2 capture recover more than 61% of the combined capital expenditures of CO2 removal systems, any required sulfur dioxide (SO2) scrubbing systems, and any new generation capacity required to replace the output lost to CO2 capture. However, there is a major economic advantage to flexible CO2 capture because flexibility eliminates the need to spend several billion dollars in capital costs to replace the output lost to CO2 capture energy requirements at maximum CO2 removal. While these particular results do not present positive economic arguments for CO2 capture investment, the general methodology can be utilized to examine what combinations of fuel prices, CO2 prices, electricity demand, and power plant fleet are conducive to flexible or inflexible CO2 capture installation. 13. Solvent Management of MDEA/Piperazine p. 180 by Fred Closmann (also supported by the Process Science & Technology Center) In this quarter, we conducted a single oxidative degradation experiment and a single thermal degradation experiment on 7 m MDEA/2 m PZ loaded to 0.24 moles CO2/mole alkalinity. The oxidative degradation experiment solvent was amended with 1 mM Fe2+ and 100 mM Inhibitor A. We measured a formate production rate of 0.011 mM/hr, which is comparable to rates with the same solvent in the absence of Inhibitor A. Hydrolyzed samples resulted in a formate production rate (0.02 mM/hr) that was approximately double the rate when hydrolysis was not performed. In hydrolyzed samples, the oxalate production rate was 0.007 mM/hr, but no other heat stable salt was consistently observed in the study. A foaminess factor of 8.65 was measured in the undegraded solvent, but a foaminess factor of >20 was observed with the degraded solvent. 7 A thermal degradation experiment on the 7 m MDEA/2 m PZ solvent was performed at temperatures of 135 and 150 °C. The loss of MDEA and PZ occurred at a greater rate at 150 °C, with complete loss of PZ occurring somewhere between days 42 and 57 at both temperatures at a loading of 0.11 moles CO2/mole alkalinity. The concentration of MDEA and PZ diminished on an equimolal basis, indicating that the MDEA and PZ are reacting together resulting in loss of both compounds until the PZ is completely degraded. We will be investigating disproportionation reactions to identify the byproducts of this mechanism. We performed wetted wall column studies on the 7 m MDEA/2 m PZ solvent at 80 and 100 °C and about 0.03 moles CO2/mole alkalinity, and measured kg’ to be 2. 76 X 10-10 mols/s-Pa-cm2 at a partial pressure of CO2 of 1,273 Pa, and 1.63 X 10-10 mols/s-Pa-cm2 at a partial pressure of CO2 of 5,212 Pa, respectively. 14. Modeling Absorber/Stripper Performance with MDEA/PZ p. 188 by Peter Frailie This study will focus on a blended amine solvent system containing piperazine (PZ) and methyldiethanolamine (MDEA). Previous studies have shown that this particular blend has the potential to combine the high capacity of MDEA with the attractive kinetics of PZ (Bishnoi, 2000). These studies supplied a rudimentary Aspen Plus®-based model for an absorber with the MDEA/PZ system. The report also makes the recommendation that more kinetic and thermodynamic data must be acquired concerning the MDEA/PZ system before the model can be significantly improved. Two researchers in the Rochelle lab are currently acquiring this data, but it has not yet been incorporated into an absorber/stripper model. One of the major goals of this study will be to improve the supplied Aspen Plus® absorber model with up to date thermodynamic and kinetic data. Another major goal of this study will be to combine absorber and stripper models to evaluate the overall system performance. 15. Measurement of kga in Packing p. 191 by Chao Wang This research is focused on the properties of dumped and structured packings in the CO2 capture with chemical absorption. The properties of the packings are the effective area ae, pressure drop, and mass transfer coefficient. According to the theory of series resistance, the mass transfer coefficient contains the gas-side coefficient kG, liquid-side coefficient kL and the overall coefficient KG. What we want to find is the method of measuring kG, kL and finally KG. Literature review about the measurement of kGa and ae has been done during this period. We propose to measure kGa with SO2-air-NaOH-water. The relation between gas diffusivity DG and gas-side mass transfer coefficient has also been k G , SO 2 D G , SO 2 discussed. It is acceptable to use the equation that according to Sharma = k G ,CO 2 DG ,CO 2 (1965). Thus we can apply the kG we measure in the SO2 absorption for the CO2 absorption. Rate-based Modeling Study of CO2 Capture with Aqueous Monoethanolamine Solution Submitted to Industrial and Engineering Chemistry Research. 8 p. 194 Oxidative Degradation and Solvent Management of Piperazine in a Pilot Plant Campaign Quarterly Report for October 1 – December 31, 2008 by Stephanie Freeman Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 3, 2009 Abstract Oxidative degradation of concentrated PZ was studied this quarter. Six conditions were studied in the presence of 1.0 mM of iron. PZ showed little production of degradation products in studies with just iron, iron with 100 mM Inhibitor A, and iron with 30 mM Inhibitor B. In the case of iron and 30 mM EDTA, the most EDA and formate was produced as compared with the other oxidative inhibitors. All degradation observed was lower than that of MEA in similar conditions although the reduction of PZ observed in all cases was not wholly matched to the amount of degradation products detected. A pilot plant campaign was conducted this quarter on 8 m PZ at Pickle Research Campus at the University of Texas. The goal of the campaign was to tackle any operational difficulties with using concentrated PZ and to demonstrate the improved mass transfer performance of the solvent. Fourteen runs were performed with sampling at five points throughout the system, with a few samples missing due to empty sample bombs. The final five runs were performed at 5 m PZ. Initial analysis shows enhanced mass transfer performance of the 8 m PZ over similar 7 m MEA systems. The equivalent work was minimized in systems with higher liquid flow rates and higher lean loadings. The optimum liquid flow rate was not found during the course of the campaign and appears to be at a value higher than 18 gallons per minute. Further analysis is ongoing, including a comparison of these pilot plant results to that of an MEA system. Introduction Concentrated aqueous piperazine (PZ) is being investigated as a possible alternative to 30 wt % (or 7 m) MEA in absorber/stripper systems to remove CO2 from coal-fired power plant flue gas. Aqueous PZ has been given a proprietary name of ROC20 for 10 m PZ and ROC16 for 8 m PZ. Previous reports include the proprietary name, while the concentration of PZ will be explicitly used in this document. Preliminary investigations of PZ have shown numerous advantages over 7 m MEA systems (Freeman, 2008). Aqueous concentrated PZ solutions have less oxidative and thermal degradation, as previously shown at concentrations of 5 and 8 m PZ (see previous quarterly reports). The kinetics of CO2 absorption are faster in concentrated PZ, as shown by Cullinane, and are currently being measured by Dugas (Cullinane and Rochelle, 2006; Dugas, 2008). The capacity of concentrated PZ is greater than that of MEA while the heat of absorption and volatilities are comparable. 9 As reported in the last quarter, the heat of absorption of concentrated PZ solutions is comparable to MEA over a range of temperatures. Above a loading of approximately 0.1 mol CO2/equiv PZ, there was little difference in the values for the heat of absorption between 80, 100, and 120°C. At all temperatures, the heat of absorption decreased as loading increased and fell off as loading reached 0.4 mol CO2/equiv PZ, producing a trend different from other amines whose heat of absorption remains constant from loadings of 0 to 0.5 mol CO2/mol amine before dropping off. The heat of absorption data are still being analyzed for any pertinent trends. This quarter was focused on short term oxidative degradation experiments with 8 m PZ and a pilot plant campaign. The oxidation experiments were conducted to determine the best operative conditions for the pilot plant runs to follow. The pilot plant campaign was conducted at the Pickle Research Campus. The primary goals of the campaign were to demonstrate enhanced mass transfer characteristics and to identify operation issues with the 8 m PZ solvent. Experimental Methods Analytical Methods Total Inorganic Carbon Analysis (TIC): Quantification of CO2 loading was performed using a total inorganic carbon analyzer. In this method, a sample is acidified with 30 wt % H3PO4 to release the CO2 present in solution (Hilliard, 2008). The CO2 is carried in the nitrogen carrier gas stream to the detector. PicoLog software is used to record the peaks that are produced from each sample. A calibration curve is prepared at the end of each analysis using a TIC standard mixture of K2CO3 and KHCO3. The TIC method quantifies the CO2, CO3-2, and HCO3present in solution. These species are in equilibrium in the series of reactions shown below. CO32− + 2H+ ↔ HCO3− + H+ ↔ H2CO3 ↔ CO2 + H2O Acidification of the sample shifts the equilibrium toward CO2 which bubbles out of solution and is detected in the analyzer. Acid pH Titration: Titration with 0.2 N H2SO4 is used to determine the concentration of amines in experimental samples. The automated Titrando apparatus (Metrohm AG, Herisau, Switzerland) is used for this method. A known mass of sample is diluted with water and the autotitration method is then used. The Titrando titrates the sample with acid while monitoring the pH. The equivalence points are recorded. The equivalence point around a pH of 3.9 corresponds to basic amine species in solution (Hilliard, 2008). The test is not sensitive to the type of amine, so if PZ has degraded to EDA, the titration test will detect the sum of contributions from the species. Anion IC: The anion IC was used to determine the concentration of glycolate, acetate, formate, chloride, nitrite, sulfate, oxalate, and nitrate in experimental samples. A Dionex ICS-3000 instrument with AS15 IonPac column, ASRS 4-mm self-regenerating suppressor, carbonate removal device (CRD), and carbonate removal from eluent generation was used as previously described by Andrew Sexton using a linear KOH eluent concentration (Sexton, 2008). No major modifications have been made to the method in this quarter. Cation IC: The cation IC was used to determine the concentration of PZ and ethylenediamine (EDA) in experimental samples. A Dionex ICS-2500 instrument with CS17 IonPac column with CSRS 4-mm self-regenerating suppressor was used as previously described by Andrew Sexton 10 with a linear increase of methanesulfonic acid (MSA) concentration in the eluent (Sexton, 2008). No major modifications have been made to the method in this quarter. NaOH Treatment for Amides: An analytical test for the formation of amides has been developed previously by Andrew Sexton and has been included in the results shown here. Experimental samples are treated with 5 N NaOH (in equal gravimetric amounts) and allowed to sit overnight. The anion IC analytical method is then used to quantify increases in the concentrations of analytes as compared to the original samples (Sexton, 2008). In most cases, the main increases are shown in the production of formate and oxalate following NaOH treatment. The addition of strong base reverses the amide formation reaction that has occurred during the experiment. As an example, the formation of N-formylpiperazine is shown in Eqn. 1 below: + (PZ) (1) → (Formate) (N-formylPZ) The addition of NaOH hydrolyzes the bond between the amine group and the carbon of the formyl group to reverse the reaction. In this way, the free formate created from reversing this reaction can be used to identify the formate bound has N-formylPZ. The same process can be used to identify the oxalate amine of PZ. Results The focus of this quarter has been on six short-term oxidative degradation experiments and a pilot plant campaign. Results of Oxidative Degradation Experiments Leading up to the pilot plant campaign on 8 m PZ, multiple short-term oxidative degradation experiments were conducted to determine the best operating conditions for the pilot plant runs. A summary of the oxidative degradation experiments conducted is shown below in Table 1. The first two experiments, OE5 and OE6, investigated the degradation effect of iron alone and whether Inhibitor A had any inhibiting effect on iron-catalyzed degradation. The next two experiments performed were OE7 and OE8 which investigated two other chemicals, Inhibitor B and EDTA, for their inhibiting effect on iron-catalyzed degradation. Table 1: Oxidative Degradation Experiments Performed on 8 m PZ Expt. OE5 OE5B OE6 OE6B OE7 OE8 Solvent System 8 m PZ 8 m PZ 8 m PZ 8 m PZ 8 m PZ 8 m PZ Additives 1 mM Fe2+ Final OE5 Solution Used 1 mM Fe2+, 100 mM “A” Final OE6 Solution Used 1 mM Fe2+, 20 mM “B” 1 mM Fe2+, 30 mM EDTA 11 Duration (hrs) 70 92 70 92 164 164 Foaming Test? Yes Yes Yes Yes No No The concentration of detected degradation products is shown for OE5, OE5B, OE6, OE6B, OE7, and OE8 in Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, and Figure 6. All values are adjusted for possible water loss using the sulfate concentration changes since sulfate added with the iron should not react during the experiment. Degradation products not detected in an individual experiment were omitted from each figure. Apart from a few suspect data points that will be re-analyzed, such as the third post-NaOH sample in OE6, the data show reasonable trends for each experiment. Overall, very low levels of acetate, glycolate, nitrate, nitrite, and oxalate were detected in the first five experiments (OE5 through OE7). OE8 showed slightly higher levels of these degradation products, as described below. Figure 1: Concentration Profile for OE5 (8 m PZ, 1 mM Fe2+, α=0.3, 55°C) 12 Figure 2: Concentration Profiles for OE5 and OE5B (8 m PZ, 1 mM Fe2+, α=0.3, 55°C). OE5 ended and OE5B started at 70 hours. Figure 3: Concentration Profiles for OE6 (8 m PZ, 1 mM Fe2+, 100 mM Inhibitor A, α=0.3, 55°C) 13 Figure 4: Concentration Profiles for OE6 and OE6B (8 m PZ, 1 mM Fe2+, 100 mM Inhibitor A, α=0.3, 55°C). OE6 ended and OE6B started at 70 hours. Figure 5: Concentration Profiles for OE7 (8 m PZ, 1 mM Fe2+, 20 mM Inhibitor B, α=0.3, 55°C) 14 Figure 6: Concentration Profiles for OE8 (8 m PZ, 1 mM Fe2+, 30 mM EDTA, α=0.3, 55°C) For PZ oxidation, the production of formate and EDA indicates degradation (see previous quarterly reports). A comparison of formate production among the six oxidative degradation experiments is shown below in Figure 7. A comparison of post-NaOH treatment formate concentrations is shown in Figure 8. OE5, OE5B, OE6, OE6B, and OE7 all have very low formate and post-NaOH treatment formate production levels of below 3 mM formate. The only experiment with significant formate production is OE8, the experiment with 1.0 mM Fe2+ and 30 mM EDTA. Even though this experiment demonstrates the highest formate levels, the levels reached are not as high as heavily degraded PZ or MEA solutions. For example, post-NaOH treatment formate levels achieved through copper-catalyzed degradation reach 100 mM of formate within 25 hours of operation. The maximum observed in the short duration of OE8 is 30 mM of formate. Therefore, EDTA at a concentration of 30 mM is not an effective inhibitor of iron-catalyzed degradation in 8 m PZ solutions. 15 Figure 7: Comparison of Formate Production Figure 8: Comparison of Post-NaOH Formation Concentrations 16 The only experiments with detectable levels of EDA were OE5B and OE8. The initial concentration in OE5B was 12 mM and did not increase significantly throughout the experiment. This indicates that there may have been a small amount formed from sitting on the counter or in the foaming experiment in between experiments OE5 and OE5B. Since the concentration did not increase during OE5B, it does not seem to indicate that it is an actual product from the degradation of PZ in this solvent system. The concentration of EDA detected in OE8 started at zero and reached over 60 mM during the course of the experiment. OE5, OE6, OE6B, and OE7 did not have detectable levels of EDA produced during the experiments. A small amount may have been produced, but was below the detection limit of the cation IC. Also interesting to note was that the EDA concentration in the OE8 experiment increased in the post-NaOH treatment sample compared to the original sample, as shown below in Figure 9. This indicates that the EDA is also reacting with the addition of NaOH. This may indicate that the EDA is forming amides with the formate or oxalate in solution as is observed with PZ. This participation in the formation of amides has not been noticed in previous experiments. Figure 9: EDA Concentration Profiles for OE8 Results of Foaming Tests Xi Chen performed foaming tests on four oxidative experiments after their completion. The intent was to determine which additives to use in the pilot plant based on foaming performance. Please see Xi Chen’s section of this quarterly report for the details of his experimental set-up. Chen’s foaming work involves measuring the foaminess coefficient of a solvent in order to compare to other solvents. The foaminess coefficient is essentially the ratio of the volume of foam produced to the gas flow rate during the experiment. A higher foaminess coefficient 17 indicates a propensity to foam, so a low coefficient is desirable. The foaminess coefficient as measured by Chen is shown in Table 2. Table 2: Results of Foaming Analysis of Oxidation Experiments Expt Solution Additives Foaminess Coefficient (m2/s) OE5 OE5B OE6 OE6B 8 m PZ 8 m PZ 8 m PZ 8 m PZ 8 m PZ None 1 mM Fe2+ 1 mM Fe2+ 1 mM Fe2+ + 100 mM “A” 1 mM Fe2+ + 100 mM “A” 86 85 >>300 92 68 Degradation Time (hrs) 0 70 162 70 162 The results of the foaming tests are not immediately clear. For PZ with iron (OE5 and OE5B), the foaminess coefficient is the same as neat PZ after 70 hours of degradation, but increases to over 300 m2/s after 162 hours of degradation. This jump in foaminess is surprising as the first test was so low. The degradation products responsible for promoting foaming must have continued reacting after the first experiment (OE5) was shut down and the foaming test was performed before being returned to the reactor for the beginning of OE5B. Also confusing is the fact that the opposite trend occurred in the experiments with iron and inhibitor A. After 70 hours, the foaminess was slightly higher than neat PZ but after 162 hours the foaminess was reduced below that of neat PZ. It appears that Inhibitor A helped decrease foaming in this experiment. Results of Pilot Plant Campaign A three week campaign was performed at the pilot plant on the Pickle Research Campus this quarter. The solvent investigated was 8 m PZ. The primary objectives of the campaign were as follows: • Identify operational issues associated with 8 m PZ o Small operation window due to solubility o Dealing with a highly viscous solvent • Demonstrate enhanced absorber performance o Enhanced reaction rates o Increased CO2 capacity • Observe stripper performance o Assess and minimize equivalent work of system o Compare to other solvents Fourteen runs were completed during the campaign and the identifying run characteristics are shown in Table 3. The gas rate was held at a constant 350 scfm while the liquid rate was varied at 12, 15, and 18 gpm. The stripper pressure was initially 20 psia while the final 8 runs were at increased pressure from 50 to 60 psia. The highest pressure of 60 psia was initially used for the high pressure runs, but this was reduced to 51 and 50 in order to maintain the temperature of the stripper below 260°F. The lean loading was variable and an attempt was made to use various lean loadings for each L/G combination that was repeated. The lean loading shown in Table 2 is the loading recorded during the pilot plant campaign. The loading as determined by TIC is 18 discussed below. In preparation for disposing of the solvent after the run, the solvent was diluted to 5 m P for the final 4 runs. Further results of the runs are shown in Table 4 including an equivalent work comparison. Table 3: Summary of Pilot Plant Run Conditions Run 1 2 3 4 5 6 7 8 9 10 11 12 13 14 PZ Conc (m) 8 8 8 8 8 8 8 8 8 8 5 5 5 5 Gas Rate (scfm) 350 350 350 350 350 350 350 350 350 350 350 350 350 350 Liquid Rate (gpm) 15 15 15 15 12 18 15 12 18 15 18 18 15 12 Stripper Pressure (psia) 20 20 20 20 20 20 60 60 51 51 51 50 50 50 Lean Loading (mol/mol alk) 0.26 0.31 0.25 0.38 0.27 0.30 0.31 0.30 0.27 0.33 0.32 0.27 0.26 0.26 Solvent Management Results The most prominent operational issue during the campaign was maintaining the solution within the small solubility envelope for concentrated, aqueous PZ solutions. Since the plant did not run 24/7, care was taken to ensure the proper CO2 concentration in the solvent before the plant was shut down on weekends and during outages. This took time to circulate the solvent to get the rich and lean loadings consistent after the simulated flue gas had been shut off. Additional time was needed to titrate the solvent to check the CO2 concentration. These concerns did take a few hours to address, but the issue of solubility was not as difficult to address as was predicted before the campaign. The use of online monitoring of the solution density was very useful in predicting the approximate CO2 concentration. This online correlation needed slight modifications as the campaign proceeded, but offered the operators a very useful estimate of the CO2 concentration and identified when the concentration was nearing a zone where solidification or crystallization was possible. There was only one incident of crystallization that affected the campaign run schedule. After the first week of operation, the plant was shut down with the CO2 concentration within the solubility envelope for the solvent. The next week, the plant was not running because of the AIChE and GHGT-9 conferences, so the plant sat idle for 9 days. When the plant was restarted on the following Monday, there was a slush observed in the lean absorber pump and absorber bottoms pump. The operator had to manually spin the pumps to loosen the solids that had built up, and the pump worked when turned on. 19 Analytical results Samples taken from the absorber lean and absorber rich from all 14 runs were analyzed for PZ concentration, CO2 concentration, and degradation product concentration. PZ concentration was measured using sulfuric acid titration in our laboratory and was compared to the hydrochloric acid titration performed at the pilot plant at the time of sampling each end run. The results of each analysis are shown in Figure 10 and Figure 11 for the absorber lean and absorber rich samples, respectively. Missing values in the absorber rich figure are due to the inability to analyze crystallized samples at the pilot plant site. The PZ concentration as measured in our laboratory appeared to be always slightly lower than that measured immediately after the runs at the pilot plant. The reason for this difference is not immediately apparent. Both protocols use the same approach and calculations. The main differences are in the acid used and the titration apparatus used. The laboratory method uses sulfuric acid and an automatic titrator with end point detection. The pilot plant method uses hydrochloric acid and a manually operated, digital titrator. It is assumed that the choice of acid does not affect the results and was originally chosen in each lab due to cost and availability. The pilot plant method titrates to a specific end point, a pH of 3.9. The laboratory method is more accurate because the exact endpoint is detected automatically where the pilot plant method assumes that 3.9 is appropriate for all samples. The exact end points do usually occur around 3.9, but can be up to 0.1 pH units different. Although this is a small difference, it may account for the slightly lower concentration detected using the more accurate laboratory method. Figure 10: PZ Concentration in Absorber Lean for All Runs 20 Figure 11: PZ Concentration in Absorber Rich for All Runs At the pilot plant, the CO2 concentration is measured using a methanol-KOH titration method. These data were compared to TIC results performed using the standard protocol in our laboratory. The CO2 concentration, reported as CO2 loading, is compared for the absorber lean and absorber rich samples in Figure 12 and Figure 13. There is some disagreement between the two methods were the TIC results are both higher and lower than the pilot plant results for some samples. There has been some disagreement about which method is more accurate and it is not currently known in a quantitative way which is correct. The methods are individually preferred in each lab, but an analysis of which is correct has not been completed at this time. Presently, the average absolute value of the difference between the values is 0.0094 mol/mol alk with the minimum and maximum differences being 0.0008 and 0.0273 mol/mol alk, respectively. The difference is not very significant, as the average difference is just 2.7% of a typical loading of 0.35 mol/mol alk. Considering the other sources of error present in a pilot plant run of this magnitude, a slight difference in the measured loading with different methods should not pose problems in analyzing the data. Unfortunately, the calculation of PZ concentration in molality rather than weight percent does rely on this loading value. Therefore, any error in this measurement is further propagated. 21 Figure 12: CO2 Loading in Absorber Lean for All Runs Figure 13: CO2 Loading in Absorber Rich for All Runs 22 Each absorber lean and absorber rich sample was analyzed using anion and cation IC to detect possible degradation products. The levels of formate, oxalate, and acetate are shown in Figure 14, Figure 15, and Figure 16, respectively. Overall, the levels of these three degradation products are all very low, below 0.4 mM. This does not indicate that any degradation of PZ has been occurring to produce these products. Also indicative of the lack of degradation is the fact that the concentrations of all three products during Run 1 are approximately the same throughout the four week campaign or even one of the highest of all the values. Figure 14: Formate Concentration in Absorber Lean and Rich for All Runs 23 Figure 15: Oxalate Concentration in Absorber Lean and Rich in All Runs Figure 16: Acetate Concentration in Absorber Lean and Rich in All Runs 24 The concentration of chloride and sulfate was also measured for these samples and is shown. The concentration of chloride remained fairly constant below 0.1 mM except for three samples which showed significantly higher concentrations. Since these samples were spread out in different campaigns, the cause of this spike in chloride concentration is not known. The concentration of sulfate fluctuated from zero to 0.28 mM without a recognizable pattern. Sulfate was not added to the process through one of the additives, so the source of this detected sulfate is not known. Figure 17: Chloride Concentration for Absorber Lean and Rich for All Runs 25 Figure 18: Sulfate Concentration for Absorber Lean and Rich in All Runs Initial PZ Performance Results The goals of the PZ pilot plant campaign were met during the course of the 14 recorded runs. The enhanced mass transfer of PZ was demonstrated through high removal rates with standard circulation rates. Detailed analysis of the results using Aspen Plus® modeling has not be completed to date, but this analysis will be done by David Van Wagener and Jorge Plaza in the future. Details of the 14 runs are shown below in Table 4. All runs were performed using 350 scfm of inlet gas and the liquid rate, stripper pressure, and lean loading are included in the table. The removal percentage of CO2 for each run is shown in the table and was calculated using data directly from the DeltaV pilot plant software. The equivalent work for each system is included in the table as well and was calculated as previously described (Freeman, 2008). This calculation uses the heat duty of the reboiler, Q, the reboiler steam temperature, Treb, the sink temperature, Tsink, the work of the pumps, Wpumps, and the work to compress the CO2, Wcomp. (2) To estimate the equivalent work of these 14 runs, the pumping work was assumed to be approximately constant for all of the cases and was ignored. Since the campaign included runs varying in stripper pressure from 20 to 60 psia, the compression work to compress the captured CO2 from the stripper pressure to 60 psia was calculated for each run. For the runs at 60 psia, no additional work was added. 26 This work was estimated by calculating the adiabatic work to compress a gas from its pressure to 60 psia. The general expression for adiabatic work, including polytropic efficiency, ηp, is shown in Eqn. 3 (Smith, Van Ness et al., 1995). (3) Additional variables in the equation are the universal gas constant, R, system temperature, T, initial pressure, P1, final pressure, P2, and the ratio of heat capacities, as defined below. To adjust for the polytropic efficiency in the pressure ratio, a switch from the variable n to the variable k is used (NGPSA, 1967). In this expression, n and k are defined in the same way, but an efficiency term is added. (4) (5) Substituting Eqn. 5 into Eqn. 3, the final equation (Eqn. 6), where the efficiency term only appears inside the brackets, is as follows: (6) The total estimated equivalent work was estimated for each run as Eqn. 2 using Eqn. 6. For CO2 compression, as estimation was made that the value of the constant volume heat capacity, CV, was equal to five times the universal gas constant, R. This is based on molecular structure and an assumption of 10 degrees of freedom within the molecule. The polytropic efficiency was estimated at 78%. Table 4: Initial Results of Pilot Plant Campaign Run 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Liquid Rate (gpm) 15 15 15 15 12 18 15 12 18 15 18 18 15 12 Stripper Pressure (psia) 20 20 20 20 20 20 60 60 51 51 51 50 50 50 Lean Loading (mol/mol alk) 0.26 0.31 0.25 0.38 0.27 0.30 0.31 0.30 0.27 0.33 0.32 0.27 0.26 0.26 CO2 Removal (%) 87 69 93 33 71 79 76 64 93 52 64 90 80 70 Estimated Eq. Work (kJ/mol CO2) 24.7 26.1 22.3 28.3 12.6 22.1 25.9 24.0 23.8 19.9 26.0 22.5 23.4 24.8 In order to understand the differences in each system, an attempt was made to plot the data on a common basis. First, the equivalent work was compared to the CO2 penetration, or the percentage of CO2 entering the system that was not absorbed by the solvent. It is calculated as 27 100 minus the CO2 removal percentage. For the runs performed with an 8 m PZ solution, the data are shown in Figure 19 below. The data are grouped by the liquid flow, in gallons per minute (gpm), and the pressure of the stripper, in psia. The data from Run 5 were strange because of crystallization in some of the sample bombs and those data have been omitted from these figures. Figure 19: Equivalent Work and CO2 Penetration for 8 m PZ Pilot Plant Runs Using the semi-log plot representation shown in Figure 19, there appears to be a trend in the run with common liquid flow rates. Exponential trend lines are shown that connect runs with the same liquid flow rates, despite differences in stripper pressure. It follows that the equivalent work is increased as the CO2 penetration decreases. This association is predicted as it takes more work to remove more CO2 from a solution, shown as an increased removal percentage, or a decreased CO2 penetration. Also, as the liquid flow rate increases, the overall curve is moved downward towards regions of lower CO2 penetration and lower equivalent work. If a system of constant CO2 penetration is compared between 15 and 18 gpm flow rates, the 18 gpm system has a lower equivalent work. One of the goals of this pilot plant project was to assess and minimize the equivalent work of the system. It was assumed that the equivalent work would reach a minimum over a variety of flow rates. It appears that the liquid flow was not increased high enough to see this minimum, as demonstrated in Figure 20. 28 Figure 20: Equivalent Work for a CO2 Penetration of 20% The figure above was created by essentially taking a slice of Figure 19 at a CO2 penetration of 20% and using the trend lines to predict what the values would be for each of the three flow rates. The equivalent work is reducing as the liquid rate increases, but it is clear that a minimum has not been reached for this solvent. The increased capacity of this solvent and high reaction rate would allow for a much higher liquid flow rate in the absorber to maintain an acceptable removal. Only four runs were completed with 5 m PZ solvent, but the equivalent work and CO2 penetration data are shown below in Figure 21. All the lower concentration runs were performed at high pressure and only one run was performed with a liquid flow rate of 12 and 15 gpm. 29 Figure 21: Equivalent Work and CO2 Penetration for 8 m PZ Pilot Plant Runs The same trends were observed in the 5 m PZ runs, except that the slope of the trend line was much steeper for the 18 gpm data. Since multiple runs were not performed for 12 and 15 gpm, it is not possible to confirm this trend for other flow rates. Also, both of the 18 gpm data points were at high pressure, where the data on the previous graph include multiple pressures without a duplicate in pressure and liquid rate. A second analysis was performed to compare the lean loading and the CO2 penetration for all of the pilot plant runs. The lean loading calculated using the TIC and titration results from our laboratory are shown with the CO2 penetration for each run in Figure 22. 30 Figure 22: Lean Loading and CO2 Penetration for all Pilot Plant Runs For this analysis, trends are not as distinguishable as with the previous analysis. Within a single PZ concentration, 5 or 8 m PZ, the higher liquid flow rates appear to shift the data downward toward lower CO2 penetration and higher lean loading. For a given PZ concentration and CO2 penetration, the 18 gpm runs have the highest lean loading. There do not appear to be enough data to draw further conclusions on what combination of variables is the most advantageous with this analysis alone. Previous energy analyses have indicated that the equivalent work is minimized with systems using higher lean loading than previously thought for multiple solvents (Van Wagener and Plaza, 2008). Discussion Oxidative Degradation Experiments The oxidative degradation experiments performed this quarter demonstrated the reduced effect of iron-catalyzed degradation on PZ as compared to other, more susceptible solvents. In the presence of 1.0 mM of iron (experiment OE5 and OE5B), little PZ degradation was observed. In the instance of OE5, the PZ concentration was reduced 0.8 m, or approximately 450 mM, through the course of the experiment. This reduction is small, but was not matched to the sum of the degradation products found with anion and cation IC analyses. A nitrogen balance on the PZ and products reveals that only 0.01% of the lost PZ is accounted for in the detected degradation products. For carbon, only 0.2% of the lost PZ is account for in the products. This analysis does not account for the volatility of the PZ solution, which would result in a loss of PZ throughout the experiment due to the gas flowing over the top of the solution. This volatility could account 31 for a significant decrease in PZ concentration, but there is a strong possibility that there are degradation products that are not detected using our standard methods. The identification of these products will be an important part of the remainder of this PZ work. Identifying the paths through which PZ is degraded and into what products is crucial to the understanding of overall PZ degradation. 8 m PZ Pilot Plant The pilot plant campaign performed this quarter demonstrated the advantages of a concentrated, PZ solvent system. Qualitative observations indicated that mass transfer was enhanced over previous solvents tested. Conclusions The oxidation behavior of PZ has been studied this semester. PZ does oxidize slightly in the presence of iron but at a very slow rate. The addition of Inhibitor A and Inhibitor B does not affect this result significantly. In the case of PZ in the presence of iron and EDTA, degradation seems increased due to the increased production of detectable degradation products. EDA has been found to interact with amide formation if present in solution as a degradation product. The pilot plant campaign performed this quarter on 8 m PZ demonstrated enhanced mass transfer capability. The overall equivalent work was minimized and CO2 removal was increased in systems with high liquid flow rates. The liquid flow rate was not increased enough to find the flow rate that minimized the equivalent work. The effect of stripper pressure on equivalent work has not been identified in this initial analysis. Future Work Work on oxidative degradation of PZ will continue in the first quarter of 2009. Identification of unknown degradation products will also continue to be a topic of interest. I will be working with Fred Closmann to develop a new apparatus to combine the effects of thermal and oxidative degradation to be able to study collaborative degradation effects. A few remaining solubility tests will be performed to complete the understanding of the PZ solid solubility window. An analysis of the pilot plant results in comparison to MEA will be pursued in the next few quarters. This work will take place in conjunction with David Van Wagener and Jorge Plaza. In addition, I will be working on my proposal for my preliminary examination which will take place in the second or third quarter of 2009. 32 References Cullinane JT, Rochelle GR. Kinetics of carbon dioxide absorption into aqueous potassium carbonate and piperazine. Ind Eng Chem Res. 2007;45(8):2531–2545. Dugas R. Absorption and desorption rates of carbon dioxide with monoethanolamine and piperazine. GHGT-9, Washington D.C. 2008. Freeman S. Carbon dioxide capture with concentrated, aqueous piperazine. GHGT-9, Washington D.C. 2008. Hilliard MD. A Predictive Thermodynamic Model for an Aqueous Blend of Potassium Carbonate, Piperazine, and Monoethanolamine for Carbon Dioxide Capture from Flue Gas. The University of Texas at Austin. Ph.D. Dissertation. 2008;1083. NGPSA. Engineering Data Book. Tulsa, OK, Natural Gas Processors Suppliers Association (NGPSA). 1967. Sexton A. Amine Oxidation in CO2 Capture Processes. The University of Texas at Austin. Ph.D. Dissertation. 2008. Smith JM, Van Ness HC, et al. Introduction to Chemical Engineering Thermodynamics. New York, McGraw-Hill. 1995. Van Wagener D, Plaza JM. Modeling CO2 capture with aqueous monoethanolamine. GHGT-9, Washington D.C. 2008. 33 Rate and CO2 Partial Pressure Measurements for Monoethanolamine and Piperazine Solutions Progress Report for October 1 – December 31, 2008 by Ross Dugas Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin January 30, 2008 Abstract The remaining measurements of CO2 partial pressure and CO2 absorption/desorption rate at 80 and 100 ˚C for monoethanolamine (MEA) and piperazine (PZ) solutions were completed. 7 m MEA/2 m PZ solutions were also tested at 40, 60, 80, and 100 ˚C with four CO2 loadings. The measured values of CO2 partial pressure match very well with available literature sources. PZ absorbs CO2 2–3 times faster than MEA. The 7 m MEA/2 m PZ solution absorbs CO2 faster than MEA and slightly slower than PZ. Even though the pseudo-first order theory for the liquid film mass transfer coefficient, kg’ contains both temperature and concentration dependent terms, the measured kg’ values are generally not affected by changes in temperature or amine concentration. Varying temperature and MEA concentrations did not affect kg’ in the wetted wall column. Piperazine experiments in the wetted wall column show lower kg’ values at higher temperature and higher CO2 loading. These conditions require greater CO2 fluxes in the wetted wall column experiments and may increase the significance of the liquid film physical mass transfer coefficient, klo. Under these conditions, diffusion limitations of reactants and products near the interface can restrict CO2 mass transfer giving reduced kg’ values. Introduction Data for CO2 partial pressure and CO2 absorption/desorption rates have previously been reported for 7, 9, 11, and 13 m MEA and 2, 5, 8, and 12 m PZ at absorber conditions (40 and 60 ˚C) using the wetted wall column (Rochelle, Dugas et al., 2008). Some stripper conditions (80 and 100 ˚C) have also been reported (Rochelle, Dugas et al., 2008). This report includes the remaining 80 and 100 ˚C data for MEA and PZ as well as 7 m MEA/2 m PZ. As in previous work, solution CO2 loading is defined on an alkalinity basis. The alkalinity is essentially the number of nitrogen atoms on the amine; 1 for MEA but 2 for PZ. The CO2 loading definition is shown in Equation 1. 34 CO2 Loading = nCO 2 n MEA + 2n PZ (1) Results and Discussion CO2 Partial Pressure The complete rate and CO2 equilibrium partial pressure data sets for MEA and PZ solutions are included in Tables 1 and 2. Table 3 contains the 7 m MEA/2 m PZ rate and CO2 partial pressure data. Due to equipment limitations, only lower loading solutions were tested at 80 and 100 ˚C. Figure 1 shows the CO2 partial pressure plot using the data from Table 1. Similarly Figures 2 and 3 graphically represent the CO2 partial pressure data from Table 2 and 3. Table 1: Rate and CO2 Partial Pressure Data for 7, 9, 11, and 13 m MEA MEA Temp CO2 Ldg mol/molalk m C 0.252 0.351 40 0.432 0.496 0.252 0.351 60 7 0.432 0.496 0.271 80 0.366 0.444 0.271 100 0.366 0.231 0.324 40 0.382 0.441 0.496 0.231 0.324 9 60 0.382 0.441 0.496 0.265 80 0.356 0.265 100 0.356 P*CO2 k g' . . 2 Pa mol/s Pa m 15.7 3.34E-06 77 1.40E-06 465 7.66E-07 4216 3.47E-07 109 2.92E-06 660 1.70E-06 3434 9.28E-07 16157 3.76E-07 1053 2.85E-06 4443 1.87E-06 18826 7.52E-07 5297 2.98E-06 19008 1.40E-06 10.4 34 1.86E-06 107 1.40E-06 417 8.36E-07 5354 3.02E-07 61 3.80E-06 263 2.44E-06 892 1.47E-06 2862 9.57E-07 21249 3.24E-07 979 3.24E-06 4797 1.75E-06 4940 3.40E-06 21534 1.33E-06 MEA Temp CO2 Ldg mol/molalk m C 0.261 0.353 40 0.428 0.461 0.261 0.353 11 60 0.428 0.461 0.256 80 0.359 0.256 100 0.359 0.252 0.372 40 0.435 0.502 0.252 0.372 13 60 0.435 0.502 0.254 80 0.355 0.254 100 0.355 35 P*CO2 k g' . . 2 Pa mol/s Pa m 14.0 3.36E-06 67 1.76E-06 434 7.14E-07 1509 4.34E-07 96 3.35E-06 634 1.80E-06 3463 8.71E-07 8171 5.02E-07 860 4.35E-06 3923 1.93E-06 4274 3.72E-06 18657 1.56E-06 12.3 3.08E-06 84 1.28E-06 491 6.96E-07 8792 1.62E-07 100 2.98E-06 694 1.54E-06 3859 7.56E-07 29427 1.93E-07 873 4.21E-06 3964 1.85E-06 3876 3.66E-06 18406 1.56E-06 Table 2: Rate and CO2 Partial Pressure Data for 2, 5, 8, and 12 m PZ PZ m 2 5 Temp CO2 Ldg mol/molalk C 0.240 0.316 40 0.352 0.411 0.240 0.316 60 0.352 0.411 0.239 80 0.324 0.239 100 0.324 0.226 0.299 40 0.354 0.402 0.226 0.299 60 0.354 0.402 0.238 80 0.321 0.238 100 0.321 P*CO2 k g' . . 2 Pa mol/s Pa m 96 3.32E-06 499 2.04E-06 1305 1.39E-06 7127 5.55E-07 559 3.33E-06 2541 2.06E-06 5593 1.38E-06 25378 3.84E-07 2492 3.34E-06 12260 1.32E-06 9569 2.40E-06 39286 9.12E-07 65 4.39E-06 346 2.57E-06 1120 1.69E-06 4563 7.93E-07 385 4.75E-06 1814 2.62E-06 5021 1.80E-06 17233 6.59E-07 2192 4.67E-06 9699 1.91E-06 8888 3.52E-06 36960 1.02E-06 PZ m 8 12 Temp CO2 Ldg mol/molalk C 0.231 0.305 40 0.360 0.404 0.231 0.305 60 0.360 0.404 0.253 80 0.289 0.253 100 0.289 0.231 60 0.289 0.354 0.222 80 0.290 0.222 100 0.290 P*CO2 k g' . . 2 Pa mol/s Pa m 68 4.27E-06 530 1.98E-06 1409 1.14E-06 8153 3.53E-07 430 4.41E-06 2407 2.02E-06 7454 9.57E-07 30783 3.20E-07 3255 3.61E-06 9406 1.97E-06 13605 2.18E-06 32033 1.20E-06 331 4.19E-06 1865 1.85E-06 6791 7.73E-07 2115 4.24E-06 9141 1.48E-06 7871 3.78E-06 33652 8.30E-07 Table 3: Rate and CO2 Partial Pressure Data for 7 m MEA/2 m PZ MEA PZ m m 7 2 Temp CO2 Ldg mol/molalk C 0.242 0.333 40 0.416 0.477 0.242 0.333 60 0.416 0.477 0.242 80 0.333 0.242 100 0.333 36 P*CO2 Pa 27 166 1425 7418 178 1256 7122 33704 1138 6174 4340 26571 k g' . . 2 mol/s Pa m 3.45E-06 1.96E-06 8.76E-07 4.32E-07 4.00E-06 2.03E-06 9.08E-07 3.75E-07 4.29E-06 2.12E-06 4.83E-06 1.23E-06 1000000 100000 Open Points – Hilliard (2008) – 3.5, 7, 11 m MEA Dashes – Jou (1995) – 7 m MEA Filled Points – Current Work – 7, 9, 11, 13 m MEA PCO2* (Pa) 10000 1000 100 10 100˚C 80˚C 60˚C 40˚C 1 0.05 0.15 0.25 0.35 0.45 0.55 CO2 Loading (mol/molalk) Figure 1: CO2 Partial Pressure for Monoethanolamine Solutions at 40, 60, 80, and 100 ˚C The current work matches the Hilliard (2008) data very well at 40 and 60 ˚C below 0.45 loading. Above 0.45 loading, the current work shows an increase in the CO2 partial pressure which seems to be amine concentration dependent. The current work also matches the 40, 60, 80, and 100 ˚C data collected by Jou (1995). 1000000 Open Points – Hilliard (2008) – 0.9, 2, 2.5 3.6, 5 m PZ Dashes – Ermatchkov (2006) – 1-4.2 m PZ Filled Points – Current Work – 2, 5, 8, 12 m PZ PCO2* (Pa) 100000 10000 100 ˚C 1000 80 ˚C 100 60 ˚C 40 ˚C 10 0.10 0.15 0.20 0.25 0.30 0.35 0.40 0.45 CO2 Loading (mol/molalk) Figure 2: CO2 Partial Pressure for Piperazine Solutions at 40, 60, 80, 100 ˚C The current PZ CO2 partial pressure measurements match 40 and 60 ˚C data obtained by Hilliard (2008). The current work also agrees with 40, 80, and 100 ˚C CO2 partial pressure data collected by Ermatchkov (2006). Only Hilliard (2008) provides CO2 partial pressure data for 7 m MEA/2 m PZ solutions. Figure 3 shows a very good agreement between the current work and Hilliard (2008). 37 100000 Open Points – Hilliard (2008) – 7 m MEA/2 m PZ Filled Points – Current Work – 7 m MEA/2 m PZ 10000 PCO2* (Pa) 100˚C 1000 100 80˚C 60˚C 10 40˚C 1 0.05 0.15 0.25 0.35 0.45 CO2 Loading (mol/molalk) Figure 3: CO2 Partial Pressure for 7 m MEA/2 m Piperazine Solutions at 40, 60, 80, and 100 ˚C CO2 Absorption/Desorption Rates Wetted wall column obtained rates are shown in Figure 4 and 5 for MEA and PZ, respectively. 1.0E-05 60˚C 80˚C 100 ˚C . . 2 kg' (mol/s Pa m ) 40˚C 1.0E-06 7 m MEA 11 m MEA 9 m MEA 13 m MEA 1.0E-07 10 100 1000 10000 100000 P*CO2 (Pa) Figure 4: CO2 Absorption/Desorption Rates for 7, 9, 11, and 13 m MEA at 40, 60, 80, and 100 ˚C. 38 kg' (mol/s.Pa.m2) 1.0E-05 40 ˚C 60 ˚C 80 ˚C 100 ˚C 1.0E-06 8 m PZ 5 m PZ 12 m PZ 2 m PZ 1.0E-07 10 100 1000 10000 100000 P*CO2 (Pa) Figure 5: CO2 Absorption/Desorption Rates for 2, 5, 8, and 12 m PZ at 40, 60, 80, and 100 ˚C. Figures 4 and 5 both show clear trend lines for each temperature. The liquid film mass transfer coefficient, kg’, can drop by a factor of more than 10 from lean to rich conditions. Much of this drop is explained by the decrease of free amine in rich solutions. The total amine concentration does not seem to significantly affect the measured mass transfer coefficient, kg’, for either the MEA or PZ solutions. Generally, the temperature does not significantly affect kg’ either. Higher temperatures in Figures 4 and 5 cause an increase in the equilibrium partial pressure. Higher temperatures do not cause a significant change in the kg’ values. If Figures 4 and 5 were plotted against CO2 loading or the equilibrium partial pressure at a given temperature, the temperature dependence of Figures 4 and 5 could be removed. Figure 6 changes the x-axis to the equilibrium partial pressure at 40 ˚C. This is similar to plotting on a loading basis but allows for the comparison of different amines. The 7 m MEA/2 m PZ data as well as all the MEA and PZ data are shown in Figure 6. 39 kg' (mol/s.Pa.m2) 1E-05 40˚C 60˚C 80˚C 100˚C 1E-06 Open Points – 7, 9, 11, 13 m MEA Filled Points – 2, 5, 8 m PZ X’s – 7 m MEA/2 m PZ 1E-07 10 100 1000 P*CO2 @ 40C (Pa) 10000 Figure 6: Absorption/Desorption Rates for CO2 in MEA and PZ Solutions Plotted Versus the Equilibrium Partial Pressure at 40 ˚C Figure 6 shows that at a given partial pressure, piperazine always has a faster kg’ than MEA. Generally, piperazine solutions seem to absorb CO2 2–3 times faster than MEA. None of the MEA kg’ values show significant deviations with changes in temperature. PZ sometimes does show significant deviations in kg’ with temperature, particularly at higher temperature and loading (equilibrium partial pressure). Recall the x-axis in Figure 6 is indicative of increasing CO2 loading. Rich CO2 loaded solutions of PZ start to show slightly smaller kg’ values at 60 ˚C than 40 ˚C. 80 ˚C PZ data show kg’ values below those of 60 ˚C at intermediate CO2 loadings. The 100 ˚C PZ kg’ data falls below the 80 ˚C data even for the leanest loading. At intermediate loading the 100 ˚C PZ rates are significantly lower than at lower temperature. This effect is likely due to diffusion of reactants and products to and from the interface. At both higher temperature and loading, wetted wall column experiments require larger CO2 fluxes and accumulation at the interface is more likely to be significant. This effect is likely more apparent in the PZ solutions because the PZ solutions are both faster reacting and more viscous than MEA. Increasing viscosity results in decreasing diffusion coefficients for the solution. However, viscosity increases drastically with increasing PZ concentration. Oddly, Figure 6 does not show an indication of the 8 m PZ kg’ values dropping more than 2 m PZ values, as would be expected if diffusion coefficients were limiting. This may suggest that the increased viscosity is offset by the increased free amine concentration of the more concentrated solutions. Diffusion 40 resistances on CO2 mass transfer in the wetted wall column will be explored more fully in upcoming modeling work. The 7 m MEA/2 m PZ data show rates that are slightly below those of PZ. This is not surprising since PZ is the dominant amine reacting with CO2 in the solution. The 7 m MEA/2 m PZ data exhibit slight signs of diffusion resistance in the wetted wall column. The 100 ˚C data point at the intermediate loading is significantly lower than the 40, 60, and 80 ˚C kg’ values. Conclusions Piperazine solutions absorb CO2 2–3 times faster than MEA solutions. 7 m MEA/2 m PZ solutions absorb CO2 faster than MEA but slightly slower than PZ solutions. Piperazine and MEA liquid film mass transfer coefficients, kg’, can vary by a factor of more than 10 over typical lean and rich CO2 loading conditions. Even though kg’ contains both temperature and concentration dependent terms, kg’ values are generally not affected by changes in temperature or amine concentration. At some conditions for PZ solutions, and to a lesser degree for 7 m MEA/2 m PZ, the solutions exhibit signs of instantaneous reaction. PZ experiments at high CO2 loading (low free amine concentration) and high temperature were operating with high CO2 fluxes. These experiments seem to be somewhat diffusion-limited. These apparent diffusion limitations in the wetted wall column have not been observed for MEA solutions. References Ermatchkov V, Perez-Salado Kamps A, et al. Solubility of Carbon Dioxide in Aqueous Solutions of Piperazine in the Low Gas Loading Region. J Chem Eng Data. 2006;51(5):1788– 1796. Hilliard M. A Predictive Thermodynamic Model for an Aqueous Blend of Potassium Carbonate, Piperazine, and Monoethanolamine for Carbon Dioxide Capture from Flue Gas, The University of Texas at Austin. Ph.D. Dissertation. 2008;1025. Jou F-Y, Mather AE et al. The Solubility of CO2 in a 30 Mass Percent Monoethanolamine Solution. Can J Chem Eng. 1995;73(1):140–147. Rochelle GT, Dugas R et al. CO2 Capture by Aqueous Absorption. 2nd Quarterly Report 2008. http://www.che.utexas.edu/rochelle_group/Pubs/2nd_quarterly_report_2008.pdf. Rochelle GT, Dugas R et al. CO2 Capture by Aqueous Absorption. 3rd Quarterly Report 2008. http://www.che.utexas.edu/rochelle_group/Pubs/Rochelle_Research_Report_Q3_2008.pd f. 41 Analysis of Degraded Piperazine Solutions that Result in Foaming Quarterly Report for October 1 – December 31, 2008 by Xi Chen Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 3, 2009 Abstract NMR and mass spectroscopy (MS) analysis were conducted on PZ solutions with and without formaldehyde added and an oxidized PZ solution. to identify contaminants that account for increased foaming tendency of degraded PZ solutions. Most peaks in the NMR spectrum were tentatively interpreted and correlated to different molecules. There were no additional peaks found in foaming solution that had been oxidized for two weeks. For another degraded PZ sample, NMR peaks with the same position were found as seen in formaldehyde-added PZ, which may indicate that formaldehyde is the cause of the increased foaming tendency. MS analysis seems not to render useful information on contaminants. Introduction In the previous report, formaldehyde (HCHO) was reported to greatly increase the foaming tendency of undegraded piperazine solutions. It was also found that degraded piperazine solutions have a serious foaming problem. Since HCHO is regarded as an important intermediate oxidation product of piperazine, it was hypothized that HCHO causes foaming of oxidatively degraded piperazine. To verify this hypothesis, 1H &13C NMR and MS analysis was performed on formaldehyde-added PZ samples as well as oxidized ones in order to identify different species derived from HCHO. All the spectra were documented in this report and will be used as a reference for future work. Experimental Materials The following materials were used in this study: PZ (anhydrous, 99%, Alfa Aesar); carbon dioxide (Coleman Intrument, 99.99%, Matheson); Deuterium oxide (≥99.9%, Cambridge Isotopes); DSS (sodium 2,2-Dimethyl-2-silapentane-5-Sulfonate, 97%, Aldrich), deionized water (Millipore, Direct-Q). Preparation of NMR samples DSS was used as a reference for the NMR analysis. A solution of D2O/DSS = 50/1 (w/w) was prepared beforehand. Then 0.5g of the D2O/DSS solution was added to a 3.5g sample under well 42 mixing. After that, experimental samples (approximately 1mL for each) were transferred into yellow top NMR sample tubes (5.00mm O.D. x 0.77 mm I.D. x 7 in. length, 300 mHz, WILMAD Labglass) and was submitted for analysis to the NMR laboratory at the Department of Chemistry and Biochemistry, the University of Texas at Austin. Mass spectroscopy analysis Samples were diluted 50 times before being injected with a needle pump into a mass spectrometer (Thermo Finnigan TSQ) located at the Department of Civil Engineering, the University of Texas at Austin. The injection rate was set at 50 ul/min. Results and Discussion Molecular structure of different PZ species present in loaded PZ solution is illustrated in 1. a) 3H H H3 H2 C3 C2 N+ H 3H b) H2 N C3 C2 H3 H2 OC22 H2 O c) Figure1: Molecular structure and active nuclei of protons and carbons associated with a) PZ/PZH+, b)PZCOO- /H+PZCOO-, c) PZ(COO-)2. Different types of nucleus are labeled with numbers to distinguish them. Figures 2 and 3 show 1H NMR and 13C NMR spectrum for 2 m PZ without CO2 loading respectively. The peaks are labeled with number of the corresponding nucleus. (NMR peak positions and areas are summarized in the Appendix for all samples anaylized in this study.) 43 water H1 Figure 2: 1H NMR Spectrum of 2 m PZ, α=0 C1 Figure 3: 13C NMR Spectrum of 2 m PZ, α=0 44 As formaldehyde is added to PZ solutions, the speciation becomes complicate. Figure 4 shows some of the possible molecular structures of products that could form as HCHO reacted with PZ as well as PZ carbamate. 45 Figure 4: Molecular structure and active nuclei of protons and carbons associated with products of CO2 loaded PZ and formaldehyde. Two samples of unloaded 2 m PZ containing 325 mM and 743 mM HCHO were analyzed and the spectra were shown in Figures 5–17. The sample with 325 mM HCHO added is still clean solution while the one with 743 mM HCHO is milky white. As the ratio of HCHO to PZ is increased, the amount of pentamers (the last one in Figure 4) was expected to increase. In the spectrum for these two samples, apparently the peaks whose area increases with HCHO concentration are associated with HCHO, methanol contained in HCHO solution or products of reactions that HCHO participated. Figure 5: 1H NMR Spectrum of 2 m PZ, α=0, [HCHO]=325mM (0–10 ppm) 46 Methanol H10 H29 Figure 6: 1H NMR Spectrum of 2 m PZ, α=0, [HCHO]=325mM (2.97–3.44 ppm) H1 H9 H8 Figure 7: 1H NMR Spectrum of 2 m PZ, α=0, [HCHO]=325mM (2.40–2.91 ppm) 47 Figure 8: 13C NMR Spectrum of 2 m PZ, α=0, [HCHO]=325mM (0–200 ppm) C1 C2 Figure 9: 13 C NMR Spectrum of 2 m PZ, α=0, [HCHO]=325mM (80.7–84.8 ppm) 48 C1 C9, C28 C8, C27 C3 Methanol Figure 10: 13C NMR Spectrum of 2 m PZ, α=0, [HCHO]=325mM (45.0–55.6 ppm) Figure 11: 2-D correlation NMR Spectrum of 2 m PZ, α=0, [HCHO]=325mM 49 Figure 12: 1H NMR Spectrum of 2 m PZ, α=0, [HCHO]=743mM (0–10 ppm) H1 H9, H8, Methanol H10 H30 H29 Figure 13: 1H NMR Spectrum of 2 m PZ, α=0, [HCHO]=743mM (2.0–3.6 ppm) 50 Figure 14: 13C NMR Spectrum of 2 m PZ, α=0, [HCHO]=743mM (0–200 ppm) C10 C29 Figure 15: 13C NMR Spectrum of 2 m PZ, α=0, [HCHO]=743mM (81.2–85.6 ppm) 51 C1 C8, C27 C9, C28 C30 Methanol Figure 16: 13C NMR Spectrum of 2 m PZ, α=0, [HCHO]=743mM (45.6–55.2 ppm) Figure 17: 2-D correlation NMR Spectrum of 2 m PZ, α=0, [HCHO]=743mM 52 The spectra for 8 m PZ with 0.3 loading are shown in Figures 18–23. Figure 18: 1H NMR Spectrum of 8 m PZ, α=0.3 (0–10 ppm) H H H H Figure 19: 1H NMR Spectrum of 8 m PZ, α=0.3 (0–3.7 ppm) 53 Figure 20: 13C NMR Spectrum of 8 m PZ, α=0.3 (0–220 ppm) C22 C23 Figure 21: 13C NMR Spectrum of 8 m PZ, α=0.3 (163.2–166.2 ppm) 54 C1 C2 C3 C4 Figure 22: 13C NMR Spectrum of 8 m PZ, α=0.3 (43.0–48.0 ppm) Figure 23: 2-D correlation NMR Spectrum of 8 m PZ, α=0.3 55 367 mM HCHO was added to loaded PZ solutions and the spectra are shown in Figures 24–31. Figure 24: 1H NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM (0–10 ppm) H1 H2 H3 H4 H17, H20 H18 H21 H19 H16 Figure 25: 1H NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM (2.4–3.6 ppm) 56 Figure 26: 13C NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM (0–200 ppm) C22 C23 C25 C26 Figure 27: 13C NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM (162.9–167.7 ppm) 57 C21 C18 Figure 28: 13C NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM (79.8–84.3 ppm) C19 C15 C16 Methanol C14 Figure 29: 13C NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM (50.2–55.0 ppm) C1 C2 C3 C4 C17, Figure 30: 13C NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM (43.8–47.7 ppm) 58 Figure 31: 2-D correlation NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM 59 Figure 32: 2-D correlation NMR Spectrum of 8 m PZ, α=0.3, [HCHO]=367mM Oxidatively degraded sample OE5 was used in this study for NMR and MS analysis. The foaming test showed that this sample had a much higher foaming tendency than undegraded PZ solution. Prior to the NMR analysis, 5 mM Na2S was added to the sample to precipitate Fe2+ and reduce interference with the NMR spectrum. The results are shown in Figures 33–37. 60 Figure 33: 1H NMR spectrum of degraded 8 m PZ, 1mM Fe2+, 55 °C, α=0.3, Fe2+ (OE5) was precipitated with 5mM Na2S before NMR analysis. (0–10 ppm) Figure 34: 1H NMR spectrum of degraded 8 m PZ, 1mM Fe2+, 55 °C, α=0.3, Fe2+ (OE5) was precipitated with 5mM Na2S before NMR analysis. (2.82–3.66 ppm) 61 Figure 35: 13C NMR spectrum of degraded 8 m PZ, 1mM Fe2+, 55 °C, α=0.3, Fe2+ (OE5) was precipitated with 5mM Na2S before NMR analysis. (0–200 ppm) Figure 36: 13C NMR spectrum of degraded 8 m PZ, 1mM Fe2+, 55 °C, α=0.3, Fe2+ (OE5) was precipitated with 5mM Na2S before NMR analysis. (44.1–47.4 ppm) 62 Figure 37: 2-D correlation NMR spectrum of degraded 8 m PZ, 1mM Fe2+, 55 °C, α=0.3, Fe2+ (OE5) was precipitated with 5mM Na2S before NMR analysis. Unexpectedly, there were no interesting additional peaks found for OE5. All the major peaks that show up in the spectrum have the same peak position as those for the neat loaded PZ solution. Because OE5 is only degraded for three days, the oxidation products might be present in such a small concentration that although they can significantly change the foaming of the amine solution, they are not able to give rise to significant peaks on NMR spectrum. Sexton had also done much NMR analysis for degraded PZ samples. The spectrum he got for degraded PZ solution was used here for comparison with the ones shown above. Figures 38–42 give the 1H NMR spectrum of degraded 2.5 m PZ with the presence of 5mM V. Those peaks which have same position as seen in Figure 25 are pinpointed with red arrows. Same peaks indicate that the degraded sample may have similar species as formaldehyde-added PZ. There are many small peaks in the 13C NMR spectrum for this degraded sample and they are hard to interpret. 63 Figure 38: 1H NMR spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM (0– 10 ppm) Figure 39: 1H NMR spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM (5.3–8.5 ppm) 64 Figure 40: 1H NMR spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM (3.64–4.36ppm) EDA H1 H3 H2 Figure 41: 1H NMR spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM (2.56–3.52 ppm) 65 Figure 42: 1H NMR spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM (0– 2.54 ppm) Figure 43: 13C NMR spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM (0–200 ppm) 66 Figure 44: 13C NMR spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM (39–51 ppm) Figure 45: 13C NMR spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM (162–178 ppm) 67 Figure 46: 2-D correlation spectrum of degraded 2.5 m PZ, 5 mM V, 55 oC, α=0.30, 1400 RPM. Mass spectroscopy was also tried to identify contaminants in the degraded sample OE4. Figures 47, 48, and 49 show the MS spectrum for neat PZ, degraded PZ solutions (OE4), and HCHOadded PZ, respectively. For all 3 samples, the only two peaks that consistently showed up in the spectrum were m/z=87 and m/z=70. Apparently the m/z=87 is corresponding to PZ. Which species the peak of m/z=70 corresponds to is still unknown at this point. The fact that here are no extra significant peaks showing up in the MS spectrum for the degraded sample might be attributed to the detection limit of MS apparatus in this study. 68 Figure 47: Mass spectrum of 8 m PZ, α=0.3 69 Figure 48: Mass spectrum of oxidized PZ solution(OE4, 8 m PZ, α=0.3, 5.0 mM Cu2+, 0.1 mM Fe2+, 100 mM “A”, degraded at 55 °C for ~4 weeks). 70 Figure 49: Mass spectrum of 8 m PZ, α=0.3, [HCHO]=270 mM Summary Speciation in formaldehyde-added PZ aqueous solution was studied by 1H, 13C and 1H-13C HSQC NMR analysis as well as mass spectroscopy. Trimer and pentamer, which are produced by condensation reaction between PZ and HCHO, were proposed to be the major new species on addition of HCHO to PZ. 2.5 m PZ degraded with the presence of 5mM V was found to have similar NMR peaks as seen in HCHO-added PZ, indicating that HCHO could be an important intermediate oxidation product and the cause of increased foaming tendency. 71 Influence of Viscosity and Surface Tension on the Effective Mass Transfer Area of Structured Packing Quarterly Report for October 1 – December 31, 2008 by Robert Tsai Supported by the Luminant Carbon Management Program Department of Chemical Engineering The University of Texas at Austin January 5, 2009 Abstract Two structured packings were evaluated: a prototype 500-series packing (P500) (ap = 500 m2/m3) and Sulzer Mellapak 2Y (M2Y) (ap = 205 m2/m3). P500 exhibited higher pressure drops (15%) than Mellapak 500Y (M500Y), a geometrically similar packing, under dry conditions and for liquid loads up to 10 gpm/ft2. At higher loads, the pressure drop behavior of the two packings became less distinct. In contrast, hold-ups for P500 were consistently lower for all investigated liquid loads (up to 20 gpm/ft2). Base case and low surface tension (σ ~ 30 dynes/cm) mass transfer tests with P500 yielded results comparable to those obtained with M500Y. The reduction in surface tension increased the effective mass transfer area, although the effect was not as pronounced as with M500Y. M2Y was compared against Mellapak 250Y (M250Y), which had a comparable albeit slightly higher geometric area (ap = 250 m2/m3). M2Y displayed lower pressure drops by about 20–25% under both dry and irrigated (up to 30 gpm/ft2) conditions. Hold-up values were comparable to M250Y. M2Y also appeared to have similar fractional areas as M250Y, despite being a coarser packing. The mass transfer area database was updated to incorporate P500 and M2Y. The current global (ae/ap) correlation, able to represent the entire database within limits of ±15%, is as follows: [ ae −1 = 1.341 (WeL )(FrL ) 3 ap ] 0.117 Introduction Packing is commonly used in industrial processes as a means of promoting efficient gas-liquid contact. One important application for which packed columns are being considered is treating flue gas for CO2 capture. The conventional method consists of an aqueous amine solvent such as monoethanolamine (MEA) contacting the gas, resulting in the absorption of CO2 (Kohl and Nielsen, 1997). The enriched solvent is sent to a stripper for regeneration and is then recycled back to the absorber. Gas-liquid contact in both the absorber and stripper is enhanced through the use of packing. 72 Reliable mass transfer models are necessary for design and analysis purposes. A critical factor involved in modeling is the prediction of the effective interfacial area of packing (ae), which can be considered as the total gas-liquid contact area that is actively available for mass transfer. The current research effort is focused on this parameter. Characterization of effective areas is vital to amine-based CO2 capture at the industrial level, because absorption rates actually become independent of conventional mass transfer coefficients (kG or kL0) but remain directly proportional to the effective area. Thus, it is highly desirable to have an accurate area model. Numerous packing area correlations have been presented in the literature, but none has been shown to be predictive over a wide range of conditions. The Rocha-Bravo-Fair (Rocha et al., 1996) and Billet-Schultes (Billet and Schultes, 1993) models, two of the more widely used correlations for structured packing, seem to be notably poor in their predictions involving aqueous systems. Wang et al. (2005) performed a comprehensive review of the available models. The various correlations predict different and sometimes even contradictory effects of liquid viscosity and surface tension, properties that would be expected to fundamentally influence the wetted area of packing. It is evident that their role is not well understood, and there is a definite need for work on this subject matter. The Separations Research Program (SRP) at the University of Texas at Austin has the capability of measuring packing mass transfer areas. Measurements are performed by absorbing CO2 from air with 0.1 M NaOH in a 427 mm (16.8 in) ID column. Unfortunately, physical parameters are limited to those of water, making it potentially inaccurate to extend these results to other fluids of interest, such as amine solvents, due to the differences in viscosity and surface tension. Limited understanding of the fluid mechanics and mass transfer phenomena in packed columns has been noted, and the need for experiments over a broader range of conditions has been identified (Wang et al., 2005). The goal of this research is to address these shortcomings and ultimately develop an improved effective area model for structured packing. The general objectives are to: • develop a fundamental understanding of the fluid mechanics associated with structured packing operation; • determine suitable chemical reagents to modify the surface tension and viscosity of the aqueous caustic solutions employed to make packing area measurements, and characterize potential impacts of such additives on the CO2-NaOH reaction kinetics; • expand the SRP database by measuring the hydraulic performance and mass transfer areas of several different structured packings over a range of liquid viscosities and surface tensions; • combine the data and theory into a semi-empirical model that captures the features of the tested systems and adequately represents effective area as a function of viscosity, surface tension, and liquid load. 73 Experimental 427 mm ID Packed Column The packed column had an outside diameter of 460 mm (18 in), inside diameter of 427 mm (16.8 in), and a 3 m (10 ft) packed height. For details regarding the apparatus and procedure for mass transfer or hydraulic tests, earlier quarterly reports may be consulted. Wetted Wall Column (WWC) The wetted wall column (WWC) was a vapor-liquid contactor with a known interfacial area (38.52 cm2) that was used for kinetic measurements. The associated equipment and experimental protocol are archived elsewhere (e.g. Q3 2006 report). Goniometer The goniometer (ramé-hart Inc., Model #100-00) included an adjustable stage, a syringe support arm, a computer-linked camera for live image display, and a light source (see Q3 2006 report). This system was used in conjunction with FTA32 Video 2.0 software (developed by First Ten Angstroms, Inc.) to make surface tension measurements via the pendant drop method. Rheometer The Physica MCR 300 rheometer (Anton Paar, USA) employed for viscosity measurements was first described in the Q4 2006 report. The apparatus was equipped with a cone-plate spindle (CP 50-1). Temperature was regulated (±0.1 °C) with a Peltier unit (TEK 150P-C) and a Julabo F25 water bath unit (for counter-cooling). Measurement profiles consisted of a logarithmically increased or decreased shear rate (100 to 500 s-1), with 10–20 data points recorded at 15 second intervals. Viscosity was determined from a plot of shear stress (measured) vs. shear rate. Materials A nonionic surfactant, TergitolTM NP-7 (Dow), was used to reduce the surface tension of solutions. POLYOX WSR N750 (Dow) – essentially, poly(ethylene oxide) with a molecular weight of 300,000 – was employed as a viscosity enhancer. Dow Corning® Q2-3183A antifoam was used for foam suppression. Results and Discussion Theoretical Analysis of Data The performance of both the WWC and the packed column was modeled by series resistance (equation 1). The overall mass transfer resistance is the sum of the gas- and liquid-side resistances. 1 1 1 = + K G kG kg ' (1) For the WWC, the overall mass transfer coefficient (KG) was determined from the CO2 flux and the partial pressure driving force. The gas-side mass transfer coefficient (kG) was a function of physical properties and was calculated using a correlation that was developed by absorption of SO2 into NaOH solution, an entirely gas-side controlled process (Bishnoi, 2000). KG and kG 74 were used to calculate kg′, which has been defined as a liquid-side mass transfer coefficient expressed in terms of a CO2 partial pressure driving force. ( ) i * i N CO 2 = k g′ PCO − PCO = k g′ PCO 2 2 2 (2) In equation 2, P*CO2 is zero because of the irreversibility of the CO2-NaOH reaction. Under the assumption of pseudo-first-order conditions, surface renewal theory may be used to present the flux as (Danckwerts, 1970): N CO 2 = k L0 1 + i k1 D CO 2 , L PCO 2 (k ) 0 2 L H CO 2 = k L0 1 + Ha 2 i PCO 2 H CO 2 (3) Ha2 was on the order of 102 in the WWC, so equation 3 was simplified to: N CO 2 = k1 DCO 2 , L H CO 2 i PCO 2 (4) Thus, from equations 2 and 4, we have: k = ' g k1 DCO 2 , L H CO 2 = k OH − [OH − ]DCO 2 , L (5) H CO 2 Measured kg′ values were compared with calculated ones, evaluated using literature values for the terms in equation 5. The correlations for the diffusion coefficient (DCO2,L), the Henry’s constant (HCO2), and the reaction rate constant (kOH-) were based on the work of Pohorecki and Moniuk (1988). For the packed column experiment, gas-side resistance was intentionally limited by using dilute caustic solution (0.1 M) and operating at high superficial air velocities (0.6, 1.0, or 1.5 m/s). This resistance was estimated to account for 1% of the overall mass transfer resistance, with kG calculated from the correlation of Rocha et al. (1996). The 1/kG term in equation 1 was ignored, and KG was assumed to be equal to kg′. This approximation enabled the effective area (ae) to be determined by separating it from the volumetric mass transfer coefficient, KGae. ⎛ y CO 2 in u G ln⎜ ⎜ y CO out 2 ⎝ ae = Z K G RT ⎞ ⎛ y ⎟ u G ln⎜ CO 2 in ⎟ ⎜ y CO out 2 ⎠ ≈ ⎝ Z k g' RT ⎞ ⎛ y ⎟ u G ln⎜ CO 2 in ⎟ ⎜ y CO out 2 ⎠= ⎝ ZRT ⎞ ⎟ ⎟ ⎠⋅ H CO 2 k OH − [OH − ]DCO 2 ,L (6) kg′ was calculated with equation 5, although the Ha2 >> 1 approximation was weaker in these experiments. Ha2 was around 15 in the worst case scenario, with kL0 estimated from the correlation of Rocha et al. (1996). Wetted Wall Column (WWC) The purpose of the WWC was two-fold. First, it served to verify the correlations (Pohorecki and Moniuk, 1988) employed to interpret the packed column mass transfer data (kg′). Second, it was utilized to evaluate the impact of additives (NP-7 or POLYOX) on the CO2-NaOH kinetics. The data from these studies have been discussed in other quarterly reports. In short, the correlations 75 of Pohorecki and Moniuk have been deemed to be acceptable. The systems containing additives have been concluded to require no modification (surfactant) or a small modification (POLYOX) to kg′. Prototype 500-Series Packing (P500) – Mass Transfer The prototype 500-series packing (P500) and Mellapak 500Y (M500Y) were similar; both had textured, perforated surfaces and were assumed to have a geometric area of 500 m2/m3. Figure 1 compares the base case and low surface tension (σ ~ 30 dynes/cm) mass transfer area data for the two packings. 1 Fractional area, ae/ap 0.9 0.8 0.7 M500Y - Baseline 0.6 M500Y - 30 dynes/cm P500 - Baseline 0.5 P500 - 30 dynes/cm 0.4 0 5 10 15 20 Liquid load (gpm/ft2) Figure 1: 500-series packing (ap = 500 m2/m3) mass transfer area data A comparison of the baseline data sets suggests the performance of P500 to be marginally better than that of M500Y, although it should be noted that the difference is on the order of the anticipated experimental error (~10%). The low surface tension data sets, for the most part, overlay cleanly. There is some deviation at the lower liquid loads, but this could perhaps be due more to the inherent control/accuracy issues associated with low-flow measurements than with actual physical phenomena. On average, the reduction in surface tension resulted in a 15% increase in effective area for P500 – noticeable, but not quite as significant as the effect observed with M500Y (20–30%). It is possible that there is some feature of P500 that gives it a slight edge over M500Y – for instance, a different surface texture that lessens liquid bridging and pooling within the packing structure, phenomena that could be detrimental to the wetted area (Tsai et al., 2008). Lowering the surface tension may alleviate this problem but only to a certain, maximum extent. This would explain why analogous results were obtained at 30 dynes/cm but not for the base case. 76 Prototype 500-Series Packing (P500) – Hydraulics Dry pressure drop data for P500 are shown in Figure 2, plotted against several replicated M500Y data sets. The results have been normalized by equation 7, a simple power law expression obtained from a regression of all of the dry M500Y data; this was done to exaggerate any differences between the two packings. ΔPdry,M500Y Z (7) M500Y - Dry (0625AWH) Dry (0806AWH) Dry (0815AWH) Dry (0817AWH) Dry (0819AWH) P500 - Dry (0829AWH) 1.3 ΔP / ΔPdry, M500Y = 0.598F 1.832 1.2 1.1 1 0.9 0.5 1.5 2.5 F-factor (Pa)0.5 3.5 4.5 Figure 2: Dry pressure drop data for M500Y and P500 Dry pressure drops were 15% higher for P500. It is not clear why such a significant difference was observed, given the supposed similarity between M500Y and P500. Pressure drop has been observed to scale with specific area, so it might be conceivable that P500, in reality, has inherently more area than its counterpart. The areas would probably need to differ by about the same ratio (15%) though, which seems unlikely, considering that the two packings are intended to be directly competitive. This would raise questions about the mass transfer results (Figure 1) as well. This 15% deviation (in the pre-loading region) persisted when water was introduced, up to loads of 10 gpm/ft2 and higher. Around this point, the P500 pressure drop behavior became less consistent, either matching M500Y (within < 10%) or in a few instances, even exhibiting lower pressure drops than M500Y (as much as 30%). This could have been a consequence of the packing channels becoming filled with water. That is, for high-area packings like M500Y and P500, liquid might start to occupy enough volume at 10 gpm/ft2 and above to make certain packing-specific characteristics (i.e. contour or texture) irrelevant. This would explain the trend toward comparable pressure drop behavior. The points displaying lower pressure drops are suspected to be aberrations, possibly related to the greater difficulty associated with controlling and reproducing upper-capacity-limit hydraulic measurements. 77 Hold-up data (water) for M500Y and P500 are presented in Figure 3. The results are displayed on a differential basis, where each measured fractional hold-up (hL) has had a baseline value (calculated from an average of the M500Y hold-up(s) at the corresponding liquid load) subtracted from it. 0.005 Differential hL 0 -0.005 f-factor ~ 0.7 (Pa)0.5 M500Y -0.01 P500 -0.015 -0.02 0 5 10 15 Liquid load (gpm/ft2) 20 25 Figure 3: Hold-up data for M500Y and P500. F-factor was low (0.7 Pa0.5) to ensure data were within the pre-loading region. The hold-ups for P500 were clearly lower than those for M500Y. This would certainly make sense if paired with lower pressure drops as well, but recall that greater pressure drops were, in fact, observed. Hence, the P500 hold-up and pressure drop data do not seem to be related. Interestingly, though, there may be some correspondence with the mass transfer results. Low surface tension tests with M500Y (performed earlier) exhibited higher effective areas and marginally depressed hold-ups relative to base case conditions; both results were attributed to an alleviation of internal liquid bridging and pooling. Since P500 displayed slightly better mass transfer performance than M500Y (Figure 1), we might expect it to have lower hold-ups for the same reasons. Mellapak 2Y – Mass Transfer Figure 4 compares the base case mass transfer area data for Mellapak 250Y (M250Y) and Mellapak 2Y( M2Y). 78 1.2 Fractional area, ae/ap 1.1 1 0.9 M250Y 0.8 M2Y 0.7 0.6 0 5 10 15 20 25 30 35 Liquid load (gpm/ft2) Figure 4: M250Y (ap = 250 m2/m3) and M2Y (ap = 205 m2/m3) mass transfer area data The measured fractional areas for M2Y were the same as for M250Y, despite the former having a lower surface area. One would anticipate a coarser (lower surface area) packing to exhibit a higher fractional area than a finer packing at a given liquid load, as observed in tests with M250Y, Flexipac 1Y (F1Y), and M500Y (ordered in terms of decreasing “efficiency”). M250Y is much closer in geometry to M2Y than to either F1Y or M500Y though, so even if M2Y technically should have followed the efficiency trend, this could have been completely lost in the experimental noise. It is also possible that M250Y represents a critical point, where “lost” surface area from liquid bridging and pooling is no longer a significant issue. While there is an advantage in going from M500Y to M250Y, for example, there may be no such benefit for going down further to M2Y, M125Y, etc. After all, it is hard to imagine fractional areas getting much higher than for M250Y. Very coarse geometries could even be detrimental, as complete wetting of the packing surface could be challenging at extreme limits (i.e. flat plates). Mellapak 2Y – Hydraulics Dry pressure drop data for M250Y and M2Y are shown in Figure 5. The results have been normalized by equation 8, obtained from a regression of all of the dry M250Y data. It should be noted that the pre-exponential coefficient in equation 8 is approximately half that in equation 7, corresponding to the geometric ratio between M500Y and M250Y. ΔPdry,M250Y Z = 0.309 F 1.856 79 (8) 1.1 ΔP / ΔPdry, M250Y 1.05 1 0.95 M250Y - Dry (0617AWH) Dry (0715AWH) Dry (0804AWH) Dry (0821AWH) Dry (0823AWH) M2Y - Dry (0831AWH) 0.9 0.85 0.8 0.75 0.5 1 1.5 2 2.5 F-factor 3 (Pa)0.5 3.5 4 4.5 5 Figure 5: Dry pressure drop data for M250Y and M2Y Pressure drops were consistently lower for M2Y by 20–25%, which was somewhat expected, given that coarser geometries tend to imply more void space and fewer directional changes for the gas flow. This difference was maintained even when the packing was irrigated, up to a liquid load of 30 gpm/ft2. A comparison of the M250Y and M2Y hold-up data are presented in Figure 6. As with Figure 3, the results have been interpreted on a differential basis. M2Y, being coarser than M250Y, was anticipated to retain a smaller fraction of liquid, but the measured hold-ups were basically equivalent. As was postulated in the mass transfer section, it is conceivable that the geometric difference between the two packings was insufficient to have an experimentally discernable impact on the hold-up (even if one might actually exist). However, why this would be true for hold-up and not for pressure drop does not really make sense. 80 0.01 Differential hL 0.005 0 -0.005 M250Y M2Y f-factor ~ 0.7 (Pa)0.5 -0.01 0 5 10 15 20 25 30 35 Liquid load (gpm/ft2) Figure 6: Hold-up data for M250Y and M2Y. F-factor was low (0.7 Pa0.5) to ensure data were within the pre-loading region. Film Saturation Test Packing is commonly pre-wetted in industrial applications in order to maximize its efficiency; the intent is to create a surface conducive to liquid spreading (liquid-liquid contact instead of liquid-solid contact). In this body of research, pre-wetting has always been incorporated into the experimental protocol in an effort to mimic industry. However, it was recently taken into consideration that this practice could be resulting in falsely high effective areas, particularly for low liquid load measurements. Such would be the case if stagnant liquid (left behind from prewetting) were contributing to the CO2 removal. To investigate this, a simple experiment was conducted with M2Y in the column. The packing was pre-wet with 0.1 M NaOH solution at a liquid load of 25 gpm/ft2 for 10 minutes (standard protocol). The pump was then shut down, and liquid was allowed to drain from the packing for 15 minutes. The blower was turned on and set at 300 ACFM, and the steady-state approach of the outlet CO2 concentration was observed. Afterward, this same procedure was repeated, except with a drainage time of only about 5 minutes. Figure 7 displays the results from these tests. 81 Outlet CO2 concentration (ppm) 450 CO2 inlet (436 ppm) 1% approach 420 5% approach 390 Second test (5 mins drainage) 10% approach 360 330 First test (15 mins drainage) 300 0 5 10 15 20 Time (min) 25 30 35 40 Figure 7: Pre-wetting film saturation data. The inlet CO2 concentration was periodically confirmed, as reflected by gaps in the data (e.g. from 10–15 mins). Static hold-up is generally quite small in structured packing (Rocha et al., 1993), so it was not surprising that the two experiments gave similar results. In other words, the packing was expected to drain quickly. Much of the active hydroxide seemed to be consumed within the first 10 minutes; the outlet CO2 concentration was already within about 5% of the inlet concentration by this point. A 1% approach to the inlet concentration was achieved after about 20–25 minutes. Since the liquid circulation was completely shut off, all of the CO2 removal was attributable to the pre-wetting process. Therefore, these tests were representative of the maximum possible impact from pre-wetting. As such, it would appear there is no major need for concern, provided that sufficient time (20 minutes) is allowed for steady-state establishment when switching from a high liquid load to a low one. In past experiments, these conditions were typically given around 15 minutes to stabilize. Although this was perhaps slightly less than ideal, the data should still be trustworthy. In the future, about 20–25 minutes will be allotted to be on the safe side. 82 Mass Transfer Area Database Figure 8 shows the structured packing mass transfer area database, updated with P500 and M2Y. 1.2 +15% Fractional area, ae/ap 1.1 1 [ ae −1 = 1.341 (We L )(FrL ) 3 ap ] 0.117 M250Y - Baseline 30 dynes/cm 4 cP, 55 dynes/cm 14 cP, 40 dynes/cm M500Y - Baseline 30 dynes/cm 4 cP, 40 dynes/cm 10 cP, 40 dynes/cm F1Y - Baseline 6 cP, 60 dynes/cm P500 - Baseline 30 dynes/cm M2Y - Baseline 0.9 0.8 0.7 0.6 -15% 0.5 0.4 0.0001 0.001 0.01 0.1 1 (WeL)(FrL)-1/3 Figure 8: Structured packing mass transfer area database, compared with global correlation (equation 9). From a purely quantitative perspective, the global correlation (equation 9) still appears to fit all of the data quite well. The P500 data are more flattened out compared to the other data sets, which could perhaps be associated with specific features (e.g. surface texture) that the model fails to account for in its current form. The M2Y results closely parallel the baseline M250Y data – no major surprise, considering their similar geometries. It is worth mentioning that prior to M2Y (ap = 205 m2/m3), the database consisted of packings with areas ranging from 250 to 500 m2/m3. The fact that the M2Y data is consistent as well further validates the model. It also suggests that, to a certain extent, we can extrapolate beyond the geometric limits of the database without fear of some drastic discontinuity. [ ae −1 = 1.341 (WeL )(FrL ) 3 ap ] 0.117 4 ⎡ 3⎤ 1 ⎛ Q ⎞ ρ = 1.341⎢ L g 3 ⎜ ⎟ ⎥ ⎜L ⎟ ⎥ ⎢σ ⎝ p⎠ ⎦ ⎣ 0.117 (9) It should be noted that the definition of wetted perimeter (Lp) has been modified from previous versions of the correlation, in order to allow for more convenient generalization of equation 9 (i.e. with different-scale columns) and also to be consistent with other proposed flow models (Sidi-Boumedine and Raynal, 2005). This change did not qualitatively affect any conclusions (i.e. regarding surface tension or viscosity), but quantitatively, it did cause a uniform shift in the data. The wetted perimeter equation is presented below: Lp = 4S A Bh 83 (10) This definition envisions the adjacent triangular flow channels in the packing structure to form a diamond (Figure 9) and essentially assumes 1) complete wetting and, 2) both top-side and underside flow on the packing sheets, a feature apparently confirmed by the X-ray images of Green (2006). h B S Figure 9: Structured packing flow channel schematic For reference, Table 1 contains a compilation of the relevant geometric dimensions of several structured packings. Table 1: Structured packing parameters Packing Specific area, ap (m2/m3) 250 Channel side, S (mm) 17 Channel base, B (mm) 24.1 Crimp height, h (mm) 11.9 Petre et al. (2003) Mellapak 500Y (M500Y) 500 8.1 9.6 6.53 Aroonwilas (2001) Mellapak 2Y (M2Y) 205 21.5 33 13.8 Sulzer contact Direct measurement Flexipac 1Y (F1Y) 410 9 12.7 6.4 Koch contact Petre et al. (2003) Mellapak 250Y (M250Y) Source(s) Conclusions Hydraulic data for P500 and M2Y were obtained and compared against their closest respective counterparts: M500Y and M250Y. Pressure drops for P500 were greater than for M500Y by about 15%, up to a liquid load of 10 gpm/ft2. Hold-ups were consistently lower for P500. Pressure drops for M2Y were lower than for M250Y by 20–25%. Hold-ups were the same. The effective mass transfer areas of P500 and M2Y were measured via absorption of CO2 into caustic solution. P500 exhibited slightly higher areas (~10%) than M500Y. On a fractional area basis, M2Y yielded the same results as M250Y, despite being a coarser packing. The mass transfer area database (updated to include P500 and M2Y) was still represented well by the correlation that was regressed as a function of (WeL)(FrL)-1/3. The contribution of pre-wetting to mass transfer performance was found to become negligible after a period of about 20 minutes. Future experiments should take this into account to avoid potential misinterpretation of mass transfer results. 84 Future Work Hydraulic and mass transfer tests with several more structured packings are planned. These include: an untextured (smooth) version of M250Y, Mellapak 250X (60˚ corrugation angle), and MellapakPlus 252Y (modified joints). The current mass transfer model will be further refined by means of these additional data, as well as theoretical considerations. Nomenclature A = cross-sectional area of packed column, m2 ae = effective area of packing, m2/m3 ap = specific (geometric) area of packing, m2/m3 B = packing channel base, m DCO2 = diffusivity of CO2, m2/s F = gas flow factor, (m/s)(kg/m3)0.5 or Pa0.5 g = gravitational constant; 9.81 m/s2 HCO2 = Henry’s constant of CO2, m3·Pa/kmol h = packing crimp height, m hL = (total) liquid hold-up, m3/m3 KG = overall gas-side mass transfer coefficient, kmol/(m2·Pa·s) k1 = pseudo-first-order reaction rate constant, s-1 kG = gas-side mass transfer coefficient, kmol/(m2·Pa·s) kg′ = liquid-side mass transfer coefficient, kmol/(m2·Pa·s) kL0 = physical liquid-side mass transfer coefficient, m/s kOH- = second-order reaction rate constant, m3/(kmol·s) Lp = wetted perimeter in cross-sectional slice of packing, m NCO2 = molar flux of CO2, kmol/(m2·s) ΔP = pressure drop, Pa P*CO2 = equilibrium partial pressure of CO2, Pa PiCO2 = partial pressure of CO2 at gas-liquid interface, Pa Q = volumetric flow rate, m3/s R = ideal gas constant; 8314.5 (m3·Pa)/(kmol·K) S = packing channel side, m T = absolute temperature, K u = velocity, m/s yCO2 in/out = mole fraction of CO2 at inlet/outlet 85 Z = packed height, m Greek Symbols δ = film thickness, m μ = dynamic viscosity, kg/(m-s) ρ = density, kg/m3 σ = surface tension, N/m Subscripts G = gas phase L = liquid phase Dimensionless Groups af = fractional area of packing, ae/ap Fr = Froude number, Ha = Hatta number, We = Weber number, u2 gδ k1 DCO 2 ,L k L0 ρ u 2δ σ References Aroonwilas A. Mass-Transfer with Chemical Reaction in Structured Packing for CO2 Absorption Process. Ph.D. Thesis, University of Regina, Regina, Saskatchewan, 2001. Billet R, Schultes M. Predicting Mass Transfer in Packed Columns. Chem Eng Technol. 1993;16(1):1–9. Bishnoi S, Rochelle GT. Absorption of Carbon Dioxide into Aqueous Piperazine: Reaction Kinetics, Mass Transfer, and Solubility. Chem Eng Sci. 2000;55(22):5531–5543. Danckwerts PV. Gas-Liquid Reactions. McGraw-Hill: New York, 1970. Green CW. Hydraulic Characterization of Structured Packing via X-Ray Computed Tomography. Ph.D. Thesis, University of Texas at Austin. 2006. Kohl A, Nielsen R. Gas Purification; Gulf Publishing Co.: Houston, 1997. Petre CF, Larachi F, Iliuta I, Grandjean BPA. Pressure Drop through Structured Packings: Breakdown into the Contributing Mechanisms by CFD Modeling. Chem Eng Sci. 2003;58(1):163–177. 86 Pohorecki R, Moniuk W. Kinetics of Reaction between Carbon Dioxide and Hydroxyl Ions in Aqueous Electrolyte Solutions. Chem Eng Sci. 1988;43(7):1677–1684. Rocha JA, Bravo JL, Fair JR. Distillation Columns Containing Structured Packings: A Comprehensive Model for Their Performance. 1. Hydraulic Models. Ind Eng Chem Res. 1993;32(4):641–651. Rocha JA, Bravo JL, Fair JR. Distillation Columns Containing Structured Packings: A Comprehensive Model for Their Performance. 2. Mass-Transfer Model. Ind Eng Chem Res. 1996;35(5):1660–1667. Sidi-Boumedine R, Raynal L. Influence of the Viscosity on the Liquid Hold-up in Trickle-bed Reactors with Structured Packings. Catal Today. 2005;105:673–679. Tsai RE, Schultheiss P. Kettner A, Lewis JC, Seibert AF, Eldridge RB, Rochelle, GT. Influence of Surface Tension on Effective Packing Area. Ind Eng Chem Res. 2008;47(4):1253–1260. Yuan, XG, Yu, KT. Review of Mass-Transfer Correlations for Packed Columns. Ind Eng Chem Res. 2005;44(23):8715–8729. 87 Modeling Stripper Performance for CO2 Removal Progress Report for October 1 – December 31, 2008 by David Van Wagener Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 3, 2009 Abstract Previous simulations using the Hilliard (2008) piperazine (PZ) thermodynamic model produced unexpected results in the optimization process for 8 m PZ, and further investigation revealed that the heat capacity predictions are wildly inaccurate at high temperatures. Heat capacity data for 8 m PZ were not yet available, so values were extrapolated from 2.0 m PZ and 3.6 m PZ data sets. Additionally, the relationship between heat capacity and temperature is strongly linear, so heat capacities at high temperatures were determined by extending the linear trend. Selected parameters in the Hilliard PZ model were regressed using two high concentration data sets extrapolated by different methods. The regressions did not improve the accuracy of heat capacity predictions by Aspen Plus®, and the VLE predictions weakened. The important parameters to be varied during regressions need to be determined in future work to generate an accurate model. In addition, high concentration heat capacity data are expected, so the questionable extrapolation can be eliminated in future model improvement. Introduction Piperazine (PZ) is of interest as a solvent because it has no detectable thermal degradation up to at least 150 °C. Many explored stripper configurations operate more efficiently at high temperatures, so it is expected that piperazine will perform better than the baseline, MEA (Freeman et al., 2008). In addition to being able to operate at high temperatures, piperazine has two amine groups. Since each molecule has twice the alkalinity of MEA, it will achieve richer solutions in the absorber with faster rates, and it will require less sensible heat in the stripper due to a higher capacity. Previously, a thermodynamic model was developed for PZ (Hilliard, 2008), and it was used to simulate a simple stripper with the accompanying rich and lean pumps, cross heat exchanger, and multistage compressor. The simulations produced results with few convergence errors; however, the behavior while varying the lean loading specification was unexpected. Typically, the calculated equivalent work of the stripper has a single distinct optimum lean loading (Oyenekan, 2007). Conversely, the PZ stimulation demonstrated both a local and global optimum (Van 88 Wagener, 2008). The local optimum was at a lean loading of 0.325, an expected value based on the measured VLE at absorber conditions. The global optimum was at a lean loading of 0.15, and the temperature profile was very hot, reaching temperatures over 120 °C. A suggested source for this unusual behavior was the accuracy of the model's predictions of thermodynamic values for the solvent. The predictions that seemed particularly questionable were the solvent heat capacity and heat of absorption of CO2. The behavior of the heat capacity across a range of temperatures for specific CO2 loadings can be seen below in Figure 1. Figure 1: Heat Capacity Predicted by Hilliard PZ Model Though perhaps slightly inaccurate, the predictions are well behaved between 40 °C and 80 °C. However, the values change wildly between 100 °C and 140 °C, especially for loadings of 0.2 and 0.3. The Hilliard model was regressed mostly with data up to 80 °C and some 120 °C data, but the solutions only contained up to 5.0 m PZ. With the types of data set used for the development of this model, it is not unexpected that the predictions are inaccurate for 8.0 m PZ at stripper conditions. This work focuses on improving the heat capacity predictions. Methods and Results In Quarter 3, the PZ solvent model was applied to a simple stripper simulation, and the lean loading was optimized to obtain a minimum equivalent work, similar to previous studies. However, the behavior of the equal the work was unexpected, and it is suspected that the thermodynamic model is not predicting thermodynamic values correctly. Currently, heat capacities of concentrated PZ solutions are not available, so estimates are made using the database of values collected by Hilliard, which were included in the regression of the original model. Data were available for 2.0 m and 3.6 m PZ for temperatures between 40 °C and 120 °C. The CO2 loadings of the solvents tested were different for the two concentrations of PZ, so the 89 data were interpolated for the 2.0 m PZ solvent to obtain heat capacities at equal loadings for the two concentrations of solvent. Table 1: 2.0 m PZ Heat Capacity from Hilliard and Interpolated Values Loading T (° C) 40 45 50 55 60 65 70 75 80 85 90 95 100 105 110 115 120 0.000 3.87 3.88 3.89 3.91 3.92 3.93 3.94 3.95 3.96 3.98 4.00 4.02 4.03 4.05 4.06 4.07 4.08 Hilliard data 0.157 0.269 Cp (kJ/kg-K) 3.75 3.64 3.76 3.65 3.77 3.67 3.79 3.69 3.80 3.71 3.81 3.72 3.82 3.74 3.83 3.75 3.85 3.76 3.87 3.78 3.88 3.80 3.90 3.82 3.92 3.84 3.93 3.86 3.94 3.87 3.95 3.89 3.97 3.90 0.401 3.58 3.62 3.65 3.67 3.67 3.67 3.69 3.70 3.71 3.73 3.75 3.77 3.79 3.81 3.83 3.85 3.87 Interpolated data 0.000 0.159 0.375 Cp (kJ/kg-K) 3.87 3.74 3.58 3.88 3.76 3.61 3.89 3.78 3.64 3.91 3.79 3.66 3.92 3.80 3.67 3.93 3.81 3.68 3.94 3.83 3.69 3.95 3.84 3.70 3.96 3.85 3.71 3.98 3.87 3.73 4.00 3.89 3.75 4.02 3.91 3.78 4.03 3.92 3.79 4.05 3.94 3.81 4.06 3.95 3.83 4.07 3.97 3.85 4.08 3.98 3.87 Data for 8 m PZ were extrapolated from these data at equal loadings and lower PZ concentration. The form of dependence of heat capacity on solvent concentration is unknown, and having only two data sets introduces guesswork. Two linear extrapolation methods were explored. The first method assumed a linear dependence on solvent molality, and the second assumed a linear dependence on solvent mole fraction. The equations for extrapolation are shown in equations 1 and 2, respectively. Tables 1 and 2 display the Hilliard experimental data as well as the interpolated values (where applicable) for this work. Tables 3 and 4 display the extrapolated values based on equations 1 and 2. (1) (2) Table 2: 3.6 m PZ Heat Capacity from Hilliard Loading T (° C) 40 45 50 55 60 65 70 75 0.000 0.159 Cp (kJ/kg-K) 3.72 3.50 3.73 3.53 3.74 3.55 3.75 3.57 3.76 3.58 3.77 3.60 3.79 3.62 3.80 3.63 90 0.375 3.29 3.34 3.38 3.41 3.42 3.43 3.45 3.46 80 85 90 95 100 105 110 115 120 3.81 3.82 3.83 3.84 3.85 3.86 3.87 3.88 3.90 3.65 3.66 3.68 3.69 3.71 3.73 3.75 3.76 3.78 3.49 3.50 3.52 3.54 3.57 3.59 3.62 3.65 3.68 In addition to extrapolating to the higher solvent concentration of 8 m, the linear trend of the calculated values was extended up to a temperature of 150 °C. Since PZ is highly stable to thermal degradation, it will be valuable to be able to create simulations with reliable results at very high temperatures. Therefore, it is desirable to have data at these high temperatures, which can be regressed to make a reliable model. Typically, the response of heat capacity to temperature (as measured by Hilliard) follows a linear trend, so this is a fair approximation. Occasionally, measured heat capacities increase more dramatically at higher temperatures, especially when testing solvents with higher loadings. This effect is proposed to be an illusion due to evaporating CO2. Table 3: 8.0 m PZ Heat Capacity, Extrapolated Linearly with Molality (Method 1) Loading T (°C) 40 45 50 55 60 65 70 75 80 85 90 95 100 105 110 115 120 125 130 135 140 145 150 0.000 3.31 3.31 3.32 3.33 3.34 3.36 3.37 3.39 3.39 3.37 3.36 3.36 3.36 3.36 3.36 3.37 3.38 3.39 3.39 3.39 3.40 3.40 3.40 0.159 Cp (kJ/kg-K) 2.83 2.88 2.91 2.94 2.98 3.02 3.05 3.08 3.10 3.10 3.10 3.10 3.12 3.16 3.19 3.21 3.23 3.26 3.28 3.31 3.33 3.35 3.37 91 0.375 2.50 2.60 2.67 2.73 2.73 2.77 2.79 2.82 2.86 2.87 2.89 2.90 2.95 2.99 3.05 3.09 3.18 3.16 3.19 3.23 3.26 3.29 3.33 Table 4: 8.0 m PZ Heat Capacity, Extrapolated Linearly with Mole Fraction (Method 2) Loading T (°C) 40 45 50 55 60 65 70 75 80 85 90 95 100 105 110 115 120 125 130 135 140 145 150 0.000 0.159 Cp (kJ/kg-K) 3.35 2.89 3.35 2.94 3.36 2.97 3.37 3.00 3.38 3.04 3.40 3.08 3.41 3.10 3.43 3.13 3.43 3.15 3.41 3.15 3.41 3.15 3.40 3.15 3.41 3.18 3.41 3.21 3.41 3.24 3.42 3.26 3.43 3.28 3.43 3.31 3.44 3.33 3.44 3.36 3.45 3.38 3.45 3.40 3.45 3.42 0.375 2.57 2.67 2.73 2.79 2.80 2.83 2.85 2.88 2.92 2.93 2.95 2.96 3.00 3.05 3.11 3.15 3.23 3.21 3.24 3.27 3.31 3.34 3.37 The graphs in the following figures detail the results of several regressions. In each regression, one or more of several choices were altered: the heat capacity data set, the VLE data sets, and the regressed parameters. The graphs display the heat capacity data set used (points) as well as the heat capacity predictions by the newly regressed model (lines). The conditions for the regressions are as follows: 1. Includes all high temperature VLE data, 80 °C and 120 °C ΔHabs data, Method 1 heat capacity data (only data for 125 °C+), excludes B parameters of τ for PZ-HPZ/HCO3, PZHPZ/PZCOO, and CO2-HPZ/HCO3. 2. Same as regression 1, but Method 2 heat capacity data is used, still only data for 125 °C+. 3. Same as regression 2, but only Cp parameters of HPZ+, +HPZCOO-, and PZ were regressed (four term expressions for heat capacity). Other variations of these regressions were also conducted. The results demonstrate that the correct approach has not yet been determined. Neither extrapolation method yields accurate predictions. In addition, the correct parameters to regress are unknown; none of the regression scenarios improved predictions. In fact, the predictions with some of the new regressions are worse than with the initial model. 92 Figure 2: Regression 1. = Extrapolation of Hilliard Data to 8 m PZ. x = Linear Extrapolation of 8 M PZ Values Past 120 °C. Lines = Aspen Prediction after Regression. Figure 3: Regression 2. = Extrapolation of Hilliard Data to 8 m PZ. x = Linear Extrapolation of 8 M PZ Values Past 120 °C. Lines = Aspen Prediction after Regression. 93 Figure 4: Regression 3. = Extrapolation of Hilliard Data to 8 m PZ. x = Linear Extrapolation of 8 M PZ Values Past 120 °C. Lines = Aspen Prediction after Regression. There are several projections as to why the PZ model is not able to accurately predict heat capacity. First, it is uncertain which parameters in Aspen Plus® should be chosen to be regressed when fitting heat capacity data. The focus has been on ΔG's, ΔH's, Cp's, and τ parameters of the most active components, but this approach has not been verified to yield the best results. Next, during Hilliard's collection of values for heat capacity, CO2 may have vaporized in the DSC. This would result in higher heat capacities, but more importantly, inaccurate measurements. Finally, the methods used for creating 8 m piperazine heat capacity data may have assumed too much simplicity in the change of heat capacity with solvent concentration. However, with data only at two concentrations, the exact relationship between heat capacity and concentration was not determined. In addition to no improvement in heat capacity predictions, the regressions performed in this quarter decreased the accuracy and VLE predictions. The previous high concentration and high temperature PZ model estimated CO2 solubility at 40 °C and 60 °C within small error. Currently, there is no data for high temperature CO2 solubility in 8 m PZ, but the model also closely fit high temperature VLE data at lower PZ concentrations. The weakening accuracy of VLE predictions indicates that the data or regression methods need improvement to generate a good model. Conclusions • Since the Hilliard PZ model has trouble predicting the heat capacity of loaded solutions, additional regressions were performed. o 8 m PZ heat capacity data are not available, so methods for linearly extrapolating from lower concentration solutions were used. 94 • No regressed models were able to accurately predict heat capacities of the solutions. Additionally, the accuracy of the VLE predictions deteriorated after defining new values for parameters. o The parameters which were regressed may not be the best for addressing concerns in heat capacity prediction. o The underlying assumptions (regarding solvent behavior) for data extrapolation may have produced inaccurate heat capacity data. o Vaporization of CO2 during the measurement of heat capacity data by Hilliard has called into question the accuracy of these data. Future Work Heat capacity of 8 m PZ is currently being measured in the labs. These data will be used for further regressions of the PZ model to improve its accuracy and validate the previous simulation results for the simple stripper. Additionally, the PZ model will be used to validate pilot plant results from a November 2008 campaign at the JJ Pickle Research Campus. Finally, an accurate PZ model could be applied to other configurations previously explored by this group. The impact of stripper complexity on performance will also be investigated. Many arrangements varying from a simple one-stage heated flash to the double matrix configuration will be simulated. It is hypothesized that increased process complexity can reduce the energy requirement in the stripper, but it would come at a cost of increased capital investment for the process. Improvement in performance will be compared against the increase in process complexity to determine whether there is an optimum level of complexity for the process. References Freeman SA, Dugas R, Van Wagener D, Nguyen T, Rochelle GT. Carbon dioxide capture with concentrated, aqueous piperazine. GHGT-9. Washington, DC: Elsevier, 2008. Hilliard MD. A Predictive Thermodynamic Model for an Aqueous Blend of Potassium Carbonate, Piperazine, and Monoethanolamine for Carbon Dioxide Capture from Flue Gas. Ph.D. Dissertation, The University Of Texas at Austin, 2008. Oyenekan B. Modeling of Strippers for CO2 Capture by Aqueous Amines. Ph.D. Dissertation, The University Of Texas at Austin, 2007. Van Wagener D. Modeling Stripper Performance For CO2 Removal. Report for Third Quarter, The University Of Texas at Austin, 2008. 95 Modeling CO2 Absorption Using Aqueous Amines Progress Report for October – December, 2008 by Jorge M. Plaza Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 13, 2009 Abstract The previously developed MEA model (Plaza, Van Wagener and Rochelle, 2008) was used by AspenTech in a rate-based process modeling study of CO2 capture with MEA. Pilot plant data from the pilot plant at the J.J Pickle Research Campus presented in Dugas (2006) was used to assess the advantages of rate-based modeling over the traditional equilibrium-stage models. Results show excellent match of rate-based model predictions against the comprehensive pilot plant data sets. During this period, collaboration with AspenTech led to the submission of this work for publication in Industrial & Engineering Chemistry Research. Work also focused on absorber intercooling and the critical L/G. Intercooling was evaluated as a process option in CO2 absorption by piperazine (PZ) promoted potassium carbonate. The system performance with 4.5 m K+/4.5 m PZ was simulated by a model in Aspen Plus® RateSepTM. The absorber was evaluated for use with a double matrix stripper by optimizing the position of the semilean feed and intercooling stages to maximize CO2 removal. Additionally, a simple absorber system was modeled to observe the effect of intercooling on systems with variable CO2 lean loading. Intercooling increases CO2 removal by as much as 10% with the double matrix configuration. With a simple absorber, the effectiveness of intercooling depends on solvent rate. Near a critical liquid/gas ratio (L/G) there is a large improvement with intercooling. This is related to the position of the temperature bulge. An approximation was proposed to estimate the critical L/G where intercooling may maximize removal. This work has also led to an article that is being finalized for publication. The final draft of this article follows this report. Future Work Work will be focused in developing a CO2 absorber model for the PZ solvent using the Hilliard (2008) thermodynamic representation. The developed piperazine model will be validated using pilot plant data from the November, 2008 campaign. Intercooling will also be evaluated with different absorber configurations. The amine water wash and the flue gas blower will be included in the developed model. Various structured packings will be tested to evaluate pressure drop. 96 References Dugas R. Pilot Plant Study of Carbon Dioxide Capture by Aqueous Monoethanolamine. Master's Thesis. The University of Texas at Austin. 2006. Hilliard MD. A Predictive Thermodynamic Model for an Aqueous Blend of Potassium Carbonate, Piperazine, and Monoethanolamine for Carbon Dioxide Capture from Flue Gas. Ph.D. Dissertation. The University of Texas at Austin. 2008. Plaza JM, D Van Wagener, et al. Modeling CO2 Capture with Aqueous Monoethanolamine. 9th International Conference on Greenhouse Gas Control Technologies. Washington D.C, Elsevier. 2008. 97 Absorber Intercooling in CO2 Absorption by Piperazine Promoted Potassium Carbonate Jorge M. Plaza, Eric Chen, and Gary T. Rochelle Department of Chemical Engineering, The University of Texas at Austin, 1 University Station, Mail Code C0400, Austin, TX 78712-0231, USA Intercooling was evaluated as a process option in CO2 absorption by piperazine (PZ) promoted potassium carbonate. The system performance with 4.5 m K+/4.5 m PZ was simulated by a model in Aspen Plus® RateSepTM. The absorber was evaluated for use with a double matrix stripper by optimizing the position of the semilean feed and intercooling stages to maximize CO2 removal. Additionally, a simple absorber system was modeled to observe the effect of intercooling on systems with variable CO2 lean loading. Intercooling increases CO2 removal by as much as 10% with the double matrix configuration. With a simple absorber, the effectiveness of intercooling depends on solvent rate. Near a critical liquid/gas ratio (L/G) there is a large improvement with intercooling. This is related to the position of the temperature bulge. An approximation is proposed to estimate the critical L/G where intercooling may maximize removal. Key words: CO2 absorption, temperature bulge, modeling, intercooling, critical L/G Introduction Carbon dioxide capture and sequestration is a major option to reduce greenhouse gases and address global climate change. Chemical absorption is the most attractive technology to reduce CO2 emissions from coal fired plants. Intercooling is a common strategy to increase the performance of absorption systems. Jackson and Sherwood1 showed that intercooling increased absorption up to 37% and even higher during the winter in refinery gas absorbers for absorption of C4+ from cracking coil gas. Linhoff2 reports the use of intercooling in a refinery with vapor recovery by an absorption oil. Sobel3 introduces the use of a computational method for absorbers which routinely include a feature for modeling intercooling. A number of authors have shown that absorber intercooling can be effective with CO2 capture by amines (Thompson & King4, Chang & Shih5, Tobiesen at al.6 and Freguia & Rochelle7). Patents have also been filed with more complex intercooling configurations to increase absorber performance8-10. Intercooling is especially useful for systems where the heat of absorption (i.e. heat of solution and/or reaction) results in an increase in temperature of the solvent affecting the vapor pressure of the dissolved species. Kvamsdal and Rochelle11 observed this behavior for the absorption of carbon dioxide from flue gas by aqueous monoethanolamine (MEA). They studied absorber parameters such as liquid/gas ratio (L/G), height of packing, and flue gas composition and its effect on the appearance of a temperature bulge in the absorber. Chen12 observed similar behavior for systems using piperazine (PZ) promoted potassium carbonate (K+). He developed a rate-based absorber model for the mentioned system. The model was originally generated from 98 work carried out by Cullinane13 and later translated into Aspen Plus® by Hillard14. Chen used the Data Regression System® in Aspen Plus® to simultaneously regress equilibrium constants and interaction parameters to predict equilibrium and speciation. This work uses the tools developed by Chen to analyze a system using a 4.5 m K+/4.5 m PZ solvent. Different absorber configurations were studied to evaluate the effect of intercooling on absorber performance. Vapor-Liquid Equilibrium (VLE) and Kinetics Model The K+/PZ solvent was introduced by Cullinane13 as an alternative to the widely used MEA. MEA has a high capacity and a fast rate of CO2 absorption. According to Cullinane, the K+/PZ solvent has a higher CO2 capacity than MEA because PZ is a diamine and potassium carbonate increases the absorption capacity. Furthermore, the rate of absorption was increased due to the presence of two amine groups in PZ, the high pKa, and the large quantity of carbonate/bicarbonate. He reported a CO2 absorption rate 1.5 to 3 times faster than with 30 wt % MEA. Cullinane13 measured the thermodynamics and kinetics of potassium carbonate, piperazine, and carbon dioxide using a wetted wall column and developed a rigorous thermodynamic model in FORTRAN using the electrolyte non-random two-liquid (e-NRTL) theory. The model predicted vapor-liquid equilibrium (VLE) and speciation for the H2O-K2CO3-PZ-CO2 system. The equilibrium constants and interaction parameters were regressed using experimental data and input into the FORTRAN model. Additionally, a rigorous kinetic model was developed that determined the rate constants and diffusion coefficients based on experimental data. Cullinane conducted experiments with 0.45–3.6 m PZ and 0–3.1 m potassium carbonate at 25– 110 °C. The absorption rate of CO2 was determined using the eddy diffusivity model developed by Bishnoi and Rochelle15 and rate constants were regressed from the experimental data using the model. The reaction of CO2 with piperazine was modeled with a termolecular, basecatalyzed mechanism. The following amine reactions were used in the Cullinane13 model. k PZ −OH − ⎧OH − ←⎯ ⎯ ⎯→ PZCOO − + H 2O ⎪ k PZ − H 2O ⎪ H 2O ←⎯ ⎯ ⎯→ PZCOO − + H 3O ⎪⎪ k PZ − PZ ⎯→ PZCOO − + PZH + PZ + CO2 + ⎨ PZ ←⎯ ⎪ k 2− − PZ −CO32− → PZCOO − + HCO3 ⎪CO3 ←⎯ ⎯⎯ ⎪ k PZ − PZCOO − ⎪⎩ PZCOO − ←⎯ ⎯⎯→ PZCOO − + H + PZCOO − 99 (1) k PZCOO − − H 2O ⎧ H O ←⎯ ⎯⎯ ⎯→ PZ (COO − ) 2 + H 3O 2 ⎪ k PZCOO − − PZ ⎪⎪ PZ ←⎯ ⎯⎯→ PZ (COO − ) 2 + PZH + PZCOO − + CO2 + ⎨ k − PZCOO − −CO32− ⎪CO3 2 − ←⎯ ⎯ ⎯⎯→ PZ (COO − ) 2 + HCO3 ⎪ k − + − PZCOO − − PZCOO − ⎩⎪ PZCOO − ←⎯ ⎯ ⎯ ⎯→ PZ (COO ) 2 + H PZCOO (2) All of the buffering reactions were considered to be in equilibrium and reversible rate expressions for CO2 with PZ and PZCOO- were developed. The catalysis of the formation of bicarbonate ion by hydroxide, piperazine, and piperazine carbamate was also included in the Cullinane model. The reactions to form bicarbonate ion were included to properly model equilibrium in the boundary layer and do not affect the CO2 absorption rate. The three reversible reactions are: k OH CO 2 + OH − ←⎯ ⎯→ HCO 3 − − k PZ PZ + CO2 + H 2O ←⎯→ ⎯ PZH + + HCO3 k (3) − PZCOO PZCOO − + CO 2 + H 2 O ←⎯ ⎯ ⎯→ H + PZCOO − + HCO 3 − (4) − (5) Hilliard14 developed a VLE model in Aspen Plus® using the thermodynamic data by Cullinane and the Data Regression System® (DRS) to simultaneously regress the interaction parameters and equilibrium constants to be used in the built-in electrolyte-NRTL model. Later, Chen12 developed a rate-based model using Aspen Plus® RateSepTM. It incorporates the Hilliard VLE model to predict equilibrium and speciation, and the rate constants developed by Cullinane to predict kinetics. The model calculates heat and mass transfer and physical properties using correlations specified by the user within the Aspen Plus® framework. Initial absorber modeling by Chen predicted an unexpected temperature profile indicating that the heat of absorption for CO2 was not being correctly predicted by Aspen Plus®. This was in part due to the fact that the simultaneous regression of the interactions parameters by Hilliard did not incorporate heat capacity data for the K2CO3-PZ-CO2-H2O system. Therefore, the temperature dependence of the regressed binary interaction and enthalpy parameters may not have been adequately captured. The heat of absorption calculated by Aspen Plus® is derived from an enthalpy balance using the heats of formation, heat capacities, and heats of vaporization of the various species. The heats of formation of the piperazine species (PZH+, PZCOO-, PZ(COO-)2, and H+PZCOO-) were adjusted to provide the same heat of CO2 absorption as that predicted from equilibrium constants used in the chemistry model. The heats of formation (liquid) at 298.15K were calculated using the parameters from the equilibrium constants and the Van’t Hoff equation. The equilibrium equations and results obtained for the four piperazine species can be found in Chen12. Since the Hilliard VLE model does not contain heat capacity parameters for the PZ species (PZH+, PZCOO-, PZ(COO-)2, H+PZCOO-) regressed entropy reference values for the four PZ species were used to calculate heat capacities. Multi-parameter heat capacity correlations were 100 developed using the equilibrium constants. Aspen Plus® does not account for the existence of net-neutrally charged zwitterions which were included in the Hillard K+/PZ VLE model. The H+PZCOO- ion was given a net charge of 0 and was thus treated as a molecule. This created a number of issues such as the skewed predictions of the heats of absorption. Therefore, the charge for the H+PZCOO- ion was changed to 0.0001. The equilibrium constants were activity-based while the rate constants developed by Cullinane used concentration-based units. Therefore, Chen implemented activity-based kinetics within the model, taking advantage of the fact that the new version of RateSepTM has the capability to handle activities, in terms of mole gamma, using the power law kinetic expression: n ⎛ −E ⎛ 1 1 ⎞⎞ ⎜ ⎜ ⎟⎟ ⎜ R ⎜ T −T ⎟ ⎟ ⎛T ⎞ α (6) r = k ⎜⎜ ⎟⎟ exp ⎝ ⎝ o ⎠ ⎠ ∏ ( xi γ i ) i ⎝ To ⎠ where k is the pre-exponential factor (independent of temperature), n is the temperature exponent, E is the activation energy, To is the reference temperature (298.15K), xi is the fraction of reactant species i, γi is the activity coefficient, and αi is the reaction order for the species. A simple algebraic manipulation was performed using the following equation: ka = kc [ PZ ][CO2 ][b] ( xPZ γ PZ )( xCO2 γ CO2 )( xbγ b )(total mol / L) (7) where ka is the activity-based rate constant, kc is the concentration-based rate constant, [i] is the concentration of species i in units of mol/L, and xi is the mole fraction and γi is the activity coefficient. The last term in the denominator represents the total molar concentration per liter of solvent and will be specific for a particular solvent composition and loading. Therefore, a representative total molar concentration was selected and assumed to be constant across the column. Kinetics developed by Cullinane contain a correction for ionic strength. However, in Aspen Plus®, this correction cannot be directly implemented. Therefore, a representative ionic strength at 50 oC and 0.5 loading (mol CO2/Total Alkalinity) was selected and assumed to be constant over the various temperature and loading ranges. Chen12 reported results for the forward and reverse activity-based rate parameters for piperazine, piperazine carbamate, and bicarbonate reactions as input into Aspen Plus® RateSep™ for systems with 5 m K+/2.5 m PZ, 6.4 m K+/1.6 m PZ. The power law constants for the 4.5 m K+/4.5 m PZ, were calculated using the methodology used by Chen and are presented in Tables 1 to 3. 101 Table 1: Activity-Based Rate Parameters for formation of PZCOO- in 4.5m K+/4.5m PZ Forward b* Reverse k x 1010 E (kJ/kmol) n k E (kJ/kmol) n OH- 28.11 -67,847 34.75 2.12 x 10-2 246,966 -38.02 H2O 0.0127 -42,414 23.48 2.93 x 1012 160,611 -26.81 PZ 3.25 -155,841 53.66 6.12 x 102 325,276 -66.41 CO3-2 17.64 -105,880 53.25 3.42 x 103 200,502 -32.53 PZCOO- 10.40 -51,821 28.72 6.12 x 102 325,276 -66.41 *b corresponds to PZ + CO2+b↔PZCOO- + bH+ Table 2: Activity-Based Rate Parameters formation of PZ(COO-)2 in 4.5m K+/4.5m PZ Reverse Forward b* k x 1012 E (kJ/kmol) n k E (kJ/kmol) n H2O 0.0039 61,606 -1.46 2.22 x 1013 78,135 -1.46 PZ 2.02 -51,821 28.72 9.45 x 103 242,800 -41.06 CO3-2 5.39 -1,860 28.31 2.59 x 104 118,027 -7.18 PZCOO- 0.0065 52,199 3.78 2.07 x 104 116,084 3.78 *b corresponds to PZCOO- + CO2+b↔PZ(COO-)2 + bH+ Table 3: Activity-Based Rate Parameters for the formation of HCO3- in 4.5m K+/4.5m PZ a* Forward * b Reverse k x 1012 E (kJ/kmol) n k E (kJ/kmol) n -- OH- 5.28 x 10-6 54,758 5.24 2.18 x 10-3 66,014 19.54 H2O PZ 2.27 x 10-8 -30,856 24.15 2.35 146,702 -8.85 H2O PZCOO- 1.04 x 10-8 73,163 -0.79 7.39 19,986 35.99 *a, b correspond to a+CO2+b↔HCO3- + bH+ . (OH- does not form bH+) 102 Absorber Modeling The absorber was simulated with a rate-based model accounting for mass transfer resistance and reaction kinetics. Kinetics, in the liquid film, were calculated by discretizing it into 5 segments with a ratio of ten for the first 4 segments starting at 0.0001 and a value of 0.4 for the segment closest to the bulk liquid. Electrolyte thermodynamics were also considered. This approach required a full characterization of mass and heat transfer, hydrodynamics, vapor-liquid equilibrium, and physical properties of the entire system. This model was set up in Aspen Plus® using RateSepTM. The previously obtained activity-based kinetics were introduced along with the VLE model generated by Hilliard14. Packing interfacial area was calculated using a correlation based on data generated at The University of Texas at Austin (UT) instead of the built-in correlations. Details of the model set-up are found in Chen12. The developed model was used to evaluate the effect of solvent loading and intercooling on the performance of the absorber. The absorber was modeled with 15 m of packing divided into 30 calculation stages. Table 4 shows the absorber design specifications. Table 5 shows the conditions of the gas feed stream. Table 4: Absorber design conditions for most modeling cases Variable Value Diameter (m) 9.8 Height (m) 15.0 Packing Characteristics Type CMR Vendor MTL Material Metal Dimension NO-2P Liquid hold up (%) 5 103 CO2 Rich from Absorber High P Semilean to Absorber Lean to Absorber Figure 1: Double Matrix Stripper Configuration Table 5: Flue gas conditions used for simulation cases Variable Value Flow (kmol/s) 5.4879 Temperature (oC) 40.0 Pressure (kPa) 111.33 Mol fraction H2O CO2 N2 O2 0.0670 0.1270 0.7569 0.0491 104 The Double Matrix System. Oyenekan16 proposed a stripper configuration for the regeneration of the solvent for the absorption of CO2 that reduces the temperature change across the stripper by using a multipressure system (see Figure 1). The rich solution from the absorber is split into two streams: one goes to a higher pressure stripper while the other goes to a lower pressure stripper. This configuration generates two return streams to the absorber (lean and semilean). Depending on the operating condition these streams will be at higher or lower loading and their flow will also vary. Van Wagener17, based on the work by Oyenekan, modeled the double matrix configuration using the 4.5 m K+/4.5 m PZ solvent. Results from this analysis provided an optimum loading (moles of CO2/moles of alkalinity) of 0.4012 for the lean stream and 0.4598 for the semilean stream. The flow split between the streams was 0.1850(mol semilean/mol lean). These values correspond to 0.5 kPa partial pressure of CO2 in the lean stream at 40 oC. These data were used to set up an absorber optimization to maximize CO2 removal using a fixed packing height and varying the position of the semilean feed and an additional intercooling point. Figure 2 summarizes the conditions used for this analysis. Initially, the position of the semilean feed was optimized without any type of cooling (Figure 3). The optimization target was to maximize CO2 removal. It is possible to see that the change in the position of the semilean feed does not significantly change the performance of the absorber. CO2 removal remains around 81% removal and decreases as the feed is placed close to the extremes of the absorber. The optimum semilean feed position was found at a third from the top of the column. Figure 4 shows the resulting liquid temperature and CO2 rate profile. Figure 2: Absorber modeling conditions for 4.5 m/4.5 m K+/PZ 105 Figure 3: CO2 removal with semilean feed with no intercooling for the 4.5 m/4.5 m K+/ PZ system. 0.5 kPa CO2 lean solvent, 15 m packing. Figure 4: Temperature and CO2 absorption rate profiles for absorber with semilean feed at 0.30 column height; no intercooling. Solvent 4.5m/4.5 m K+/ PZ. 0.5 kPa CO2 lean solvent. 15 m packing. 106 Intercooling was defined to achieve 40 °C at the selected stage, which should be feasible with cooling water. When intercooling was used at the same point as the semilean feed, the optimum semilean feed position changed from the upper half of the column to the lower third. (See Figures 5 and 6.) Figure 7 shows the profiles obtained with an additional intercooling stage. The semilean feed with intercooling was simultaneously optimized along with the additional intercooling point. The optimum locations for the semilean and the additional intercooling were found at 0.63 and 0.87 of the column height, respectively. Figure 5: CO2 removal with intercooled semilean feed for 4.5m/4.5 m K+/PZ. 0.5 kPa CO2 lean solvent. 15 m packing. 107 Figure 6: Temperature and CO2 rate profiles for absorber with semilean feed and intercooling at 0.70 column height. Solvent 4.5m/4.5 m K+/PZ. 0.5 kPa CO2 lean solvent. Figure 7: Temperature and CO2 rate profiles for absorber with intercooled semilean feed at 0.63 and intercooling at 0.87 column height. Solvent 4.5m/4.5 m K+/PZ. 0.5 kPa CO2 lean solvent. 108 Van Wagener reported conditions for the stripper using a higher loading lean solvent corresponding to 0.7 kPa CO2 partial pressure. The lean solvent loading was 0.4208, the semilean loading was 0.4743 and the split was 0.1453. An analysis analogous to the 0.5 kPa loading was conducted, obtaining similar results. A summary of the results for the various operating conditions modeled is presented in Table 6. A single intercooling at the point of the semilean feed is very effective. Additional intercooling provides only marginal improvement. Table 6: CO2 removal results for K+/PZ absorber configurations CO2 Pressure in lean Solvent @ 40oC 0.5 kPa Intercooling None Single with semilean feed Double 0.7 kPa CO2 Removal (%) 81.4 91.3 92.8 71.6 82.9 84.2 Effect of Intercooling on solvent capacity and rich loading The lean solvent loading was varied to determine its effect on solvent capacity and rich loading for a simple absorber system with a single feed while maintaining removal at 90%. Solvent capacity is defined as the kmol of CO2 removed per kg lean solvent feed. Intercooling was set up in a similar matter to the previous analysis. It was placed in the middle of the column and at the optimum point (minimum amount of solvent for the level of lean loading). The flue gas and absorber specifications are the same as in Tables 4 and 5 but packing height was increased to 20 m. Figures 9 and 10 show the results obtained for this analysis. The optimum curve and the Z/ZTotal = 0.5 curve overlap along the studied range. Thus, the position of the intercooling stage is not critical with 20 m of packing. At these absorber conditions, the use of intercooling significantly improves solvent capacity for lean loading from 0.27 to 0.40. Figures 11 and 12 show that intercooling at high loading lean feed is not very beneficial because there is a limited temperature increase (7 oC) in the absorber. The higher solvent flow needed at high lean loading buffers any temperature increase due to reaction. The heat is absorbed by the increased solvent flow. Thus, mass transfer is not limited by the increase of temperature. On the other hand, lean loading solvent feeds (Figures 13 and 14) show a large increase in solvent temperature towards the top of the column. The low CO2 content in the solvent offers an initial high driving force that allows for increased reaction rates at the top of the column causing a noticeable temperature increase. The lower solvent rates are not capable of absorbing all the generated heat and there is a top of column temperature bulge (around 70 oC). As temperature increases the equilibrium becomes a limiting factor, yet most of the CO2 has already been 109 absorbed so the bottom of the column does not react much. Figure 14 shows that the use of intercooling does not provide a considerable benefit in performance. Figure 13 shows that the CO2 rate profile obtained without intercooling differs little from the former. However, absorbers operating at conditions within the observed loading bracket benefit from intercooling. The temperature bulge is located near the center of the column and limits mass transfer rates. By adding intercooling it is possible to boost this phenomenon thanks to a lowering of the temperature of the bulge and of the column in general. (See Figures 15 and 16.) This bracket is defined as the critical L/G region. Looking at the behavior of the rich loading with respect to lean loading (Figure 10), intercooling proves especially beneficial between in the critical L/G (0.27–0.38) for stripper performance. The higher loading from the absorber allows for lower energy consumption in the stripper. Figure 8: Change in solvent capacity vs. lean loading. 4.5 m K+/4.5 m PZ. 90% CO2 removal with 20 m of CMR#2 packing. 110 Figure 9: Variation of rich loading with lean loading. 4.5 m K+/4.5 m PZ. 90% CO2 removal with 20 m of CMR#2 packing. Figure 10: Temperature and CO2 rate profiles for absorber. 4.5 m K+/4.5 m PZ with a lean loading of 0.44. 111 Figure 11: Temperature and CO2 rate profiles for absorber with intercooling at 0.50 column height. 4.5 m K+/4.5 m PZ with a lean loading of 0.44. Figure 12: Temperature and CO2 rate profiles for absorber. 4.5 m K+/4.5 m PZ with a lean loading of 0.21. 112 Figure 13: Temperature and CO2 rate profiles for absorber with intercooling at 0.50 column height. 4.5m/4.5 m K+/PZ with a lean loading of 0.21. Figure 14: Temperature and CO2 rate profiles for absorber. 4.5m/4.5 m K+/PZ with a lean loading of 0.315. 113 Figure 15: Temperature and CO2 rate profiles for absorber with intercooling at 0.50 column height. 4.5m/4.5 m K+/PZ with a lean loading of 0.315. Determining the critical Liquid-Gas ratio (L/G)c The benefit of using intercooling is maximized when the selected operating conditions cause the temperature bulge to coincide with a mass transfer pinch. This relates to the capacity of the gas and the liquid to carry heat out of the column. At the critical L/G the heat generated by the absorption of CO2 is removed evenly between the liquid and the gas producing a temperature bulge towards the middle of the column. In order to determine the critical L/G it is useful to set up global mass and energy balances and balances between the top of the column and the location of the temperature bulge. The global energy balance can be written as follows: (8) where: L and G are the liquid and gas flow rates respectively (moles/s); H is the enthalpy of the stream; L,G are superscripts for gas and liquid properties; in, out label inlet and outlet streams around the absorber. Likewise, an energy balance around the top of the absorber and the location of the temperature bulge results in the following equality: (9) where b stands for conditions at the temperature bulge location. 114 The following approximations and assumptions will be made to determine (L/G)c based on engineering criteria and observations from the presented modeling cases: • In the global energy balance enthalpies are determined using the inlet liquid temperature (TinL) as reference temperature (To). This eliminates the inlet liquid term from equation 8. • The energy contribution due to CO2 absorption and vaporization of water is included in the outlet gas enthalpy as follows: (10) where: n is the flow rate (moles/s) of CO2 or H2O respectively in the gas; To is the reference temperature; habs│To is the CO2 heat of absorption at To; hvap│To is the heat of vaporization of water at To; CpoutG is the heat capacity of the gas at outlet conditions. • Replacing equation 10 in the global energy balance and including the expressions for outlet liquid and inlet gas enthalpy results in: (11) where: CpoutL, is the heat capacity of the liquid at outlet conditions; CpinG is the heat capacity of the gas at inlet conditions. • We have observed from the rigorous simulations that the maximum temperature at the bulge is approximately equal to the temperature that the gas would have if all of the heat of absorption were carried out with the gas. The global energy balance (equation 11) for a system in which all the heat leaves with the gas out the top of the absorber (ToutL = TinL) is reduced to: (12) The right side of equation 12 is set to zero because for all of the cases the inlet liquid temperature was set equal to the gas inlet temperature (40 oC) • By defining a desired removal (R) the outlet gas water content is calculated. Assuming that the outlet gas leaves in equilibrium with the inlet solvent it is possible to determine the outlet temperature using equations of state or steam tables. This value is used as an approximation to the bulge temperature (Tb): 115 (13) Here the outlet gas flow (Gout) is calculated using the inlet gas composition and taking into account the outlet water content and the removed carbon dioxide. xinH2O is the mole fraction of water in the inlet solvent. • If in equation 9 the reference temperature (To) is set to the bulge temperature (Tb) the resulting energy balance is: (14) • The change in liquid flow rate across the column is neglected and the gas flow rate is defined as: (15) Here Gi, represents the inert species present in the gas stream: oxygen and nitrogen • Introducing equation 15 into equation 14 and after some manipulation: (16) • where: (L/G)ci is the critical ratio of liquid to inert gas species; YH2O, YCO2 are the fractions of water and carbon dioxide respectively to inert species in the gas stream (nCO2/Gi, nH2O/Gi). The outlet gas water content can be calculated using an equilibrium relation: (17) where: youtH2O, xinH2O are mole fractions of water in the gas and liquid; PsatH2O is the vapor pressure of water at the inlet liquid temperature (TinL). • The CO2 content at the temperature bulge is approximated based on results for simulations at various removals. Figure 16 gives total CO2 removal as a function of the removal obtained at the bulge. Results fit the following equation depending only on the desired final removal: (18) 116 where Rb is the removal at the bulge. Equation 18 fits the data adequately independent of the packing height used. Figure 16: CO2 removal at the bulge as a function of total removal. Points are simulation results. All cases with 10 m packing height. For 90% removal two additional points are included at 20 m and 5 m of packing. It is worth noticing that equation 16 is independent of the height of packing and was generated from simple energy and heat balances. Its validation was done at constant removal (R) varying the height of packing, and at constant packing height but variable removal (Table 7). The proposed approximation gives a relatively good estimate of the critical L/G (less than 10% deviation) for all cases except at 60% removal. As for the temperature bulge, it is adequately predicted with a maximum deviation of 4 oC. 117 Figure 17: Heat and material balance around the top of the absorber and the temperature bulge Figure 18: Heat and material balance around the absorber. Table 7: Temperature bulge and critical L/G predictions. Variable packing and removal. CO2 Removal (%) Tb (oC) (L/G)c youtH2O ToutG (oC) Packing Height (m) Total Below Bulge Aspen Approx. Aspen Approx Aspen Approx Aspen Approx. 5 10 20 10 10 90 90 90 80 60 61.9 63.8 61.2 47.0 18.1 3.9 4.1 4.1 3.8 3.5 4.0 4.0 4.0 4.1 4.5 69 72 72 69 61 68 68 68 66 62 0.09 0.10 0.09 0.10 0.09 0.06 0.06 0.06 0.06 0.06 46 46 46 48 47 40 40 40 40 40 118 Conclusions Absorber intercooling increases CO2 removal by as much as 10% in the double matrix example. Adequate positioning of the intercooled semilean feed and the additional intercooling assures the best possible performance of the column in this configuration In simple absorbers where the temperature of the solvent is increased by heat of absorption, as with the K+/PZ system, intercooling appears beneficial if the temperature bulge is located towards the middle of the column and coincides with a mass transfer pinch (critical L/G). In this case it will allow for higher absorption by reducing the magnitude of the bulge temperature and increasing solvent capacity by as much as 45%. Additionally, intercooling will offer a benefit in energy consumption in the stripper because of the richer feed from the absorber. However, for systems with low L/G where the bulge is located towards the top of the column and away from the mass transfer pinch, intercooling will offer limited improvement. In the same way, with high L/G the temperature bulge is small and its location matches the pinch at the bottom of the column, so intercooling will also offer very limited enhancement. In both cases intercooling may prove to be impractical because of capital costs required for its implementation. An approximation of the critical L/G for the absorption of CO2 has been developed. It requires knowledge of the heat of absorption of the solvent and approximate values for the heat capacities of the gas and the liquid. Its accuracy is strongly dependent on the estimation of the CO2 content at the temperature bulge. For the expected operating removal (>80%) it is capable of calculating (L/G)C with less than 10% error for the K+/PZ system. The temperature bulge can also be predicted to 4 oC. This approximation also gives some insight into the possible behavior of the critical L/G with different solvents. A solvent with a heat of absorption 30% lower than the studied K+/PZ will result in a 15% lower critical L/G. Similarly, a solvent with a heat of absorption 30% higher will result in a 15% higher critical L/G. The developed approximation may prove to be a valuable tool when optimizing lean loading operation since it gives an idea of conditions to avoid where intercooling is not an option, without running any simulation models. References 1. 2. 3. 4. Jackson, R. M.; Sherwood, T. K., Performance of Refinery Gas Absorbers with and without Intercoolers. American Institute of Chemical Engineers Transactions 1941, 37, 959-978. Linhoff, H. R., Intercoolers used in absorber at vapor-recovery plants. National Petroleum News 1930, 22, (20). Sobel, B. A., Iterative processes: a relaxation operator and its application to the the computation of absorber columns. American Chemical Society, Division of Petroleum Chemistry 1968, 13, (3), 5. Thompson, R. E.; King, C. J., Energy conservation in regenerated chemical absorption processes. Chemical Engineering and Processing 1987, 21, (3), 14. 119 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. Chang, H.; Shih, C. M., Simulation and optimization for power plant flue gas CO2 absorption-stripping systems. Separation Science and Technology 2005, 40, (4), 877-909. Tobiesen, F. A.; Svendsen, H. F.; Mejdell, T., Modeling of blast furnace CO2 capture using amine absorbents. Industrial & Engineering Chemistry Research 2007, 46, (23), 7811-7819. Freguia, S.; Rochelle, G. T., Modeling of CO2 capture by aqueous monoethanolamine. Aiche Journal 2003, 49, (7), 1676-1686. Mark, S. Purification and Separation of Gaseous Mixtures. 1934. Geleff, S. Method and Device for Recovery of Thermal Energy from an Exothermic Carbon Dioxide Absorption Process 10.04.2003, 2003. Satish, R.; Scherffius, J.; Gilmartin, J.; Freguia, S. Split Flow Process and Apparatus. 2006. Kvamsdal, H. M.; Rochelle, G. T., Effects of the temperature bulge in CO2 absorption from flue gas by aqueous monoethanolamine. Industrial & Engineering Chemistry Research 2008, 47, (3), 867-875. Chen, E. Carbon Dioxide Absorption into Piperazine Promoted Potassium Carbonate using Structured Packing. Ph.D. Dissertation, The University of Texas at Austin, Austin, Texas, 2007. Cullinane, J. T. Thermodynamics and Kinetics of Aqueous Piperazine with Potassium Carbonate for Carbon Dioxide Absorption. Ph.D. Dissertation, The University of Texas at Austin, Austin, Texas, 2005. Hilliard, M. Thermodynamics of Aqueous Piperazine/Potassium Carbonate/Carbon Dioxide Characterized by the Electrolyte NRTL Model within Aspen Plus®. M.S. Thesis, The University of Texas at Austin, Austin, Texas, 2005. Bishnoi, S.; Rochelle, G. T., Absorption of carbon dioxide in aqueous piperazine/methyldiethanolamine. AIChE Journal 2002, 48, (12), 2788-2799. Oyenekan, B. A.; Rochelle, G. T., Alternative stripper configurations for CO2 capture by aqueous amines. AIChE Journal 2007, 53, (12), 3144-3154. Rochelle, G.; Seibert, F.; Closmann, F.; Cullinane, J. T.; Davis, J.; Goff, G.; Hilliard, M.; McLees, J.; Plaza, J. M.; Sexton, A.; Van Wagener, D.; Xu, Q.; Veawab, A.; Nainar, M. CO2 Capture by Absorption with Potassium Carbonate; DE-FC26-02NT41440; DOE: 2007. 120 Thermal Reclaiming of 8 m PZ CO2 Capture Solvent Quarterly Report for October 1 – December 31, 2008 by Qing Xu Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 2, 2009 Abstract This report shows the results of an investigation into thermal reclaiming for 8 molal piperazine (PZ) solution at 1 atmosphere. The main impurities considered here include sulfate and formate. Sulfate comes from sulfur in coal and formate comes from oxidative degradation of amines. Both of them exist as heat stable salts in the solvent, thus reducing the amine capacity, and may cause problems such as foaming and corrosion. Apparatus was developed and modified through a series of experiments. The results show that a sharp separation of anion impurities was possible: only 0.41% of formate and 5.13% of sulfate was present in the vapor phase. All the experiments were run by Martin W. Metzner, an undergraduate research assistant, under the supervision of Dr. Gary Rochelle and Qing Xu. This report was prepared based on the project report by M. W. Metzner (2008). Introduction In previous work, sulfate was removed from CO2 capture solvents by crystallization (Xu, Rochelle, 2008; Xu, 2008). Other impurities like formate cannot be removed through that process so a thermal reclaimer, probably of a smaller size, is still needed. In this period a thermal reclaiming apparatus under 1 atm was developed with 8 m PZ solution. This solvent was chosen because of the heat stabilization of PZ and because there are few data for 8 m PZ solution under reclaiming conditions. This experiment also tested how sharp a separation can be accomplished by thermal reclaiming. In a typical thermal reclaimer in industry the pressure is about 2 atm. A slip stream from the bottom of the stripper is heated in the reclaimer, where amine, water, and CO2 are vaporized. Contaminants (heat stable salts, amine polymers, and other impurities) accumulate in the bottom and are removed when a certain level is reached. Thus this is a semi-continuous process. However, because of apparatus limitation, our experiment was batch evaporation at 1 atm. Due to time limitations, two of the experimental runs are analyzed in this report. Four other runs were completed; however either partial or no data have been obtained for them because of time and malfunctioning analysis equipment. Analysis of the samples was done by anion chromatography, cation chromatography, and total inorganic carbon analysis. 121 Theory In these experiments thermal reclaiming was used to separate sulfate and formate salts from CO2 loaded PZ solution. It was expected that CO2 would be easy to evaporate because of its high partial pressure. Figure 1 shows that PCO2 increases as loading increases. At about 0.3 loading and 80 °C PCO2 is 10 kPa; the experiment was run at 0.3 loading and above 80 °C so PCO2 is even higher than that. Figure 1: CO2 partial pressure vs. loading in ~2 m PZ at 40, 60, and 80 °C Points: ◊ (40 °C), Δ (80 °C), and squares (60 °C) (graph obtained from Hilliard, 2008) One variable that was taken into consideration is the solubility of PZ in water at different weight fractions. The starting weight fraction used in this experiment was about 0.34 weight fraction PZ. 122 Figure 2: Solubility temperature versus piperazine weight fraction of the solution Points: ◊ (Bishnoi, 2002) and squares (Hilliard, 2008) (graph obtained from Hilliard) Figure 2 gives the solubility temperature as a function of the PZ weight fraction. It can be seen that if the solution were not loaded, the temperature would have to be about 45 °C for the solution to remain liquid. Water has greater volatility than PZ, thus the weight fraction of PZ kept increasing during the experimental process, driving up the boiling points of the solution. Experimental Methods Apparatus A 100 mL round bottom flask was modified by the UT Glass shop. The top spout was replaced with a 25cm long spout. A sample draw was installed half-way up the length of the spout and equipped with a silicon bung which is safe up to 200 °C. A K type Omega® thermo couple was used to measure the inside temperature of the flask. Figure 3 below gives a detailed picture of the experimental apparatus. 123 Figure 3: Experimental Apparatus An electric hot plate was used to heat the flask along with an aluminum block which had a groove for the flask cut into the top of it to help with the heat transfer. The vapor line was stainless steel tubing, which led the vapor into a condensate collector. Changes to the vapor line between runs will be explained in the results section of this report. The collector was held at a constant temperature (varying from 40 to 50 °C for different runs) by submerging the bottom part into a water bath. In order to minimize the heat loss from the entire apparatus, everything was wrapped using Pyrex-wool insulation which was held on using aluminum foil. Two improvements were made for run 3: both the boiling flask and the vapor lines were insulated to keep the temperature about the melting point of the vapor, and the condensate collector was placed in a water bath instead of an ice bath to avoid crystallization in the vapor line. Procedures Before each experiment, aqueous PZ solution was loaded with CO2, and additives such as potassium sulfate and potassium formate were added. 124 About 100–150 ml of the desired solution was placed inside the flask with 3–4 boiling stones and heated. As the experiment progressed, liquid samples were taken by a 10 cm stainless steel needle with a 0.5 mm bore on a plastic syringe through the side draw of the flask. The vapor sample was collected by pouring the accumulated condensate into a pre-weighed flask which contained any previous accumulations. After weighing the accumulated condensate, a sample was taken. The mass of each sample was also measured. One of the sample-taking difficulties was the crystallization of PZ. As the experiment was close to its end, the liquid samples sometimes had a PZ weight fraction near 0.8, the melting point of which is about 70 °C. This sometimes caused the needle tip to clog and the experiment to be suspended. To avoid plugging, the needle was poked into the flask about 30 seconds to a minute before each sample was taken to allow it to heat up. The syringe was not completely empty before the sample was drawn, but instead some head space was left so that when the sample was ejected from the syringe, some air helped push out any PZ left in the needle tip. The sample was drawn and immediately placed into the sample vial and water was drawn through the needle tip immediately to clean it. The syringe and needle were rinsed multiple times between samples and then allowed to dry so that no cross-contamination occurs. Anion ionic chromatography, cation ionic chromatography, and the total inorganic carbon analyzer were used to analyze the concentrations of PZ, sulfate, formate, and CO2 in the samples. Results and discussions At this time, two runs of 8 m PZ have been performed and all of the data analyzed. Four other runs have also been performed, but either the data analysis is not complete or a different solvent was used. Thus this report will only analyze the results for runs 1 and 3. The procedures were improved after run 1, in which vapor samples were not taken as a cumulative sample, and the mass of the vapor was not recorded. The results of the two runs were analyzed separately in this report. Run 1: 0.3 loaded 8 molal piperazine with potassium sulfate and sodium formate TIC analysis result for run 1 is shown in Figure 4. It indicates that towards the end of the experiment, the vapor contained more CO2 than the liquid and CO2 in the liquid seemed constant after about 100 minutes. The samples for this run were not cumulative; they only indicate the concentration of the vapor since the last sample was taken. 125 Figure 4: CO2 concentrations for run 1 It should be noted that Run 1 took longer than all other runs because the apparatus was not well insulated. Figure 5 gives the liquid and vapor PZ concentrations as a function of time. PZ concentration in the vapor samples continues to increase at an exponential rate. However in this case, the PZ concentration in the liquid does not level out. The trends of both the vapor and liquid are as expected. The PZ concentration in the liquid continued to increase since the initial partial pressure of water was higher than that of PZ and water was removed faster than PZ. As the experiment progressed, the partial pressure of PZ increased, and more PZ was vaporized. Figure 5: Piperazine concentrations for Run 1 Unlike the cation IC and TIC data, the anion analyses came out with no noticeable trends. Figures 6 and 7 plot the sulfate concentrations of the vapor and liquid samples, respectively. 126 The high concentration of sulfate initially in the vapor was not expected. Over time, however, the sulfate concentration in the vapor decreased. It may come from error or fluctuation in the IC analysis since the sulfate concentration in vapor was very small, compared with that in the liquid phase. Figure 6: Sulfate vapor concentration vs. time for Run 1 Figure 7: Sulfate liquid concentrations vs. time for Run 1 Analysis of the formate levels in the vapor samples in Figure 8 indicates a similar lack of predictability to that found in the sulfate levels. Though initially very low (about 3ppm), the final vapor sample was found to contain about 360ppm formate. A possible reason for this lack of correlation may be the very low levels of formate in the samples. The anion IC gave a range of 0.14 to 1.84ppm for the diluted samples. Since the standards used were in a range from 10– 50ppm, measurements at the level of 1ppm may not be accurate. Therefore, it would be reasonable to assume that the measured concentration of formate in the vapor samples was incorrect. 127 Figure 8: Formate vapor concentrations vs. time for Run 1 Figure 9: Formate liquid concentrations vs. time for Run 1 Formate levels in the liquid samples did not follow the expected trend, either. From the start of the experiment until about 120 minutes, the formate levels decreased; this can be seen in Figure 9. The final formate concentration was not significantly higher than in the initial solution; only a 29.2% increase, indicating that the separation was not as sharp as was hoped for. Run 3: 0.3 loaded 8 molal piperazine with potassium sulfate and formic acid The results from Run 3 were better than those from Run 1 but still had some inconsistencies. The mass balance for Run 3 indicates that almost all of the CO2 was removed over the course of the experiment. Table 1 tabulates the mass balance of Run 3. 128 Table 1: Mass balance of Experiment Run 3 Initial Mass Total Vapor Final Slurry Total Mass Recovered Recovery 182.84g 138.67g 23.44g 162.12g 88.66% The total mass lost during the experiment was 20.72g, compared to 21.6g of CO2 loaded into the solution. Since CO2 was the most volatile compound in the solution, it is reasonable to assume that it represents most of the mass lost. This is supported by a mass balance of the CO2 for the experiment which indicates that 11.92g of CO2 was lost in the course of the experiment; this is 57.54% of the total mass lost. Figure 10: PZ concentration in vapor and liquid in run 3 Figure 10 plots the vapor and liquid CO2 concentrations. The trends of both lines are as expected; the CO2 in the liquid dropped as loaded piperazine was vaporized and CO2 was freed from the solution. Liquid samples at 70 and 80 minutes are not included because TIC analysis indicated that very little or no CO2 was left in the solution. Figure 11 indicates that for the first 40 minutes of the experiment water was vaporized along with very little PZ. After this, the vapor PZ concentration increased as well as the liquid concentration. This is because, although PZ was being vaporized at an increased rate, the liquid was becoming more and more concentrated as the residual water was also vaporized. Though the concentration was higher in the liquid than in the vapor, 70.6% of the PZ recovered was in the vapor. Most of the lost PZ was due to precipitation of PZ inside of the vapor line, which became clogged; eventually causing a blow out in the apparatus. 129 Figure 11: PZ concentrations for run 3 Figure 12: Formate concentration in liquid samples in run 3 130 Figure 13: Formate concentration in vapor samples in run 3 Figures 12 and 13 above show the formate concentrations in the liquid and vapor samples, respectively. The concentration increased as the experiment went on. Instability in the third data point in Figure 13, the vapor concentration, indicates that perhaps potassium formate precipitated within the condensate sample. Despite this, the formate concentration stayed relatively stable for 40–80 minutes. Together, the figures suggest that the desired separation occurred. Figure 14: Sulfate concentration in liquid samples in run 3 The inconsistency of the fourth data point on Figure 14 may be due to an incorrect or miscalculated dilution of the sample. If this point were left off, then the data would have the expected trend of increasing sulfate concentration. The sulfate concentration in the vapor 131 (Figure 15) indicates a similar trend as for the formate vapor concentration; that the sulfate concentration levels off. Figure 15: Sulfate concentrations in vapor samples in run 3 Conclusions The results show that thermal reclaiming can be an effective method for the removal of sulfate and formate. Formate levels were reduced by a factor of 200 and sulfate levels were reduced by a factor of 20. A recovery of 70.6% of the piperazine was accomplished in Run 3; it may be possible to achieve a higher recovery, which was hindered by precipitation of piperazine in the vapor line. The apparatus needs to be modified to solve this problem. Most of the CO2 loaded into the solution was lost from heating. Because the experiments indicate that no CO2 was left in the liquid after 80 minutes, further experiments will use unloaded solutions. References Garland CW, Nibler JW, Shoemaker DP. Surface Phenomena. In Experiments in Physical Chemistry (7th ed.). New York, NY: McGraw-Hill Higher Education. 2003. McCabe W, Smith J, Harriott P. Unit Operations of Chemical Engineering (E Glandt, M Klein, T Edgar, Eds., 7th ed). McGraw-Hill International Edition: Boston; 2005 Metzner MW. Thermal Reclamation of 8 molal Piperazine Carbon Capture Solvent. Project Report. December 2008. Xu Q, Rochelle GT. Solvent reclaiming by crystallization of potassium sulfate, GHGT-9 Washington, D.C. 2008. Xu Q. Solvent Reclaiming by Crystallization of Potassium Sulfate. M.S. thesis. University of Texas at Austin. 2008 132 Appendices Appendix 1: Raw Data Run 1: Piperazine Sample Name Blank Blank 10ppm PZ 20ppm PZ 30ppm PZ 40ppm PZ 50ppm PZ Control Sample Sample D-1 Sample C-1 Sample D-3 Sample C-3 Sample D-5 Sample C-5 Sample D-7 Sample C-7 Sample C-9 Sample C-11 Final Slurry Sample Amount (ppm PZ) 0.2349 0 10.2089 20.4568 30.6305 40.7724 52.7397 34.7599 32.4493 1.1858 32.5034 1.5456 34.6647 1.102 39.6386 2.5472 9.5513 26.9014 0 g PZ/g Dil II 2.349E-07 0 1.02089E-05 2.04568E-05 3.06305E-05 4.07724E-05 5.27397E-05 3.47599E-05 3.24493E-05 1.1858E-06 3.25034E-05 1.5456E-06 3.46647E-05 0.000001102 3.96386E-05 2.5472E-06 9.5513E-06 2.69014E-05 0 g Dil II/g Dil I N/A N/A N/A N/A N/A N/A N/A 99.51133005 101.0109671 96.8925144 99.10784314 101.1243781 101.2645291 101.374498 100.7914172 100.2554455 100.965 98.00096899 99.21603128 g Dil I/g Sample N/A N/A N/A N/A N/A N/A N/A 99.0215475 101.1873747 100.1298315 99.89349112 100.2648221 101.3016032 98.06796117 100.5243781 99.61242604 102.0732932 99.61614173 100.309217 Dilution Factor g SO4/g Sample g PZ/g Sample N/A N/A N/A N/A N/A N/A N/A 0.342515917 0.331665417 0.011504431 0.321791085 0.015671175 0.355599476 0.010955633 0.401618078 0.025438092 0.098434075 0.262624337 0 Anions – sulfate Sample 10ppm Sulfate 20ppm Sulfate 30ppm Sulfate 40ppm Sulfate 50ppm Sulfate Control Sample Sample D-1 Sample C-1 Sample D-3 Sample C-3 Sample D-5 Sample C-5 Sample D-7 Sample C-7 Sample C-9 Sample C-11 Final Slurry Sample Sulfate (ppm) 10.0537 19.808 29.9241 41.1337 48.9559 22.9769 5.6808 1.1068 20.401 1.0773 12.7087 0.3315 18.2888 0.4613 0.219 0.3037 g SO4/g Dil I 1.00537E-05 0.000019808 2.99241E-05 4.11337E-05 4.89559E-05 2.29769E-05 5.6808E-06 1.1068E-06 0.000020401 1.0773E-06 1.27087E-05 3.315E-07 1.82888E-05 4.613E-07 0.000000219 3.037E-07 32.5593 3.25593E-05 N/A N/A N/A N/A N/A 99.51133005 101.0109671 96.8925144 99.10784314 101.1243781 101.2645291 101.374498 100.7914172 100.2554455 100.965 98.00096899 N/A N/A N/A N/A N/A 0.002286462 0.000573823 0.000107241 0.002021899 0.000108941 0.001286941 3.36056E-05 0.001843354 4.62478E-05 2.21113E-05 2.97629E-05 99.21603128 0.003230405 133 Expected N/A N/A N/A N/A N/A N/A N/A 0.365941423 N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A Anions – Formate Sample Name 10ppm Anions 20ppm Anions 30ppm Anions 40ppm Anions 50ppm Anions Control Sample Sample D-1 Sample C-1 Sample D-3 Sample C-3 Sample D-5 Sample C-5 Sample D-7 Sample C-7 Sample C-9 Sample C-11 Final Slurry Sample Formate (ppm) N/A 25.0418 37.1379 51.4796 62.1195 79.9835 63.373 0.1399 61.0354 0.8122 49.4906 0.4125 54.6127 0.3082 0.3018 1.8371 104.1238 g Formate/g Dil III 2.50418E-05 3.71379E-05 5.14796E-05 6.21195E-05 7.99835E-05 0.000063373 1.399E-07 6.10354E-05 8.122E-07 4.94906E-05 4.125E-07 5.46127E-05 3.082E-07 3.018E-07 1.8371E-06 0.000104124 g Dil III/g Dil I N/A N/A N/A N/A N/A 2.00611345 2.008654994 1.970554161 2.004191198 2.00009992 2.00509898 2.008708709 1.999301327 2.001595373 2.001195934 1.999601673 1.997403635 g Dil I/g Sample N/A N/A N/A N/A N/A 99.51133005 101.0109671 96.8925144 99.10784314 101.1243781 101.2645291 101.374498 100.7914172 100.2554455 100.965 98.00096899 99.21603128 g Formate/g Sample N/A N/A N/A N/A N/A 0.015967187 0.01285814 2.67114E-05 0.012123527 0.000164275 0.010048839 8.39981E-05 0.011005137 6.18468E-05 6.09789E-05 0.000360003 0.020634678 CO2 - Analysis Sample Mass (g) Area CO2(g) g CO2/g dil sample Dilution Factor gCO2/gSample Average 1-Control-I 0.0758 0.0764 0.0755 0.0761 0.0759 0.0763 0.0757 0.0763 0.076 0.0759 0.0765 0.0759 0.0762 0.076 0.0762 0.076 0.0765 0.0761 1.8839 1.9034 1.8876 1.82025 1.81395 1.8271 0 0 1.93485 1.93945 1.9454 0 0 1.63855 1.6611 1.6572 0 1.6934 4.64801E-05 4.6908E-05 4.65613E-05 4.50831E-05 4.49449E-05 4.52335E-05 5.1341E-06 5.1341E-06 4.75983E-05 4.76992E-05 4.78298E-05 5.1341E-06 5.1341E-06 4.10954E-05 4.15903E-05 4.15047E-05 5.1341E-06 4.22991E-05 6.1319E-04 6.1398E-04 6.1671E-04 5.9242E-04 5.9216E-04 5.9284E-04 6.7822E-05 6.7288E-05 6.2629E-04 6.2845E-04 6.2523E-04 6.7643E-05 6.7377E-05 5.4073E-04 5.4580E-04 5.4611E-04 6.7112E-05 5.5584E-04 99.51133005 99.51133005 99.51133005 101.0109671 101.0109671 101.0109671 96.8925144 96.8925144 99.10784314 99.10784314 99.10784314 101.1243781 101.1243781 101.2645291 101.2645291 101.2645291 101.374498 100.7914172 6.1020E-02 6.1098E-02 6.1369E-02 5.9841E-02 5.9815E-02 5.9883E-02 6.5714E-03 6.5197E-03 6.2071E-02 6.2284E-02 6.1965E-02 6.8404E-03 6.8134E-03 5.4757E-02 5.5271E-02 5.5302E-02 6.8035E-03 5.6024E-02 6.1162E-02 0.0762 0.0758 0.0763 0.0761 0.1988 0.1989 1.696 1.68895 0.1633 0.16245 0.9511 0.9459 4.23562E-05 4.22015E-05 8.71805E-06 8.69939E-06 2.60079E-05 2.58938E-05 5.5586E-04 5.5675E-04 1.1426E-04 1.1432E-04 1.3082E-04 1.3018E-04 100.7914172 100.7914172 100.2554455 100.2554455 100.2554455 100.2554455 5.6025E-02 5.6115E-02 1.1455E-02 1.1461E-02 1.3116E-02 1.3052E-02 1-D-1-I 1-C-1-I 1-D-3-I 1-C-3-I 1-D-5-I 1-C-5-I 1-D-7-I 1-C-7-I 134 5.9846E-02 6.5456E-03 6.2106E-02 6.8269E-03 5.5110E-02 6.8035E-03 5.6055E-02 1.2435E-02 0.1987 0.9482 2.59442E-05 1.3057E-04 100.2554455 1.3090E-02 1-C-11-I 0.1997 0.1979 0.1981 0.1985 0.0759 0.076 0.0759 2.0914 2.08135 2.0698 5.4968 2.38305 2.38315 2.3919 5.10341E-05 5.08135E-05 5.056E-05 0.000125772 5.74349E-05 5.74371E-05 5.76291E-05 2.5555E-04 2.5676E-04 2.5522E-04 6.3361E-04 7.5672E-04 7.5575E-04 7.5928E-04 100.965 100.965 100.965 98.00096899 98.00096899 98.00096899 98.00096899 2.5802E-02 2.5924E-02 2.5769E-02 6.2095E-02 7.4159E-02 7.4064E-02 7.4410E-02 7.1182E-02 1-Final Slurry-I 0.0754 1.73465 4.32045E-05 5.7300E-04 99.21603128 5.6851E-02 5.7063E-02 0.0753 0.076 1.73835 1.76615 4.32857E-05 4.38958E-05 5.7484E-04 5.7758E-04 99.21603128 99.21603128 5.7034E-02 5.7305E-02 1-C-9-I 2.5832E-02 Run 3: Piperazine Sample Name 10ppm PZ 20ppm PZ 30ppm PZ 40ppm PZ 50ppm PZ 3-V-1 3-V-2 3-V-3 3-V-4 3-Control 3-L-1 3-L-2 3-L-3 3-L-4 ppm PZ/Dilution II 10.4604 20.8933 30.239 40.3078 53.148 0.3112 3.0972 22.4158 26.0585 36.0979 34.432 50.1635 7.6943 1.1994 g PZ/gDil II 1.04604E-05 2.08933E-05 0.000030239 4.03078E-05 0.000053148 3.112E-07 3.0972E-06 2.24158E-05 2.60585E-05 3.60979E-05 0.000034432 5.01635E-05 7.6943E-06 1.1994E-06 Dilution N/A N/A N/A N/A N/A 10000 10000 10000 10000 10000 10000 10000 10000 10000 g Dil II/gDil I N/A N/A N/A N/A N/A 101.2808425 101.2985972 101.0619381 101.3269809 101.6797189 101.5567839 101.4264264 100.9320679 101.7361531 g Dil I/g Sample N/A N/A N/A N/A N/A 101.8467742 100.9162512 98.89138943 101.3813814 92.36347032 99.5083089 101.5628141 100.9929577 97.81994192 g PZ/g Sample N/A N/A N/A N/A N/A 0.003210068 0.031661668 0.22402699 0.267690353 0.339013128 0.347960971 0.516741903 0.078431294 0.011936218 Additional Dilutions: 3-L-3 3-L-4 g PZ/g Sample 0.078431294 0.011936218 g Solid 1.2302 0.1552 g Sample/g Solid 9.23191351 66.12242268 g PZ/g Solid 0.724070919 0.789251679 Anion – Sulfate Sample Name Sulfate (ppm) g Sulfate/g Dil III g Dil III/g Dil I g Dil I/g Sample g Sulfate/g Sample 3-Control-III 17.7001 1.77001E-06 4.970662112 92.36347032 0.000812625 3-L-1-III 10.6577 1.06577E-06 5.021002914 99.5083089 0.000532492 3-L-2-III 15.1858 1.51858E-06 5.028591493 101.5628141 0.000775566 3-L-3-III 17.9048 1.79048E-06 4.979216627 100.9929577 0.000900371 3-L-4-III 0.3726 3.726E-08 4.99670033 97.81994192 1.82118E-05 3-V-1-I 0.6149 6.149E-08 1 101.8467742 6.26256E-06 3-V-2-I 0.8674 8.674E-08 1 100.9162512 8.75348E-06 3-V-3-I 0.9487 9.487E-08 1 98.89138943 9.38183E-06 3-V-4-I 0.9069 9.069E-08 1 101.3813814 9.19428E-06 135 Additional Dilutions: Sample g Formate/g Name Sol'n 3-L-3-III 0.000900371 3-L-4-III 1.82118E-05 g Sol'n/g Solid 9.23191351 66.12242268 g Formate/g Solid 0.008312149 0.00120421 Anion – Formate Sample Name Formate (ppm) g Formate/g Dil III g Dil III/g Dil I g Dil I/g Sample g Formate/g Sample 3-Control-III 13.3896 1.33896E-06 4.970662112 92.36347032 0.000614727 3-L-1-III 11.8519 1.18519E-06 5.021002914 99.5083089 0.000592158 3-L-2-III 14.4794 1.44794E-06 5.028591493 101.5628141 0.000739489 3-L-3-III 7.3251 7.3251E-07 4.979216627 100.9929577 0.000368354 3-L-4-III 0.2493 2.493E-08 4.99670033 97.81994192 1.21852E-05 3-V-1-I 0 0 1 101.8467742 0 3-V-2-I 0.203 2.03E-08 1 100.9162512 2.0486E-06 3-V-3-I 0.1435 1.435E-08 1 98.89138943 1.41909E-06 3-V-4-I 0.1934 1.934E-08 1 101.3813814 1.96072E-06 Additional Dilutions: g Formate/g g Formate/g Sample Name Sol'n g Sol'n/g Solid Solid 3-L-3-III 0.000368354 9.23191351 0.003400614 3-L-4-III 1.21852E-05 66.12242268 0.000805716 CO2 Analysis: Sample 3-Control-I 3-L-1-I 3-L-2-I 3-V-1 3-V-2 3-V-3-I 3-V-4-I Mass (g) 0.0801 0.0808 0.0804 0.0804 0.0802 0.0806 0.0807 0.0801 0.0799 0.0409 0.0403 0.0409 0.0104 0.0103 0.0103 0.1990 0.2004 0.1992 0.1520 Area 2.15 2.152 2.141 1.577 1.582 1.593 1.447 1.442 1.443 3.16 3.052 3.065 3.162 3.184 3.159 2.651 2.712 2.693 2.194 CO2(g) 0.000099515 9.96125E-05 0.00009906 0.00007084 0.000071085 0.000071655 0.00006433 0.000064085 0.000064165 0.000150003 0.000144583 0.00014525 0.000150118 0.000151198 0.000149953 0.000124568 0.00012758 0.00012663 0.000101708 g CO2/g dil sample 0.00124 0.00123 0.00123 0.00088 0.00089 0.00089 0.0008 0.0008 0.0008 0.00367 0.00359 0.00355 0.01443 0.01468 0.01456 0.00063 0.00064 0.00064 0.00067 136 Dilution Factor 92.36347 92.36347 92.36347 106.31769 106.31769 106.31769 101.56281 101.56281 101.56281 1 1 1 1 1 1 98.891389 98.891389 98.891389 101.38138 gCO2/gSample 0.114750946 0.11386827 0.113800067 0.093675938 0.094234329 0.09451854 0.080960791 0.081256591 0.081561677 0.003667543 0.003587655 0.003551345 0.014434375 0.014679369 0.014558495 0.06190278 0.062956904 0.062864541 0.06783715 Average 0.1141 0.0941 0.0813 0.0036 0.0146 0.0626 0.0682 0.1510 0.1554 2.2 2.252 0.000101995 0.000104583 0.00068 0.00067 101.38138 101.38138 0.06847943 0.068228561 Appendix 2: Sample Calculations Calibration curve gives: y = 2.1947E-05x+5.1341E-06 Where y is the grams of CO2 and x is the peak area. For 1-Control-I, the peak area was 1.8839 and the injected mass was 0.0758g; this gives: This is the CO2 concentration of the diluted sample; this must be multiplied by the dilution factor to get the concentration of the actual sample. 137 Experimental Heat Capacity of Concentrated Piperazine Solution Progress Report for October 1 – December 31, 2008 by Thu Nguyen Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 5, 2009 Abstract This report discusses the heat capacity, Cp, of concentrated piperazine solution (8 m–12 m PZ). Heat capacity is analyzed as a function of CO2 loading (nominal loadings of 0.3 and 0.4 mol CO2/equivalent PZ) and temperature (40 ºC–120 ºC). The range of heat capacity values, expressed in J/g*K, at different concentrations and loadings are: 8 m (α = 0.21): 3.19–3.58; 8 m (α = 0.29): 3.11–3.56; 8 m (α = 0.4): 3.02–3.59; 10 m (α = 0.31): 2.89–3.33; 12 m (α = 0.29): 2.74–3.19. While Cp increases with temperature, it decreases with increasing loading and amine concentration. It is also determined that CO2 has a finite, though negligible, heat capacity. Finally, it must be noted that Cp measurements at high temperatures (90 ºC or greater) ought to be used with care as the magnitudes of these values also reflect CO2 and H2O vaporization in addition to the true heat of the solution. Introduction This report discusses the heat capacity, Cp, of concentrated piperazine (8 m–12 m). Heat capacity is analyzed as a function of CO2 loading (nominal conditions of 0.3 and 0.4 mol CO2/equivalent PZ) and temperature (40 ºC–120 ºC which spans the range of operating temperatures of both the absorber and stripper units). Experimental Methods The experimental procedure used for heat capacity determination is performed in accordance with ASTM E 1269-05. This is the method that was implemented in the dissertation by Hilliard (2008). A 304 stainless steel pan is filled to its capacity with 60 uL of solution sample before being sealed with its lid and O-ring. A vapor headspace of roughly 5–10% in volume is estimated to exist in the sealed unit. The sample pan is then placed against an empty reference pan inside a Differential Scanning Calorimeter (DSC) machine to measure the difference in the amount of heat absorbed by the two pans. This amount of heat differential is subsequently used to determine the heat capacity of the solution. 138 Below is a snapshot inside the DSC sample cell where the differential heat absorbed by a reference vs. sample cell is measured. Figure 1: DSC-Q100 Sample Cell Holding Reference and Sample Containers The cell constant and temperature response of the DSC instrument have to be calibrated using Indium metal with known melting point (156.6 ºC). The cell constant is an internal machine parameter that is used to adjust for subtle differences in the unit’s calorimetric response. In addition, the temperature calibration is done to ensure that the sample thermocouple is reading correctly under experimental conditions. In determining the heat capacity of the solution sample, the machine calorimetric sensitivity constant E has to be computed using the known Cp of Al2O3 (Equation 1). where Dst is the vertical displacement (heat flow difference) between the empty sample pan and the cell with Al2O3 any given temperature Wst is the mass of the Al2O3 sample The E parameter then factors into the determination of the heat capacity of the solution sample (Equation 2) Where: B is the heating rate used (5 ºC/min); Ds is the vertical displacement (heat flow difference) between the empty sample pan and the solution sample at any given temperature; Ws is the mass of the sample in mg; ∆W is the difference in the mass of the reference pan and sample pan. 139 The following plot illustrates the different heating curves for an empty baseline cell, Al2O3, and a representative sample for comparison purposes. Figure 2: Typical DSC Curves for Specific Heat Capacity Measurements Data Table 1: Heat Capacity of Concentrated PZ Solution T (º C) 8m (α = 0.21) 8m (α = 0.29) 8m (α = 0.40) 10m (α = 0.31) 12m (α = 0.29) 40 45 50 55 60 65 70 75 80 85 90 95 100 105 110 115 120 3.19 3.22 3.25 3.28 3.31 3.33 3.35 3.38 3.40 3.42 3.45 3.47 3.49 3.52 3.54 3.56 3.58 3.11 3.15 3.19 3.22 3.24 3.27 3.29 3.31 3.34 3.36 3.39 3.41 3.44 3.47 3.50 3.53 3.56 3.02 3.07 3.11 3.13 3.15 3.18 3.20 3.23 3.26 3.29 3.32 3.36 3.39 3.43 3.48 3.53 3.59 where α is the CO2 loading in mol CO2/equivalent PZ. 140 2.89 2.92 2.95 2.98 3.01 3.03 3.05 3.08 3.11 3.13 3.15 3.18 3.20 3.23 3.26 3.30 3.33 2.74 2.79 2.82 2.85 2.87 2.90 2.93 2.95 2.98 3.00 3.03 3.05 3.08 3.11 3.14 3.16 3.19 Results Figure 3 below shows the heat capacity trends for piperazine as a function of loading, amine concentration, and temperature. 3.6 3.5 3.4 Cp (J/g*K) 3.3 3.2 3.1 3 8mPZ_0.21 8mPZ_0.29 2.9 8mPZ_0.40 2.8 10mPZ_0.31 12mPZ_0.29 2.7 40 50 60 70 80 90 100 110 120 Temperature (C) Figure 3: Cp of Concentrated PZ as a Function of Temperature and Loading The plot above shows the solution heat capacity at 8 m, 10 m, and 12 m PZ concentrations. At a given PZ concentration, Cp decreases as the loading increases. This phenomenon is believed to be due to the possibility that CO2 itself has a negligible heat capacity and thereby lowering the averaged Cp of the solution as the loading or CO2 content of the system is increased. In addition, at a given concentration and loading, Cp increases with temperature as more heat is required to raise the temperature of the sample at higher temperatures. At the very high temperatures shown above, the 8 m trends appear to converge while it is not expected to do so. The phenomenon seen is possibly due to the vaporization of CO2 and H2O into the headspace of the cell (occupying about 5–10% of the total cell volume) and thereby skewing the measured heat capacity. If this truly is what is happening, then one must be careful in using the Cp results past ~85ºC from the above. Additionally, this apparent effect is exacerbated at higher loadings as there is more CO2 present in the solution to be vaporized. This phenomenon is to be investigated in greater detail subsequently. An attempt is made to validate the hypothesis that CO2 and H2O vaporization at higher temperatures does indeed skew the apparent Cp measured. An empirical model was constructed using only the 8 m PZ (α = 0.4) above as vaporization is seen to be most severe at high loadings. 141 By assuming a 10% headspace above the solution in accounting for both the CO2 and H2O vaporization, the error between the predicted value and actual value is found to be 0.8% as opposed to 2% if this had not been incorporated into the model. When Cp is reported on a CO2-free basis (J/(gamine+water*K)) as in Figure 4 there is little effect of CO2 loading. Figure 4: Cp of 8 m PZ Plotted on a CO2-free Basis Conclusion Cp of the PZ-CO2-H2O solution is a function of the solution composition, loading, and temperature. While it increases with temperature, it decreases with greater amine concentration and loading. The latter phenomenon can be understood given the definition of Cp. Since Cp = dQ / dT (unit of J/g*K), it follows that a given quantity of heat Q used to heat a greater amount of mass will result in a lower heat capacity. Cp of concentrated PZ solution (8 m–12 m) at nominal lean and rich loadings range from ~ 2.7–3.2 J/g*K at 40 ºC. At high stripper operating temperature of ~ 120 ºC, Cp for the same composition ranges vary between ~3.1–3.6 J/g*K. Note that the Cp measurements made at higher temperatures (90 ºC–120 ºC) should be used with care as these values also reflect the heats of vaporization of CO2 and H2O. As previously noted, an attempt to account for these components’ vaporization by assuming a 10% headspace reduces the error between the modeled vs. experimental values by ~1.2%. Additional work is being carried out in attempt to better study the CO2 vaporization phenomenon toward constructing a robust model for heat capacity of concentrated PZ solution. 142 Future Work An attempt to build a predictive model of Cp for PZ-CO2-H2O solution is in progress. A thorough investigation of the CO2 and H2O vaporization phenomenon is needed in order to accurately model Cp. Also, more experimental data for 10 m and 12 m PZ at different loadings is required to be integrated into the model. The Cp of the mixture will ideally be modeled as the summation of the individual species Cp weighted by its respective mass fraction. In addition to Cp modeling, efforts will be resumed toward measuring the volatility of various amine solutions at absorber operating condition. 143 Dynamic Modeling of Flexible Operation of Stripper Integrated with a Multi-Stage Compressor Quarterly Report for October 1 – December 31, 2008 By Sepideh Ziaii Fashami Supported by the Luminant Carbon Management Program And the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 5, 2009 Abstract This quarter’s work focused on dynamic simulation of flexible CO2 capture in response to variation in electricity load. The rate-based dynamic model of the stripper, which was created in Aspen Custom Modeler® (ACM®) for 30 wt % MEA, was combined with the steady state performance model of a multistage compressor. Simple ratio-control strategy was implemented to control the rich solvent rate proportional to the reboiler heat rate. When the heat rate is suddenly decreased by 10%, the reboiler pressure and temperature change approach the new steady state in 10 and 30 minutes respectively, while the lean loading remains almost constant. With a constant speed compressor, the stripper pressure varies in dynamic operation. In order to avoid the maximum discharge temperature with a constant speed compressor, the compressor should be designed based on minimum load. Background Flexible operation of CO2 capture permits capture at an economical operating condition based on hourly variation of electricity demand and pricing. In a flexible system, some or all the steam used for solvent regeneration and CO2 compression can be diverted back to the low-pressure steam turbines to generate electricity at lower cost of energy. The need to build new capacity for the peak load by generating more electricity from existing systems with flexible capture would also be avoided. Flexible systems require dynamic operation and investigation of the dynamic behavior of the system in response to the steam reduction requires a dynamic model of the system which is accurate enough to reflect important dynamic information and simple enough to be used for dynamic simulation and control. Using the dynamic model and simulation helps us estimate the response time of the plant to the load change, find optimum operating conditions, and operate the system at a safe condition For example, the following conditions should be avoided: 144 • • • flooding and excessive pressure in the columns surge in multi-stage compressor exceeding maximum discharge temperature and temperature rise rate in all stages of the multi-stage compressor Dynamic Modeling In this work, the rate-based dynamic model of a packed column was created in ACM® and the dynamic behavior of the stripper using 7 m MEA was simulated in response to the steam reduction. The following conditions are carried out for steady state design and dynamic simulation: • • • • • • • CO2 removal at 100% load: 90% Packing height: 2 m; column diameter: 4.6 m 5 °C temperature approach CO2 lean loading: 0.42 (@ 100% load) CO2 rich loading: 0.527 Liquid hold-up in the reboiler: 5 min (@ 100% load) Liquid level in the reboiler is controlled at a constant level This report evaluates the dynamic behavior of the stripper by implementing ratio-control strategy for CO2 capture. In this strategy, the absorber would operate continuously at its full load, but the reboiler steam rate would be reduced. The rich solvent stream is split into two streams proportional to the reboiler heat rate reduction. One of the streams is directed to the stripper to be regenerated and then is mixed with non-regenerated stream after coming out of the stripper and before returning to the absorber. With this strategy, the lean loading in the stripper is kept almost constant while the absorber provides variable CO2 removal. In this option, no additional inventory is needed for rich and lean solvents and the only input variable that significantly changes in the absorber would be the lean loading (Figure 1). 145 PC Constant rich loading MEA solution CO2 P=160 KPa QRe Lean solvent To the ΔT=5°C Stripper t MV Rich solvent From the LC Steam MV ⎛ Lrich ⎞ ⎜ ⎟ L rich ,base ⎠ ⎝ =1 ⎛ QRe b ⎞ ⎜ ⎟ Qreb,base ⎠ ⎝ Figure1: Ratio-Control Strategy In this study, a simple stripper dynamic model is coupled with a steady state model of a multistage compressor. Multi-Stage Compressor Model Each compressor stage model incorporates the regressed data from the steady state performance curve of a centrifugal compressor working at constant speed (100%). This general compressor performance curve was extracted from typical centrifugal compressor curves given by Ludwig (1995). The steady state design of multi-stage compressor for the simulated plant was based on 100% load of the capture and the number of stages was calculated based on 149 °C maximum discharge temperature. The calculated number of stages is 6 with an intercooler for each stage to cool the gas to 40 °C. Figure 2 shows the general performance curves of a single stage compressor with three different speeds. The 100% speed curve was used for the model of each stage of the compressor in this study. 146 fraction of rated head or pressure ratio 1.4 110% 1.2 1 100% 0.8 90% 0.6 0.4 0.6 0.8 1 1.2 fraction of rated volumetric inlet flow Figure 2: Typical centrifugal compressor performance curves (Ludwig, 1995) Dynamic Results In order to demonstrate how the stripper responds to the flexible operation, the following changes were made and compared: 1. - 10% step change in the rich solvent flow rate 2. ± 10% step change in the reboiler heat rate 3. - 10% step change in both rich solvent flow rate and reboiler heat rate simultaneously (ratio-control) Figures 3 and 4 show the response of reboiler pressure and lean loading. Decreasing reboiler duty by 10% gives 3.8% change in the reboiler pressure. However, change in the rich solvent rate does not change the reboiler pressure significantly. Because the pressure drop across the packed column is a strong function of vapor rate, the ratio-control strategy cannot keep the reboiler pressure constant. This strategy makes the response of the pressure much faster. As shown in figure 4, the lean loading changes in opposite directions when rich solvent flow rate and heat rate change individually. However, it remains almost constant as both liquid and heat rate change in ratio-control strategy. 147 reboiler pressure, KPa 165 163 -10% Lrich 161 3.8% 159 Ratio-Control 157 -10% Qreb 155 -5 0 5 10 15 20 25 30 35 40 time, min Figure 3: Reboiler pressure responses to -10% change of heat and rich solvent rate 0.435 lean loading 0.43 -10% Qreb 0.425 0.42 Ratio-Control 0.415 -10% Lrich 0.41 0.405 -5 0 5 10 15 20 25 30 35 40 time, min Figure 4: Lean loading responses to -10% change of heat and rich solvent rate Figure 5 shows the response of reboiler temperature to the step changes. The combination of effects of lean loading and reboiler pressure changes influence the reboiler temperature. As demonstrated in figure 5, the reboiler pressure change could change the reboiler temperature slightly (less than 1 °C) with ratio control while the lean loading remains constant in this scenario. 148 reboiler temperature, C 105 -10% Lrich 104 103 <1°C -10% Qreb Ratio-Control 102 101 -5 0 5 10 15 20 25 30 35 40 time, min Figure 5: Reboiler temperature responses to -10% change of heat and rich solvent rate fraction of rated head or pressure ratio Figure 6 demonstrates the movement of operating points from initial point (red point) in the first stage of multi-stage compressor in response to the step changes on the performance curve. It is shown that the change in the liquid rate does not move the operating point significantly relative to the heat rate. 1.05 -10% QReb -10% LRich 1 100% -10% QReb&LRich 0.95 0.95 1 1.05 fraction of rated volumetric inlet flow Figure 6: Operating point movement in response to the step changes As mentioned earlier, in the selection of a dynamic operation we should make sure that the equipment operates in a safe mode even during transition time. For the compressors, one of the checkpoints is discharge temperature at all stages. Figure 7 shows how the discharge 149 temperature in the compressor changes in response to the step changes in the heat rate and liquid flow rate in ratio-control strategy. 153 Stage5 T discharge , C 152 151 Stage1 150 149 Tmax 148 -5 0 5 10 15 20 25 30 time, min Figure 7: Discharge temperature responses in the multi-stage compressor for ratio-control strategy The discharge temperature increases as the heat rate and liquid rate decrease. Because the discharge pressure in the last stage is kept constant and the suction pressure in the first stage decreases, the pressure ratio and discharge temperature increase in each stage. If we want to operate the stripper at partial load, we need to design the multi-stage compressor based on minimum load to avoid exceeding the maximum discharge temperature. Conclusions and Future Work In this quarter, the dynamic model of the stripper was integrated with the steady state performance curves of a constant speed multi-stage compressor. Dynamic simulation was done to simulate the flexible operation with step changes in the reboiler heat rate and rich solvent flow rate to evaluate the ratio-control strategy. The results show that by ratio control strategy with -10% load change, reboiler pressure and temperature change and reach to the steady state (97% approach) in 10 and 30 min. However, the lean loading remains almost constant. In this study, the compressor was assumed to be operated at a constant speed. The results show that with constant speed, the stripper is operated at variable pressure in a dynamic operation. The important point that should be considered is the design of the compressor based on minimum load if the capture is flexible. In the next quarter, the dynamic model of the absorber will be created and integrated with the stripper model to include the interaction of stripper and absorber columns in dynamic operation. In addition, the model of the variable speed multi-stage compressor will be investigated and its effects on the dynamic behavior of the system will be studied. 150 References Ludwig. EE. Applied Process Design for Chemical and Petrochemical Plants. Vol 3, 3rd ed., Burlington: Elsevier, 1995. 481. 151 Electric Grid Level Implications of Flexible CO2 Capture Operation Progress Report for October 1 – December 31, 2008 by Stuart Cohen Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 3, 2009 Abstract Cost analysis of coal-based power plants with flexible CO2 capture finds that the operating point with the lowest marginal cost is highly dependent on CO2 price. There is a $5/MWh incremental cost of flexibility that is primarily the result of pessimistically assuming that maintenance costs for a flexible CO2 capture system are more than double the maintenance costs of inflexible systems. To investigate the long-term economics of flexible CO2 capture systems in the Electric Reliability Council of Texas (ERCOT) electric grid, this study uses projected fuel and CO2 prices from analysis of the Lieberman-Warner Climate Bill, ERCOT expected future electricity demand, and the current and planned ERCOT power plant fleet as inputs into a dynamic model of electricity dispatch that calculates annual generation, profits, and CO2 emissions in the years 2012-2031. A scenario without CO2 capture is compared to scenarios with flexible and inflexible CO2 capture. Over the 20 years, changes in the contribution of each plant type to meeting electricity demand and in their operating profits are primarily associated with the relative marginal costs of electricity production for each plant type. Utilization of coal-based plants remains high despite rising CO2 costs even without CO2 capture because coal remains much less expensive than natural gas. Coal-fired plants become less expensive to operate with CO2 capture than without in the late 2010s to early 2020s. Flexible CO2 capture systems become more economical to operate at maximum CO2 removal by the mid- to late-2010s, but annual operating profits are greater for inflexible systems due to the incremental cost of flexibility. After maximum CO2 removal is most economical, coal-fired plants earn much greater annual operating profits if CO2 capture is available. When capital cost considerations are included in a discounted cash flow analysis over the 20year study period, no scenarios with CO2 capture recover more than 61% of the combined capital expenditures of CO2 removal systems, any required sulfur dioxide (SO2) scrubbing systems, and 152 any new generation capacity required to replace the output lost to CO2 capture. However, there is a major economic advantage to flexible CO2 capture because flexibility eliminates the need to spend several billion dollars in capital costs to replace the output lost to CO2 capture energy requirements at maximum CO2 removal. While these particular results do not present positive economic arguments for CO2 capture investment, the general methodology can be utilized to examine what combinations of fuel prices, CO2 prices, electricity demand, and power plant fleet are conducive to flexible or inflexible CO2 capture installation. Utilizing a Dynamic ERCOT Model for a Plant Life Assessment of Flexible CO2 Capture The major research activity during the fourth quarter of 2008 was to develop and employ a methodology that uses the previously developed model of electricity dispatch and the ERCOT electricity market to perform a plant life assessment of flexible CO2 capture facilities. Using long term projections of fuel prices, CO2 price, electricity demand, and the ERCOT power plant fleet, the model was used to determine annual performance, economics, and CO2 emissions of ERCOT power plants over a 20-year period. This methodology was developed in order to determine the long-term implications of flexible CO2 capture in the context of both hourly and annually varying energy market conditions. Another activity was to perform an initial analysis of possible cost implications of flexibility to determine any additional costs that may be associated with installing or operating a flexible CO2 capture system rather than an inflexible system designed for continuous performance at one CO2 capture operating point. The remainder of this report subsequent to the Future Work section presents the details of this plant life assessment in a standalone document modified from a term paper written for an Energy and Resource Economics course. The introductory section mostly repeats information covered in previous quarterly reports. Cost analysis of flexible CO2 capture systems is discussed next, followed by the methodology and results of the plant life analysis using the dynamic ERCOT model. Results are presented for only one set of inputs, but the true utility of this work is to demonstrate a methodology that could be utilized with various input combinations of projected fuel and CO2 prices, electricity demand, and future power plant installations. Doing so could examine which overall electricity market conditions best support widespread implementation of CO2 capture, and if there are circumstances that make flexible systems more or less favorable than inflexible systems. Individual plant parameters could be varied over time to investigate changes to CO2 capture performance and cost as a result of experience and learning. The implications of different CO2 capture deployment schedules could also be examined using this approach. In general, the initial analysis presented below establishes a versatile framework that allows the dynamic grid level model to produce useful results when evaluating CO2 capture systems in a time frame relevant for investment decisions. Future Work Having developed the long-term analysis methodology, next steps will include identifying specific research questions for this modeling approach and executing the required analysis. 153 Work will also continue to investigate the costs associated with flexibility to compare more accurately flexible and inflexible systems. Continuing the work presented at the November 2008 research review meeting, the effects of different combinations of coal and natural gas prices on the operation of flexible CO2 capture systems will be investigated. Results of this analysis will be submitted to the American Society of Mechanical Engineers 3rd International Conference on Energy Sustainability in July 2009. Other work presented at the GHGT-9 conference and the November 2008 research review meeting will be expounded on to produce a comprehensive look at flexible operation in response to hourly demand variations. Several plant types will be considered for replacing some or all of the output lost to CO2 capture energy requirements, and novel econometrics will allow an examination of the costs per quantity of CO2 avoided from the perspectives of the plant operator, the electric grid, and a wholesale purchaser of electricity. 154 A Plant Life Economic Analysis of Flexible CO2 Capture Based on Short-term Electric Grid Dynamics January 4, 2009 Introduction Carbon Dioxide Capture for Climate Change Mitigation Despite broad scientific consensus that climate change is occurring due to anthropogenic greenhouse gas emissions, the debate continues over how to effectively mitigate climate change in an economically efficient manner. At the heart of such deliberation is how best to account for the uncertain future externalities associated with carbon dioxide (CO2) emissions, the largest contributor to anthropogenic greenhouse gases. Estimates of an economically efficient CO2 price vary widely because of the inherent uncertainty in estimating damages caused by CO2 emissions and the sensitivity of such estimates to economic assumptions such as discount rate (Roughgarden and Schneider, 1999). Regardless of the policy and regulatory measures that are implemented in climate change mitigation efforts, it is widely accepted that a broad range of technical approaches will be required to successfully stabilize atmospheric greenhouse gas concentrations and prevent significant changes to global climate. The popular concept of stabilization “wedges” reflects this approach, proposing that each major emissions abatement strategy can cut a “wedge” out of the projected CO2 emissions that would occur without any abatement efforts (Pacala and Socolow, 2004). Most “wedge diagrams” of CO2 emissions target power generation for several emissions reduction strategies because over 60% of worldwide CO2 is emitted from power generation systems (Metz, 2005). Coal-fired power plants alone account for 60% of this total, amounting to nearly 11.5 billion metric tons of CO2 (tCO2) emitted worldwide in 2005 and 2.1 billion tCO2 in the U.S. (Metz, 2005; USEIA, 2007) Eliminating coal from our energy mix may be a plausible long-term strategy, but coal provides roughly half of U.S. electricity, facilities require 3–6 years to build, and plants remain in service for several decades, making it likely that coal will remain a vital part of electricity generation for several decades to come (IEA and NEA, 2005; USEIA, 2007). Coal is also an abundant, politically secure, and relatively inexpensive fuel, making it attractive in spite of the environmental externalities associated with flue gas emissions from coal-based power plants. In order to continue and possibly expand the use of coal for electricity generation in an environmentally acceptable manner, it will be critical to implement CO2 capture and sequestration (CCS) technology that can significantly reduce CO2 emissions from coal-fired facilities. 155 Post-Combustion CO2 Capture with Amine Absorption and Stripping One leading option for CO2 capture at coal-based power plants is post-combustion capture, meaning that CO2 is removed from the exhaust gases produced when coal is combusted in a boiler unit. Chemical absorption and stripping using an amine solvent is one promising postcombustion process because there are already several decades of experience with the technology at smaller scales in industries such as natural gas purification and urea production. The chemical absorption/stripping process could be integrated into a coal-fired power plant in the manner shown in Figure 1. Flue gas exiting the boiler is sent through other pollution control systems before entering an absorber column, where it contacts an amine solvent that chemically reacts with CO2 at temperatures around 40°C. The solvent exiting the absorber, termed “rich” solvent because of its comparably high CO2 content, passes through a heat exchanger to a stripper column where it is heated to temperatures of about 120°C. At this temperature, the CO2/solvent reaction is reversed, and nearly pure CO2 is released out the top of the stripper column. The now “lean” solvent exiting the stripper is sent back to the absorber, and the process repeats. CO2 exiting the stripper must then be compressed for pipeline transportation to a suitable storage site. A typical design of this system can achieve 90% CO2 removal from the incoming flue gas stream (Rao and Rubin, 2002). The heat required to strip CO2 from the rich solvent is provided by extracting steam from between the intermediate and low pressure turbines being used to generate electricity. This steam can be expanded in a let-down turbine to drive the CO2 compressor before being sent to the stripper; however, the combined energy requirement for stripping heat and CO2 compression can still result in a power plant output reduction around 30% (Rubin, 2007). Without an economic value to reducing CO2 emissions, the energy requirement of CO2 capture would result in higher operating costs and lower electricity sales. Coupled with capital costs in the realm of $908 per kilowatt (kW) of rated capacity1, there are formidable technological, economic, and regulatory challenges to overcome before widespread CCS can become a reality (Rubin, 2007). 1 Dollar value is adjusted to US$2006 using the consumer price index from the U.S. Bureau of Labor and Statistics. All dollar values in this report are presented in US$2006. 156 Figure 1: Simplified process diagram of a post-combustion CO2 capture system using amine absorption/stripping, showing integration of the CO2 capture system into the base power plant. Despite the economic challenges of post-combustion amine absorption/stripping, the process remains an attractive CO2 capture option. Commercial scale experience in some industry applications provides a sound knowledge base from which to scale up and tailor the technology for coal-fired power plants. Unlike other CO2 capture options that are more intimately integrated in a plant’s power generation system, post-combustion CO2 capture is an add-on technology that is available for retrofitting the current fleet of coal-based facilities. Motivation for Studying Flexible CO2 Capture Another advantage of amine absorption/stripping is the potential to exploit flexibility in the CO2 capture process. Typical techno-economic assessments of CO2 capture assume that the CO2 capture system operates continuously throughout the plant lifetime at a single operating point optimized for high CO2 removal (Rao and Rubin, 2002; USNETL, 2007). Assuming a static operating point implies that the energy used for CO2 capture is unavailable for power generation and may have to be replaced by installing new generation capacity. However, the add-on nature of an amine absorption/stripping system offers the possibility that the CO2 capture system could operate flexibly between various operating points, allowing some or all of the energy required for CO2 capture to be recovered by diverting the stripper steam back to the power generation turbines for electricity production. Reducing steam flow into the stripper would decrease the flow of CO2 into the CO2 compressor, so the compressor work is also decreased. There are several plausible configurations for such a flexible system, but a worst-case environmental scenario would allow any CO2 that is not captured at a reduced CO2 capture load2 to be vented to the atmosphere. 2 “Load” of a CO2 capture system refers to the ratio of steam flow into the stripper at the current operating point to the maximum amount of steam that enters the stripper at maximum CO2 removal. 157 By moderating the use of steam for electricity versus CO2 removal, flexible CO2 capture operation allows additional control over power plant output and electricity production costs, both of which have economic implications at the power plant and electric grid levels. From the standpoint of the electric grid, choosing to operate CO2 capture systems at reduced or zero-load during periods of annual peak electricity demand could eliminate the need to replace the generation capacity lost to CO2 capture energy use. Previous work has shown that in the Electric Reliability Council of Texas (ERCOT) electric grid, removing the need for new capacity could save billions of dollars with relatively little part- or zero-load operation (Cohen, Rochelle et al., 2008). Depending on the time response of flexible CO2 capture systems, flexibility could also improve a power plant’s ability to perform important and potentially lucrative grid reliability services by expanding output range and offering an additional measure of output control (Chalmers, Gibbins et al., 2007). This paper focuses on the utility of using flexible CO2 capture to choose the most economical CO2 capture load based on current electricity market conditions that include electricity demand, fuel prices, and the CO2 price that may result from a given regulatory scenario. Electricity demand changes continuously, so CO2 capture flexibility is investigated on an hourly time scale to observe how flexible CO2 capture may affect real-time power plant operation and economics. However, a complete economic understanding of flexible CO2 capture must account for longterm market trends and the resulting power plant behavior. Changes to fuel prices, CO2 prices, electricity demand, and a grid’s fleet of power plants will all affect the ultimate long-term economic performance of a CO2 capture system. The next section discusses the costs associated with CO2 capture systems with particular attention paid to any additional costs associated with flexibility. Section 3 describes a modeling framework used to investigate the behavior of flexible CO2 capture systems in response to hourly variations in electricity demand. Section 4 describes how the short-term model is used to perform a long-term economic analysis, and Sections 5 and 6 respectively discuss the results and conclusions of this study. Cost Analysis of CO2 Capture Systems General CO2 Capture Cost Considerations A detailed technical and economic assessment of a particular system configuration is required to determine the capital costs of a CO2 capture system. Such an analysis is outside the scope of this work, so a representative value of $908/kW is taken as the capital cost of an amine absorption/stripping system (Rubin, 2007). Because electricity is dispatched based on the marginal cost of electricity production, it is important to consider a more detailed description of the operating cost implications of CO2 capture. At a base plant without CO2 capture, the primary contributors to marginal costs are fuel costs and any CO2 costs associated with a CO2 regulatory structure. Fuel costs are determined from power plant efficiency, often represented as a heat rate,3 and CO2 costs can be calculated from the CO2 emissions rate. Additional base plant operating costs account for other material inputs, labor, and maintenance and can be represented by an additional cost factor. On average, 3 Heat rate is defined as the energy content in fuel that is consumed per unit of electricity output. Typical units are million British thermal units per megawatt hour (MMBTU/MWh). 158 coal-fired power plants in the ERCOT grid have a heat rate of 11MMBTU/MWh and an emissions rate of 1tCO2/MWh4 (USEPA, 2007). Additional base plant operating costs at a coalfired facility are approximately $5.4/MWh (NEI, 2007). Thus, at a coal price of $1.5/MMBTU and a CO2 price of $10/tCO2, the marginal cost of electricity production would be $31.9/MWh. Operating CO2 capture reduces the efficiency of a power plant while reducing the CO2 emissions rate, resulting in higher fuel costs and lower CO2 emissions costs per quantity of electricity produced. The magnitude of these effects can be determined from the energy required per unit of CO2 captured (MWh/tCO2) and the percent of CO2 removed from the power plant exhaust. Other fixed operating costs for a CO2 capture system include the additional labor to operate the CO2 capture systems and the associated supervisory and administration costs. Maintenance of the additional equipment is also considered a fixed operating cost. In any amine absorption/stripping system, a certain rate of solvent degradation is expected, where the amine solvent undergoes irreversible chemical reactions to waste byproducts due to the presence of oxygen and other impurities or from exposure to high temperatures. Some degraded solvent can be recovered by a solvent reclamation process that uses sodium hydroxide (NaOH) injection, but this practice also produces a hazardous waste stream. Consequently, there must be variable operating costs associated with replacing degraded solvent, consumed NaOH, and waste disposal. At 100% load, the CO2 capture process could also more than double cooling water requirements compared to a plant without CO2 capture, so additional water costs must be accounted for (USNETL, 2007). Finally, there will be a cost for transport and subsequent injection of CO2 into a suitable storage site. Costs Associated with CO2 Capture Flexibility Diverting steam from the stripper back to the power plant turbines will increase plant output as well as the CO2 emissions rate. Depending on the relative prices of fuel and CO2, the combined effect of part- or zero-load CO2 capture on fuel and CO2 costs could either increase or decrease the marginal cost of electricity production. The cost implications of a flexible CO2 capture system will depend on the particular plant design and configuration. A simple configuration is chosen for consideration in this report to determine some baseline estimates of the additional costs of a flexible CO2 capture system over one designed for continuous 100% load operation. Shown in Figure 2, this design operates flexibly by simultaneously varying the steam and rich solvent flow rates into the stripper while maintaining constant flow through the absorber. CO2 flow to the CO2 compressor will then be reduced because less CO2 is removed from the flue gas. 4 CO2 emissions rates are given in metric tons of CO2 per megawatt hour. 159 Figure 2: Process diagram of a flexible CO2 capture system that simultaneously varies steam and rich solvent flow. Process modeling of the stripper in this configuration suggests that the stripping heat required per quantity of CO2 removed is nearly constant across the operating range (Ziaii, Cohen et al., 2008). However, some pumps and fans continue to operate at 100% while the CO2 removal rate is reduced, so total energy used per unit of CO2 captured increases at reduced CO2 capture load. As CO2 capture load is reduced, the solvent spends more time at the high temperatures associated with thermal degradation, so solvent consumption increases per unit of captured CO2 (Ziaii, Cohen et al., 2008). From a practical standpoint, frequently cycling the CO2 capture system between multiple operating points could cause additional stress on mechanical components, increasing maintenance costs. Capital costs are not expected to increase significantly in this configuration. Aside from some additional piping and a valve for bypassing rich solvent away from the stripper, all other valves and control systems would already be in place for startup/shutdown and normal 100% load operation. Quantifying Marginal Generation Cost for a Plant with Flexible CO2 Capture To demonstrate the effects of CO2 capture load on the marginal cost of electricity production, representative quantities are chosen for each of the above contributions to total operating cost. A 400MW coal-fired plant is considered with efficiency and CO2 emissions typical of a coal-based facility in the ERCOT grid. A coal price of $1.5/MMBTU is used in all calculations in this section. Monoethanolamine (MEA) is the only solvent considered in this report because it is relatively well understood and is often considered the baseline amine solvent for CO2 capture. CO2 capture behavior across the operating range is determined using a linear fit between the performance at 20% and 100% load as calculated in a dynamic process modeling study (Ziaii, Cohen et al., 2008). CO2 capture at 100% load achieves 90% CO2 removal. Efficiency and cost curves for CO2 capture and power system components other than the stripper are not considered because detailed consideration of these components is outside the scope of this work. Parameters necessary to determine additional capture-specific operating costs are taken from National Energy Technology Lab (NETL) Design Guidelines and other techno-economic analyses from the literature. These parameters are summarized in Table 1. Contributors to variable operating costs that are not represented in terms of a quantity per ton of CO2 captured are assumed to vary directly with the CO2 capture load. Thermal degradation at reduced load is calculated by assuming that 0.1kgMEA/tCO2 degrades due to thermal effects at 100% load 160 (Davis, 2008). While NETL recommends a maintenance cost factor of 2.2% of the total capital cost of a CO2 capture system, 5% is used for this flexible system as a pessimistic estimate of increased maintenance requirements (NETL, 2005). Table 1: Parameters used to determine the marginal cost of electricity production across the CO2 capture operating range (Rao and Rubin, 2002; Rao, Rubin et al., 2004; NETL, 2005; Rubin, 2007; USNETL, 2007). Fixed Operating Cost Parameters Variable Operating Cost Parameters Parameter Value Parameter Value Operating Labor (jobs/shift) 2 MEA Consumption (kgMEA/tCO2) 1.5 Hourly Wage ($/hr) 33 NaOH Consumption (kgNaOH/tCO2) 0.075 5 MEA Cost ($/kgMEA) 2.36 12% Caustic Cost ($/kgNaOH) 0.46 30% Waste Disposal Cost ($/kg) 0.20 Water Cost ($/m3) 0.27 CO2 Transport/Storage Cost ($/tCO2) 9.08 Maintenance Cost (% of total CO2 capture plant cost) Maintenance Cost Allocated to Labor (% of total maintenance cost) Administration & Support Labor Cost (% of total labor cost) Figure 3 displays the marginal cost of electricity production across the CO2 capture operating range for three CO2 prices, and Table 2 displays power plant performance at the 0% and 100% CO2 capture load. Because plant performance parameters follow a nearly linear relationship between the two operating points, the marginal cost curves are almost linear across the CO2 capture operating range. The average slopes of these curves, however, are highly dependent on the CO2 price. In order for marginal cost to be lower with 100% load CO2 capture, the CO2 price must be slightly above $20/tCO2. However, even above this point, it could be more profitable to operate at reduced CO2 capture load because increased electricity output at a sufficiently high electricity price may offset higher marginal cost. Such a scenario is more likely when there is little variation in marginal cost across the CO2 capture operating range. 161 Marginal Cost of Electricity Production ($/MWh) 100 80 60 $50/tCO2 $20/tCO2 40 $0/tCO2 20 0 0% 20% 40% 60% 80% CO2 Capture Load 100% Figure 3: Marginal cost of electricity production across the CO2 capture operating range for a representative coal-fired power plant with post-combustion CO2 capture using MEA. Table 2: Performance of a coal-fired power plant with post-combustion CO2 capture using MEA at 0% and 100% CO2 capture load. Parameter 0% Load 100% Load Output (MW) 400 303 Heat Rate (MMBTU/MWh) 11.0 14.5 CO2 Emissions Rate (tCO2/MWh)5 1.0 0.13 Due to the additional maintenance costs assumed for flexible systems, the marginal cost of a flexible CO2 capture system at 100% load is approximately $5/MWh greater than that of a system designed for continuous operation at 100% load. This marginal cost difference, while small, could have implications for the relative competitiveness of flexible and inflexible CO2 capture systems in the electricity market. Modeling Flexible CO2 capture in response to hourly variations in the electricity market The marginal cost of electricity production at coal-based plants with CO2 capture can then be used in a basic model of electricity dispatch and the electricity market in order to assess how flexible operation in response to hourly variation in electricity demand will affect plant and electric grid performance, CO2 emissions, and economics. The ERCOT electric grid is considered exclusively as a case study. 5 Although CO2 removal is 90% when CO2 capture is at 100% load, the increased fuel consumption per energy output (heat rate) due to the energy required for CO2 capture results in a net CO2 avoided of approximately 87%. 162 Electricity Dispatch and ERCOT Market Operations The model inputs hourly electricity demand, fuel and CO2 prices, and performance parameters of each plant in the ERCOT grid, then calculates the marginal cost of electricity production for each power generation facility. Fuel and CO2 prices may vary throughout the year depending on the market structure for each commodity, but prices are kept constant in this analysis to represent an annual average value. For each hour over a one-year period, power plants are sorted by their marginal generation cost and dispatched from least to most expensive until demand is met. Individual coal and natural gas-fired plants are represented based on individual plant performance, nuclear plants are assigned a representative cost of nuclear electricity generation, and other plant types are assigned generation costs representative of wind generation because wind capacity is the predominant contributor to the remainder of ERCOT generation (ERCOT, 2007). In order to account for the average availability of each power plant, maximum rated capacity is adjusted down, and the resulting “installed” capacity is used in model calculations. Plant availability is influenced by factors such as the frequency of maintenance outages and unreliable production inputs (such as wind speed in the case of wind turbines). Aside from some regulated areas, ERCOT is a competitive market for electricity, so the model determines a market electricity price by setting price equal to the marginal cost of the last (and most expensive) plant that is dispatched in a given hour. In practice, generator bids are used to price electricity rather than actual marginal costs, but this analysis assumes that bids equal marginal costs. Marginal cost is assumed constant across the operating range for all plants except for flexible CO2 capture plants. The calculated electricity price represents the wholesale market value for electricity, not the average retail consumer price that would include additional charges for transmission and retail service. Having apportioned plant output and determined an electricity price, the model can then calculate the operating profits of each plant in a given hour. Hourly output at each plant can also be used to determine CO2 emissions and the utilization patterns of CO2 capture systems. This basic representation of plant dispatch and the ERCOT electricity market does not account for transmission constraints or plant level constraints such as ramp rates, minimum output, and actual efficiency curves. The effects of other auxiliary electric grid service markets on plant generation are also ignored. These limitations prevent the model from determining a highly accurate dispatch order, but its simplified representation still provides an effective framework to analyze flexible CO2 capture in the ERCOT grid and make general comparisons between scenarios with and without CO2 capture that may or may not be able to operate flexibly. Model Representation of Flexible CO2 Capture Systems It is likely infeasible to design a CO2 capture system that operates efficiently across its operating range, so flexible CO2 capture facilities in the model are restricted to operate at either 20% or 100% load. To represent a worst-case environmental scenario, CO2 that is not captured at the 20% load condition is assumed to be vented to the atmosphere. CO2 removal and capture system energy performance at each operating point are summarized in Table 3. System response time is not incorporated explicitly in the model, but it is assumed that results from one-hour calculation intervals will approximate those found using the 1–2 hour response time found in process modeling results (Ziaii, Cohen et al., 2008). 163 Table 3: Performance of CO2 capture systems at 20% and 100% load. Parameter CO2 Capture Energy Requirement (MWh/tCO2) CO2 Removal (%) 20% Load 100% Load 0.281 0.269 18 90 Scenarios Considered The following scenarios are considered. (1) BAU: The business as usual scenario does not allow for any CO2 capture facilities. (2) CCS Base: For the base case CO2 capture scenario, CO2 capture systems are operated at 100% load continuously throughout the year. (3) FLEX Op Costs: In this flexible scenario, plants with CO2 capture choose the operating condition (20% or 100% load) that has the lowest marginal cost of electricity production. As mentioned in Section 2, the assumed CO2 price has a major influence on the operating point that has the lowest marginal cost. (4) FLEX Profit: This flexible scenario operates under the assumption of perfect knowledge of electricity demand and other generator costs prior to deciding whether to operate CO2 capture at 20% or 100% load. In every hour, each plant with CO2 capture calculates its hourly profits for two scenarios: if all plants with CO2 capture operate at (A) 100% load or (B) 20% load. If profits are greater for a particular plant for Option A, that plant will operate capture at 100% load; otherwise, it will operate at 20% load. Given enough CO2 capture facilities, there is a large reduction in output when all plants with CO2 capture operate at 100% load, so Option A is likely to require higher cost generators to come online to meet demand, resulting in a higher electricity price. Incorporating dynamic model results into a plant life economic analysis The dynamic model calculates annual totals of plant generation and CO2 emissions as well as operating profits that can be treated as annual cash flows in a discounted cash flow analysis. In order to use the dynamic model to calculate these annual flows over the lifetime of a power plant, model inputs must be tailored for the specific year of interest. Required inputs include fuel and CO2 prices, electricity demand, and the current fleet of available power plants. Cash Flow Analysis Methodology Using NETL guidelines, the dynamic model is run for the suggested plant lifetime of 20 years (NETL, 2005). The year 2012 is chosen as the starting date because it is the first year in which CO2 prices appear in the representation of CO2 prices described below. This starting point is perhaps too early to expect several commercial-scale CO2 capture facilities to be built; however, the specific choice of starting date is not expected to significantly change the general qualitative results. 164 Cash flow analysis is performed for each coal-fired power plant being considered for postcombustion CO2 capture. The analysis uses the NETL suggested discount rate of 7.1% for a high risk project and an inflation rate of 3%, resulting in an effective discount rate of 10.3% (NETL, 2005). Any capital expenditures are depreciated on a 20-year MACRS6 schedule, and profits after capital deductions are taxed at a rate of 38%, which includes both state and federal income taxes (NETL, 2005). The year 2012 is the first year of annual cash flows, and all capital expenditures are applied in the previous year. Capital and annual cash flows are used to calculate the net present value (NPV) and annual worth for power plants being considered for CO2 capture. Fuel and CO2 Price Paths Rather than arbitrarily project fuel and CO2 price paths over the plant lifetime, prices are taken from the Core case of the Energy Information Administration’s (EIA) analysis of the LiebermanWarner Climate Bill (S.2191). The Core case considers a cap and trade system for CO2 emissions allowances that includes bonus incentives for CCS technologies and assumes that critical low emissions technologies such as CCS are deployed in a time frame consistent with the bill’s emissions reduction goals. Though this bill did not pass, and commodity prices are exceedingly difficult to predict, the EIA analysis represents a reasonable estimate of market prices based on a contemporary model of CO2 legislation. These price projections are shown in Figure 4 below7. Delivered fuel prices reported in the raw EIA data already include the CO2 penalty, so fuel prices without additional CO2 costs are displayed below as estimated from EIA’s minemouth coal and wellhead natural gas price data. Both coal and natural gas prices are projected to remain relatively stable, and coal prices generally decrease over time because increasing CO2 cost leads to decreased coal demand. CO2 allowance prices enter the market at $16.9/tCO2 in 2012 and increase to $65.5/tCO2 in 2031. 70 8 60 Natural Gas Price 50 6 4 40 30 CO2 Price 20 2 10 Coal Price 0 2012 CO2 Price ($/tCO2) Coal and Natural Gas Price ($/MMBTU) 10 0 2016 2020 2024 Year 2028 Figure 4: Fuel and CO2 price projections from the EIA analysis of the Lieberman-Warner Climate Bill (S.2191) (USEIA, 2008). 6 MACRS: Modified Accelerated Cost Recovery System EIA projections end at 2030. Values for 2031 are extrapolated from a linear fit of the data points in 2029 and 2030. 7 165 ERCOT Electricity Demand Historical ERCOT hourly electricity demand in 2006 is used to characterize hourly demand variation over the course of a year. From these data, demand in each hour is then scaled up using the estimated long-term demand growth projected by ERCOT. Actual short-term demand variation will depend primarily on weather patterns that vary by year, but using a single data set is sufficient to characterize typical diurnal and seasonal demand variations. ERCOT’s 2008 long-term energy forecast projects a demand growth of 1.79% per year until 2018; for purposes of this study, that demand growth rate is continued until 2031 (ERCOT, 2008). The resulting estimate is a total of 342 terawatt hours (TWh) of electricity consumed in 2012, increasing to 480TWh in 2031. Electricity demand is considered to be inelastic to any electricity price changes that may result from changes in fuel price, CO2 price, or power plant utilization. The wholesale market price for electricity is not reflected directly in the price to most electricity consumers, so the extent to which a change in wholesale electricity prices would affect demand is unclear. ERCOT has found it difficult to relate wholesale electricity price to electricity demand, so no model of demand elasticity is incorporated into the analysis presented here (ERCOT, 2008). ERCOT Power Plant Fleet The Environmental Protection Agency’s (EPA) eGRID power plant database is used for performance parameters of all plants installed through 2004. To represent the ERCOT plant fleet through 2031, additional power plants are added based on installations since 2004 and planned installations through 2018 contained in reports by the Public Utilities Commission of Texas (PUC) and the Texas Water Development Board (TWDB). These reports present only rated power plant capacities, so any required performance parameters are assumed to be similar to those of typical plants of the same type. This practice results in a large number of coal and natural gas-based plants having roughly the same performance characteristics. The positions of these groups of similar plants in the dispatch order could have significant effects on model results, so a more accurate methodology may seek to logically differentiate performance of new fossil fuel-fired plants. Figure 5 displays the resulting estimate of the ERCOT fleet through 2031 in terms of installed capacity in gigawatts. There are several coal-fired plant installations planned through 2013, three of which are Integrated Gasification Combined Cycle (IGCC) facilities. Three new petroleum coke-fired plants are also included in the total coal-fired capacity because these plants have similar performance characteristics to coal-based plants. Because wind turbines, the major contributor to the “Wind/Other” category, are typically built on a relatively short-term planning period, there are currently no planned wind turbine installations after 2012. Existing nuclear power plants in Texas are scheduled for a capacity doubling in 2015, and two new nuclear plants are in early planning stages to come online in 2018. Obvious regulatory and economic hurdles remain for any large scale nuclear power projects in the U.S., but this analysis assumes that these plants are installed as planned. Some additional natural gas-fired facilities are also to be installed through 2014. The current planning horizon does not extend past 2018, but new natural gas-fired capacity is included in 2029–31 so that there is enough capacity available to meet peak demand. Natural gas is chosen for this new capacity in an attempt to minimize the potential effect on coalfired generation, which is the main focus of this study. 166 Study Period Installed Capacity (GW) 100 80 Natural Gas 60 Wind/Other 40 Nuclear 20 Coal 0 2006 2010 2014 2018 2022 Year 2026 2030 Figure 5: Estimate of the installed capacity of each power plant type in the ERCOT grid through 2031 (USEPA, 2007; King, Duncan et al., 2008; ERCOT, 2008). For scenarios that allow CO2 capture operation, all standard8 coal-fired power plants installed after 2006 are assumed to have post-combustion CO2 capture systems installed by 2012. In addition, CO2 capture is assumed to be retrofitted on 8 of the 15 coal-based plants installed prior to 2006. Plants in service prior to 2006 that utilize CO2 capture are chosen based on the capital cost of the CO2 removal system, electricity production costs when CO2 capture operates at 100% load, and the capital costs of any required sulfur dioxide (SO2) removal systems. High SO2 removal is a prerequisite to amine absorption/stripping because the presence of SO2 in the flue gas entering the CO2 capture system will quickly degrade the amine solvent (Davidson, 2007). Eight plants are chosen so that operating CO2 capture continuously at 100% load will reduce the emissions rate of the 2006 coal-fired fleet by 50%, resulting in a fleet average CO2 emissions rate near that of typical natural gas-fired facilities. The EPA’s New Source Performance Standards (NSPS) require SO2 removal systems to be installed on any new power plants, so it is assumed that additional SO2 removal is not required on these facilities9 (USEPA, 1990). Proximity to a suitable CO2 storage site will also be required for economical installation of CO2 capture systems. Because of the amount and distribution of CO2 storage capacity across Texas in the form of Gulf Coast brine reservoirs and oil wells that are candidates for CO2 enhanced oil recovery (EOR), this constraint is not used to eliminate any plants from candidacy for CO2 capture (Ambrose, Breton et al., 2006). Land availability for CO2 capture systems is another concern, but no data on land availability at each plant have been collected at this time. The magnitude of CO2 capture deployment considered in this report may not be consistent with EIA’s assumptions regarding installation of CCS technology and could be considered more ambitious than even the boldest rollout schedules, but the purpose for including a large amount 8 “Standard” refers to all coal-based power plants except those using IGCC technology. Amine absorption/stripping may require more SO2 removal than is required by the NSPS, but this analysis does not consider the requirement to upgrade SO2 removal systems at plants with planned or existing SO2 scrubbing equipment. 9 167 of CO2 capture capacity is to clearly show the general plant and grid level impacts of flexible CO2 capture. Results and Discussion Generation Patterns and CO2 Emissions The calculated electricity generation by plant type allows initial insight into the economic performance of power plants with CO2 capture. Figure 6 displays generation by plant type for the Business as Usual scenario throughout the time horizon considered. Wind/Other generation remains constant because there are no new capacity installations after 2012 and wind turbines always have the lowest marginal cost of generation. Nuclear capacity, the next lowest cost generation, is always utilized to its maximum installed capacity, so the two large planned nuclear installations in 2015 and 2018 result in two major increases in nuclear generation. On average, coal-fired generation remains less expensive than natural gas-based generation throughout the time horizon despite rising relative CO2 costs, so new nuclear generation comes more at the expense of natural gas-fired generators than those which burn coal. Because rising CO2 costs have already caused the least efficient coal-fired plants to be displaced in the dispatch order by efficient natural gas-fired generators by 2018, coal-based generation remains relatively steady after this date, with increasing electricity demand met by natural gas-fired facilities. Despite CO2 prices rising to over $65/t in 2031, low coal prices relative to natural gas prices coupled with the relatively small fraction of total ERCOT capacity consisting of coal-based generation means that fuel switching in ERCOT from coal to natural gas is relatively modest. As inefficient coal-fired plants are utilized less, new coal-based plants take their place, resulting in relatively steady total coal-based generation across the entire time horizon.The generation mix over time for the three CO2 capture scenarios shares the general features of Figure 6. Coal-based generation is 19–24 million MWh less in the CCS Base scenario until 2017, prior to which marginal costs are lower for coal-fired plants without CO2 capture than those operating CO2 capture at 100% load. Annual coal-fired generation remains 11–13 million MWh lower for all CO2 capture scenarios than in BAU until about 2023 because even though coal-fired plants with CO2 capture have lower marginal costs, utilization is still nearly as high without CO2 capture, and these plants have greater output. 168 Generation (million MWh) 500 400 Natural Gas 300 200 Coal 100 Nuclear Wind/Other 0 2012 2016 2020 2024 Year 2028 Figure 6: Generation by plant type over the 20-year study period in the BAU scenario. Figure 7 displays the cumulative generation of all coal-fired facilities being considered for CO2 capture across the 20-year study period for each of the four scenarios. These plants generate more electricity in the BAU scenario throughout the time horizon because plants have greater output without CO2 capture and are utilized almost as often as when CO2 capture is installed. Even when marginal costs are higher without CO2 capture than with CO2 capture operating, plants in BAU are still dispatched primarily at base load because costs at the majority of these coal-fired facilities are still lower than costs of most natural gas-fired plants. All four curves display declines in coal-based generation when new nuclear capacity comes online. Prior to 2015, plants with CO2 capture generate more electricity in flexible CO2 capture scenarios than in CCS Base because prior to this date, marginal costs are lower with CO2 capture at 20% load for flexible CO2 capture facilities. Marginal costs are still lower for inflexible CO2 capture facilities at 20% load in 2015, but utilization begins to decrease relative to the inflexible CO2 capture plants in CCS Base because of the $5/MWh additional cost of flexibility. The decrease in utilization is especially significant considering that plants at 20% load have greater output than the inflexible CO2 capture plants operating at 100% load. All flexible CO2 capture plants have lower marginal costs at 100% load beginning in 2017. Utilization at flexible plants is noticeably less than at inflexible plants from 2017–22 because of the incremental cost of flexibility. As CO2 price and electricity demand increase over time, utilization of facilities in both flexible and inflexible CO2 capture scenarios approach a maximum, resulting in annual generation after 2022 being approximately equal across all CO2 capture scenarios. Overall, there is little difference between the FLEX Op Costs and FLEX Profit scenarios, suggesting that there is little opportunity to find appropriate combinations of output, cost, and price that achieve substantially higher profits than those earned when operating at the lowest marginal cost. Generation is slightly lower for FLEX Profit than FLEX Op Costs until 2016, suggesting that there is actually opportunity to improve profits by restricting plant output through 100% load CO2 capture operation. From 2017–22 plants generate slightly more electricity in FLEX Profit because there are some hours when increased electricity sales at 20% load offset the increased cost of CO2 emissions. 169 The utilization of CO2 capture in the CCS Base and flexible scenarios is made clear in Figure 8, which plots only the subset of generation at facilities with CO2 capture when the CO2 capture system operates at 100% load. CCS Base can only operate at 100% load, so its curve is the same as in Figure 7. FLEX Op Costs clearly shows the transition in 2016 and 2017 when plants with flexible CO2 capture systems become less expensive to operate at 100% load than at 20% load. Before 2016, FLEX Profit finds some combinations of output, cost, and electricity price that make it profitable to operate at 100% load even though marginal costs are higher at this operating point. This result indicates that there may be times when it is profitable for plants with CO2 capture to use 100% load CO2 capture to withhold capacity and facilitate higher electricity prices. This behavior suggests that flexible CO2 capture could potentially be used to exercise market power. In 2017–19, FLEX Profit chooses to operate CO2 capture at 100% load much less often than FLEX Op Costs because during these years, fuel and CO2 prices allow for times when profits are greater with CO2 capture at 20% load despite higher marginal costs. After 2019, FLEX Profit closely follows FLEX Op Costs. Annual Generation - CO2 Capture Plants (million MWh) BAU CCS Base FLEX Op Costs FLEX Profit 120 100 80 60 40 20 0 2012 2016 2020 Year 2024 2028 Figure 7: Cumulative generation at coal-fired plants being considered for CO2 capture across the 20-year study period for each scenario. 170 Annual Generation With Capture at 100% (million MWh) CCS Base FLEX Op Costs FLEX Profit 100 80 60 40 20 0 2012 2016 2020 Year 2024 2028 Figure 8: Subset of the cumulative generation at coal-fired plants being considered for CO2 capture when the CO2 capture system is operating at 100% load. The generation patterns described above are reflected in the annual CO2 emissions at all coalfired facilities, shown in Figure 9. New nuclear installations result in some emission reduction at coal-fired facilities, most noticeably in the BAU scenario. The degree of utilization of CO2 capture, however, has the most significant effect on CO2 emissions at coal-based plants. Until plants with CO2 capture are less expensive to operate at 100% load, CO2 emissions are higher for flexible scenarios because CO2 that is not captured at 20% load is assumed to be vented to the atmosphere. After 2020, emissions remain relatively stable. While emissions from coal-fired plants trend slightly downwards for each scenario throughout the 20-year study period, total emissions in the ERCOT grid tend to increase in most years when there is not a major addition to nuclear capacity (Figure 10). The only major exceptions are the years when flexible CO2 capture scenarios are transitioning from the period where 20% CO2 capture load has lowest marginal costs to where 100% load is most economical. In years without nuclear installations, any decrease in coal-based generation must be met with increased use of natural gas. Though plants using natural gas emit roughly half the CO2 of a coal-fired facility per unit of electricity generated, this offset, combined with increased natural gas use to meet growing electricity demand, causes ERCOT grid emissions to increase after 2018 except for a few years in the flexible CO2 capture scenarios. For BAU, annual ERCOT emissions in 2031 are only 3.5% below annual emissions in the initial year 2012, though the lowest emitting year 2018 has 22% less CO2 than in 2012. Scenarios with CO2 capture allow much greater emissions cuts; relative to annual BAU emissions in 2012, CCS Base achieves over a 56% reduction in 2018 and 38% in 2031. Annual emissions are significantly greater in flexible CO2 capture scenarios than in CCS Base until 2017, causing cumulative 20-year CO2 emissions in flexible scenarios to be 13–14% greater than those in the CCS Base scenario. However, cumulative emissions in flexible scenarios are still 32–33% less than those in BAU, compared with 40% less in CCS Base. 171 Annual Coal CO2 Emissions (million tCO2) BAU 200 CCS Base FLEX Op Costs FLEX Profit 160 120 80 40 0 2012 2016 2020 Year 2024 2028 Figure 9: Annual CO2 emissions from all coal-fired facilities across the 20-year study period for each scenario. Annual ERCOT CO2 Emissions (million tCO2) BAU CCS Base FLEX Op Costs FLEX Profit 200 160 120 80 40 0 2012 2016 2020 Year 2024 2028 Figure 10: Annual CO2 emissions in the entire ERCOT grid over the 20-year study period for each scenario. Electricity Price Implications Figure 11 plots annual average electricity prices in each scenario across the 20-year period. In a given year, the electricity price is nearly the same for all four scenarios, indicating that CO2 capture has a relatively small effect on electricity prices. Outside of major changes to base load generation capacity, electricity prices generally follow trends in CO2 prices (Figure 4). Electricity prices decrease in 2013 and 2014 when new low-cost coal-fired generation comes online and in 2015 and 2018 when new nuclear capacity becomes operational. These new lowmarginal cost plants allow less expensive power plants to be dispatched as the marginal generating facility. Because coal and natural gas prices are stable relative to CO2 prices, 172 increasing electricity costs after 2018 reflect additional CO2 costs at the marginal generating facilities. The only effect CO2 capture has on electricity price is to reduce coal-based output during CO2 capture operation, thus requiring slightly more expensive generators to be dispatched as the marginal generating facility. However, any such effect is exceedingly small by the early 2020s. Avverage Annual Electricity Price ($/MWh) BAU 100 CCS Base FLEX Op Costs FLEX Profit 90 80 70 60 50 2012 2016 2020 2024 Year 2028 Figure 11: Annual average electricity prices for each scenario over the 20-year study period. Economic Performance of Plants with CO2 Capture Electricity price trends and the generation at plants being considered for CO2 capture are reflected in annual operating profits before the application of profit taxes or any deductions from capital depreciation. Figure 12 displays these results in current dollars. Operating profits in the BAU scenario are several hundreds of millions of dollars greater than in all CO2 capture scenarios through 2015, when fuel and CO2 prices are too low to justify 100% load CO2 capture. Profits for CCS Base exceed those in the BAU scenario starting in 2018, and flexible scenarios follow in 2021, which reflect the dates when plants in these CO2 capture scenarios generally have lower marginal costs at 100% CO2 capture load than plants without CO2 capture. In CO2 capture scenarios, profits climb steadily after 2018 because electricity prices rise with the emissions costs of higher emitting natural gas and coal-based generators without CO2 capture. Costs at facilities with CO2 capture do not rise as quickly as electricity prices, and utilization remains high, so annual profits increase. Because the highest emitting coal-based facilities in BAU set the electricity price 39–56% of the time in the years after 2018, profits for the coalbased facilities being considered for CO2 capture increase modestly in most years after 2021. Decreasing coal prices also help offset high CO2 prices when facilities do not have CO2 capture. However, high CO2 prices in later years allow profits to be several hundreds of millions or even billions of dollars greater when plants utilize CO2 capture. Flexible scenarios only have greater operating profits than CCS Base in 2012, when marginal costs are lower for flexible plants at 20% load than inflexible plants at 100% load, and because plant output is greater at 20% CO2 capture load. After inflexible plants at 100% load have lower 173 marginal costs than flexible plants at either operating point, profits in flexible scenarios are less than those of CCS Base because of the incremental marginal cost of a flexible system. While previous work has found that some combinations of fuel and CO2 prices allow the behavior implemented in FLEX Profit to be economically advantageous, those conditions do not occur often enough to provide a major profit improvement with the inputs used in this analysis (Ziaii, Cohen et al., 2008). Annual Before-Tax Operating Profit (Billion $) BAU 4 CCS Base FLEX Op Costs FLEX Profit 3 2 1 0 2012 2016 2020 2024 Year 2028 Figure 12: Annual before-tax operating profits at plants considered for CO2 capture in each scenario across the 20-year study period. Each of the four scenarios has capital cost implications that include the costs of installing CO2 capture, installing any required SO2 scrubbing equipment in a retrofit application, and replacing generation capacity lost to CO2 capture energy requirements. These capital costs are summarized in Table 4. While CCS Base requires enough new generation capacity to replace the output lost to operating CO2 capture at 100% load, flexible scenarios only require new capacity to replace the output lost at 20% load CO2 capture. The particular choice of replacement capacity will depend on electric grid requirements as well as investment decisions made by utility companies. Rather than attempt to predict such tendencies, replacement capacity is assumed to be new coal-fired generation with SO2 scrubbing and CO2 capture installed. In order to maintain this study’s focus on the implications of installing CO2 capture on plants that currently exist or appear in recent planning documents, the replacement capacities indicated below are not actually incorporated into the plant fleet in the model. Nevertheless, these capital costs are included in the discounted cash flow analysis to represent a system level cost associated with the output reduction caused by CO2 capture operation. 174 Table 4: Required equipment and the total capital requirement in each scenario10 (Rubin, 2007; USNETL, 2007). CO2 Capture Capacity (MW) Retrofit SO2 Capture Capacity (MW) New Generation Capacity (MW) Total Capital Requirement (Billion $) 0 0 0 0 CCS Base 14852 1640 3694 24.5 FLEX Op Costs 14852 1640 776 16.1 FLEX Profit 14852 1640 776 16.1 Scenario BAU When taxes and tax deductions from capital depreciation are applied to the annual cash flows discussed above, plants pay much higher taxes in BAU than in CO2 capture scenarios. As a result, after-tax cash flows in CCS Base are higher than in BAU for all years, and cash flows in flexible CO2 capture scenarios are nearly the same as in BAU until 2018 and much higher thereafter. Table 5 summarizes the cumulative net present value and annual worth for plants considered for CO2 capture using after-tax annual cash flows and the capital costs in Table 4. The only scenario with positive values of these economic indicators is BAU, which has no capital expenditures. All CO2 capture scenarios have large negative NPV and annual worth; however, flexible scenarios are much less negative than CCS Base because there is less new generation capacity required in these scenarios. Despite decreasing coal prices and high utilization of plants with CO2 capture, CO2 prices are insufficient to allow power plants with CO2 capture to come close to recovering their capital investment within the 20 years considered here. Only 50% of the capital cost is recovered in CCS Base and 61% in flexible scenarios, indicating that several more decades of operation or major changes to technology, policy, or electricity market conditions may be required to completely recover initial capital expenditures. Even with significantly improved economics for coal-fired facilities with CO2 capture, discounting the cash flows occurring beyond this study’s 20-year time frame would greatly reduce their present value and contribution to annual worth. FLEX Profit has better long-term economics than FLEX Op Costs, but the difference is quite small. Table 5: NPV and Annual Worth for each scenario Net Present Value (Billion $) Annual Worth (Billion $) BAU 7.10 0.852 CCS Base -12.1 -1.46 Scenario 10 The capital costs used to calculate the total capital requirement include: CO2 capture equipment that costs $908/kW, SO2 scrubbing equipment that costs $221/kW, and new coal-based capacity with SO2 and CO2 removal that costs $2895/kW. 175 FLEX Op Costs -6.29 -0.755 FLEX Profit -6.21 -0.745 As mentioned above, it is unclear what contribution the operators of the CO2 capture plants considered here would make to any replacement capacity and what type of power plant would be used for replacement capacity. If replacement capacity were excluded from the cash flow analysis described above, flexible scenarios would have a NPV loss about $1.2 billion greater than the $3.16 billion loss in CCS Base. Conclusions At a coal-fired power plant with flexible post-combustion CO2 capture using amine absorption and stripping, the marginal cost of electricity production varies almost linearly with CO2 capture load, and the average slope of this curve is highly dependent on CO2 price. Even after CO2 prices are sufficient for 100% load to have the lowest marginal costs, a plant may be more profitable at reduced load if the increase in cost is offset by increased output at a sufficiently high electricity price. At 100% CO2 capture load, the cost analysis in this study finds that a flexible CO2 capture system has a $5/MWh incremental increase in marginal cost over an inflexible CO2 capture plant. This increase is due largely to an assumed 230% increase in maintenance costs, which may be an overly pessimistic estimate of the additional maintenance required for a flexible system. A modeling approach was employed that used fuel and CO2 price projections under the Lieberman Warner Climate Bill, ERCOT projected electricity demand, and the current and planned ERCOT power plant fleet as inputs in a dynamic model of hourly electricity dispatch in order to investigate the long-term implications of flexible CO2 capture. The approach examines the behavior of all power plant types from 2012–31 for scenarios with no CO2 capture, inflexible CO2 capture, and flexible CO2 capture. In general, long-term changes to each power plant type’s contribution to meeting electricity demand reflect the relative costs of each plant type. Coal-based generation remains less expensive on average than natural gas-based generation regardless of CO2 capture, so new nuclear installations and rising CO2 prices cause only modest reductions in the annual generation at coal-fired plants. As a result, annual emissions reductions at coal-fired facilities and across the electric grid are only significant when CO2 capture is available, and annual electric grid emissions increase after 2018 due to increased use of natural gas to meet rising electricity demand. Depending on base plant efficiency and whether or not CO2 capture is flexible, marginal costs at a base plant are less than those with CO2 capture until the late 2010s and early 2020s. At plants being considered for CO2 capture, greater output allows for greater annual generation in the scenario without CO2 capture than in all CO2 capture scenarios throughout the time horizon, but annual profits in the non-CO2 capture scenario exceed those with CO2 capture only until 2018 with inflexible CO2 capture and 2020 with flexible CO2 capture. After these crossover dates, annual profits are several hundreds of millions of dollars greater in CO2 capture scenarios than in the scenario without CO2 capture. Flexible CO2 capture plants have greater generation than in 176 inflexible scenarios until 2016, after which it is more economical to operate CO2 capture at 100% load most or all of the time. However, the $5/MWh incremental cost of flexibility results in lower annual profits in flexible CO2 capture scenarios that with inflexible CO2 capture after 2012. Relative to the scenario without CO2 capture, cumulative CO2 emissions across all 20 years are 40% less with inflexible CO2 capture and 32–33% less for flexible CO2 capture scenarios. No plants with flexible CO2 capture have lower marginal costs at the 100% load operating point before 2016, but there are times before this date when combinations of marginal cost, output, and electricity price allow plants to earn additional profits at 100% load by withholding output and driving up electricity prices. There could be market conditions such as these that create incentives to use flexible CO2 capture to exercise market power, so it may be important for the electricity market operator to ensure that flexible CO2 capture is used in correspondence with market protocols. Despite the large deployment of CO2 capture systems in this analysis, the existence and operation of CO2 capture has little influence on electricity prices, which are affected primarily by new base load plant installations and trends in CO2 prices. When all capital costs are considered, a 20-year cash flow analysis for plants being considered for CO2 capture finds that the cumulative net present value across all plants is positive only in the scenario when no CO2 capture systems are installed. When plants have flexible CO2 capture systems, they only recover 61% of total capital expenditures over the 20-year period, and a case with inflexible systems recovers just 50%. Flexible CO2 capture scenarios experience over $6 billion in net present value loss, though this quantity is significantly better than the $12 billion loss when CO2 capture is not flexible. Under the model inputs considered here, there is only a small long-term economic advantage to operating flexibly in response to hourly profits as opposed to choosing the CO2 capture operating point that has the lowest marginal cost. Flexibility can provide substantial capital cost savings by eliminating the need to replace the output lost to CO2 capture, but any additional marginal production costs associated with flexibility can lead to lower utilization and operating profits in a competitive market for electricity. A more rigorous consideration of the costs of flexibility would provide a better representation of the operational differences between flexible and inflexible CO2 capture systems. Repeating this methodology for alternative projections of electricity demand, fuel and CO2 prices, and capacity installation would provide additional insight into the conditions when CO2 capture is economically feasible and if flexibility provides a significant economic advantage. References Ambrose WA, Breton CL, et al. Source-Sink Matching and Potential for Carbon Capture and Storage in the Gulf Coast. Austin, GCCC, BEG, Jackson School of Geosciences, The University of Texas at Austin. 2006. Chalmers H, Gibbins J, et al. Initial Assessment of Flexibility of Pulverised Coal-Fired Power Plants with CO2 Capture. 3rd International Conference on Clean Coal Technologies for our Future. Sardinia, Italy. 2007. 177 Cohen SM, Rochelle GT, et al. Turning CO2 Capture on & off in Response to Electric Grid Demand: a Baseline Analysis of Emissions and Economics. ASME 2nd International Conference on Energy Sustainability. Jacksonville. 2008. Davidson RM. Post-combustion carbon capture from coal fired plants – solvent scrubbing, IEA Clean Coal Centre. 2007. Davis J. Input on degradation in a flexible CO2 capture system. S. Cohen. 2008. ERCOT. 2007 Annual Report. 2007. ERCOT 2008 Capacity, Demand, Reserves Report. 2008_Capacity_Demand_Reserves_Report_FINAL.xls. Taylor, TX, System Planning. 2008. ERCOT. 2008 Long-Term Hourly Demand Energy Forecast. ERCOT 2008 Planning. 2008. IEA and NEA. Projected Costs of Generating Electricity: 2005 Update. Paris, France, OECD. 2005. King, C, Duncan I, et al. Water Demand Projections for Power Generation in Texas. Austin, TX, Bureau of Economic Geology. 2008. Metz B, Davidson O et al. IPCC Special Report on Carbon Dioxide Capture and Storage. 2005. NEI. U.S. Electricity Production Costs and Components. u.s._electricity_production_costs_and_components.xls. Washington, D.C. 2007. NETL. Carbon Capture and Sequestration Systems Analysis Guidelines. USDOE. 2005. Pacala S, Socolow R. Stabilization Wedges: Solving the Climate Problem for the Next 50 Years with Current Technologies. Science. 2004;305(5686):968–972. Rao AB, Rubin ES. A Technical, Economic, and Environmental Assessment of Amine-Based CO2 Capture Technology for Power Plant Greenhouse Gas Control. Env Sci Tech. 2002; 36(20):4467–4475. Rao AB, Rubin ES, et al. An Integrated Modeling Framework for Carbon Management Technologies. Final report to DOE/NETL (Contract no. DE-FC26-00NT40935). Pittsburgh, PA, Center for Energy and Environmental Studies, Carnegie Mellon University. 2004. Roughgarden T, Schneider SH. Climate change policy: quantifying uncertainties for damages and optimal carbon taxes. Energy Policy. 1999;27(7):415–429. Rubin, ES, Chen C, Rao AB. Cost and performance of fossil fuel power plants with CO2 capture and storage. Energy Policy. 2007;35:4444–4454. USEIA. Electric Power Annual 2006. Washington, DC, US Department of Energy. 2007. USEIA. World Carbon Dioxide Emissions from the Use of Fossil Fuels. Int En Ann. 2005 Retrieved Oct. 26 2007, from http://www.eia.doe.gov/emeu/iea/carbon.html. 2007. USEIA. Energy Market and Economic Impacts of S. 2191, the Lieberman-Warner Climate Security Act of 2007. Washington, DC, Office of Integrated Analysis and Forecasting, USDOE. 2008 USEPA. Clean Air Act. 1990;40 CFR 60:106–121. 178 USEPA. Emissions & Generation eGRID2006_Version_2_1. 2007. Resource Integrated Database (eGRID). USNETL. Cost and Performance Baseline for Fossil Energy Plants. Bituminous Coal and Natural Gas to Electricity. J. M. Klara. 2007; 1. Ziaii S, Cohen SM, et al. Dynamic operation of amine scrubbing in response to electricity demand and pricing (in review). 9th International Conference on Greenhouse Gas Technologies. Washington, DC, Elsevier. 2008. 179 Oxidative Degradation and Thermal Degradation Experiments Quarterly Report for October 1– December 31, 2008 by Fred Closmann Supported by the Luminant Carbon Management Program, the Industrial Associates Program for CO2 Capture by Aqueous Absorption, and the Process Science & Technology Center Department of Chemical Engineering The University of Texas at Austin February 2, 2009 Abstract In this quarter, we conducted a single oxidative degradation experiment and a single thermal degradation experiment on 7 m MDEA/2 m PZ loaded to 0.24 moles CO2/mole alkalinity. The oxidative degradation experiment solvent was amended with 1 mM Fe2+ and 100 mM Inhibitor A. We measured a formate production rate of 0.011 mM/hr, which is comparable to rates with the same solvent in the absence of Inhibitor A. Hydrolyzed samples resulted in a formate production rate (0.02 mM/hr) that was approximately double the rate when hydrolysis was not performed. In hydrolyzed samples, the oxalate production rate was 0.007 mM/hr, but no other heat stable salt was consistently observed in the study. A foaminess factor of 8.65 was measured in the undegraded solvent, but a foaminess factor of >20 was observed with the degraded solvent. A thermal degradation experiment on the 7 m MDEA/2 m PZ solvent was performed at temperatures of 135 and 150 °C. The loss of MDEA and PZ occurred at a greater rate at 150 °C, with complete loss of PZ occurring somewhere between days 42 and 57 at both temperatures at a loading of 0.11 moles CO2/mole alkalinity. The concentration of MDEA and PZ diminished on an equimolal basis, indicating that the MDEA and PZ are reacting together resulting in loss of both compounds until the PZ is completely degraded. We will be investigating disproportionation reactions to identify the byproducts of this mechanism. We performed wetted wall column studies on the 7 m MDEA/2 m PZ solvent at 80 and 100 °C and about 0.03 moles CO2/mole alkalinity, and measured kg’ to be 2. 76 X 10-10 mols/s-Pa-cm2 at a partial pressure of CO2 of 1,273 Pa, and 1.63 X 10-10 mols/s-Pa-cm2 at a partial pressure of CO2 of 5,212 Pa, respectively. Oxidative Degradation Experiment (OD-5) In this quarter we concluded an oxidative degradation experiment (OD-5) with 7 m MDEA/2 m PZ with 1 mM FeSO4 and 100 mM Inhibitor A, and loaded to 0.24 moles CO2/mole alkalinity. The experiment was performed in a low gas flow apparatus under well-mixed conditions. When sparged with 98% O2 at 55 oC, 7 m MDEA/2 m PZ with 1 mM Fe2+ and 100 mM Inhibitor A produced 0.01 mM formate/hr. At the same conditions, previous experiments with the same 180 solvent blend in the absence of Inhibitor A resulted in a comparable formate production rate (0.011 ± 0.001 mM formate/hr). Using the LINEST function, we determined the straight line fit and error of formate production to be 0.01 ± 0.001 mM/hr. The initial conclusion is that the presence of Inhibitor A did not change the oxidative degradation results measurably. Sexton (2008) measured the rate of production of formate to be 0.39 mmoles/L-hr in 7 m MEA, which is an order of magnitude greater than the amount measured in the solvent blend both with and without Inhibitor A present. When we took into account the amount of amide formed by hydrolyzing the samples with 5N NaOH and subsequently measuring formate concentration, the 7 m MDEA/2 m PZ with 1 mM Fe2+ and 100 mM Inhibitor A produced formate at a rate of 0.02 ± 0.004 mM/L-hr. In summary, when hydrolysis was performed, we observed a doubling in the formate production rate. Those results are indicative of other degradation byproducts being formed. Other heat stable salts including glycolate, acetate, and oxalate were measured in the oxidative degradation samples, but not on a consistent basis, making the estimate of production rates difficult. However, in the hydrolyzed samples, the oxalate production rate was estimated at 0.007 ± 0.001 mM/hr. Peaks possibly corresponding to acetate and glycolate appeared on an inconsistent basis in experimental samples, with the greatest concentration determined to be 335.8 ppm for acetate in the Day 14 sample. As presented in Figure 1, the formate production rate resulting from the hydrolysis of samples with 5N NaOH is twice the rate without performing the hydrolysis step. We also observed the presence of formate in the undegraded solvent after performing the hydrolysis step, possibly indicating that the sample treatment (hydrolysis) converts a compound(s) other than a heat stable salt into measurable formate. The data in Figure 1 also demonstrate that oxalate was formed at much lower concentrations than formate. Note that due to background peak interference issues, we were unable to definitely determine the concentration of oxalate in non-hydrolyzed samples in this experiment. 181 Heat Stable Salt Concentration: OD‐5 Concentration (ppm) 800.0 700.0 600.0 500.0 Formate Concentration Formate With Hydrolysis Oxalate With Hydrolysis 400.0 300.0 200.0 100.0 0.0 0 5 10 15 20 Time (days) Figure 1: OD-5, Oxidation of 7 m/2 m PZ, 1 mM Fe+2, 100 mM Inhibitor A Figure 2 presents the concentration of amines (MDEA and PZ) measured in samples collected in experiment OD-5 using cation chromatography. Important trends/changes in the overall concentrations of MDEA and PZ are generally indiscernible due to the small effect the formation of heat stable salts (ppm range) has on the overall concentration of amines (% range). We can, however, use the cation measurements to track the ratio of MDEA to PZ at each sampling point in time to ensure that the water balance and experimental control were maintained throughout the twenty-day experiment. The light blue lines and symbols in Figure 2 demonstrate that the initial ratio of MDEA to PZ was 3.5, and was maintained throughout the experiment. 182 Figure 2: Cations in OD-5 Density Measurements In addition to the degradation studies, we measured the density of both the undegraded stock solution and the solvent removed from the reactor after 20 days of oxidative degradation at 55 °C. The density measurements were made over the temperature range of 20 to 60 °C. At 50 °C, the density of the undegraded and degraded solvents was determined to be 1.079 and 1.071 g/ml, respectively. Figure 3 presents the density data with a three term polynomial fit. The data demonstrate a consistently lower density for the degraded solvent at all temperatures. Foaminess Factor Measurements The foaminess factor of the undegraded stock solvent as well as the oxidatively degraded solvent blend removed from the reactor were also determined using the apparatus set up and modified for measurements by Xi Chen. The foaminess factor for the undegraded 7 m MDEA/2 m PZ solvent was measured as 8.65, whereas, due to a large amount of foam formed within 1 minute of sparging with nitrogen in the foaming measurement apparatus, the foaminess factor for the oxidatively degraded solvent was immeasurable (>20). The level of foaming observed in the oxidatively degraded solvent from experiment OD-5, when compared to the undegraded solvent, suggests that oxidation of the solvent blend is resulting in the formation of compounds which have a tendency to cause foaming. However, when reconciled with the heat stable salt measurements, wherein the greatest amount of formate 183 measured after 20 days was 574 ppm (with amide reversal step), we conclude that a compound or compounds such as amides are being formed in the oxidative degradation of the solvent blend. From MEA, amides can be formed through the reaction of formic acid and MEA. However, amides generally do not form from tertiary amines (Morrison and Boyd, 1978), indicating that if an amide is being formed, it is likely the result of a reaction with PZ in the solvent blend. Other compounds possibly resulting in increased foaminess include glycine, bycine, and/or acetaldehydes (α-amino acetaldehyde is known to form as a step in the oxidative degradation of MEA); it is unlikely that glycine would be present in the oxidized solvent removed from the reactor due to its reactiveness with oxygen and/or the hydroxyl free radical (OH·). Figure 3: Density of 7 m MDEA/2 m PZ in Degraded and Undegraded Solvents Thermal Degradation (Thermal No. 7) We conducted a thermal degradation experiment (Thermal No. 7) on the blended solvent (7 m MDEA/2 m PZ) at loadings of 0.11 and 0.26 moles CO2/mole alkalinity, with bombs placed in ovens maintained at 135 and 150 °C. That experiment will be completed by February 2009. Preliminary results confirm the findings of earlier studies on the same solvent system. The concentration of amines (MDEA and PZ), as measured by cation chromatography (Figure 4), tend to diminish with time on an equimolal basis, indicating that the MDEA and PZ are reacting together resulting in loss of both compounds, until the PZ is completely degraded. The loss of MDEA and PZ occurred at a greater rate at 150 °C, with complete loss of PZ occurring 184 somewhere between days 42 and 57 at a loading of 0.11 moles CO2/mole alkalinity at both temperatures (135 and 150 °C). Replicate sample bombs were collected on a subset of sample collection events in order to utilize statistics (average and standard deviation) and understand repeatability of measurements. Because of the small data sets (three data points), the standard deviations (s) tend to be high in comparison to the absolute value of the measured concentrations. For example, we collected three samples from the 135 °C oven at a loading of 0.11 moles CO2/mole alkalinity on Day 28 and measured MDEA and PZ concentrations. From this data, we calculated the average and svalues to be 7.54 ± 0.36 molal and 1.57 ± 0.07 molal, respectively, for MDEA and PZ. In general, the standard deviation size with respect to the absolute value of the average concentration tended to be smaller for the more degraded samples (greatest time of degradation), as can be seen by the low s-value for the average MDEA concentration (4.99 ± 0.04 molal) for bombs loaded to 0.11 moles CO2/mole alkalinity and placed in the 135 °C oven. The replicate measurements were made on separate sample bombs prepared using the same methodologies, with cation IC runs performed in the same batches for each replicate set. The calculation for average and standard deviation assesses the measurement deviation for all sample handling, dilution, and instrument measurement steps. The reaction mechanism involving interaction between MDEA and PZ, which was observed in earlier experiments with the same solvent blend, is being studied so that degradation intermediates and byproducts will be identified as time progresses. Possible byproducts of the observed reaction mechanism include hydroxyethyl piperazine (HEP), dihydroxyethyl piperazine (DHEP) (also known as dimethylpiperazine), diethanolamine (DEA), methylmonoethanolamine (MMEA), triethanolamine (TEA), and diethylenetriamine (DETA). Chromatograms from Thermal No. 7 exhibit several peaks eluting in the time vicinity of PZ or immediately afterwards, strongly suggesting that these byproduct compounds are in fact diamine compounds. Standards of each will be used to spike experimental samples to assist in identifying unknown peak compounds, and quantify whenever possible. In the past, we calculated the diamine appearance rate from thermal degradation experiments, and calculated an activation energy for this mechanism of 114 kJ/mol. From conversations with Steve Bedell of Dow Chemical, we believe that the primary mechanism for thermal degradation below 150 °C of MDEA and PZ is based on disproportionation reactions. It is unlikely that significant thermal degradation of the amines occurs from homolytic cleavage steps, as these typically occur at much higher temperatures. We also do not expect that hydrolysis or elimination reactions would be significant at our conditions. Given that disproportionation is the most likely mechanism for degradation, we will focus our efforts on looking for compounds which result from the amines reacting with each other (HEP). Wetted Wall Column Studies We performed wetted wall column studies at temperatures of 80 and 100 °C with the 7 m MDEA/2 m PZ solvent blend loaded to ~0.03 moles CO2/mole alkalinity. At 80 °C, kg’ was 2.76 X 10-10 mols/s-Pa-cm2 and the partial pressure of CO2 was 1273 Pa. At 100 °C, kg’ was 1.63 X 10-10 mols/s-Pa-cm2and the partial pressure of CO2 was 5212 Pa. 185 Figure 4: Thermal No. 7 - 7 m MDEA/2 m PZ Degraded at 135 °C and 150 °C Future Work Future work will include the determination of the unknown peaks observed in the thermal degradation experiments, as discussed above. Standards for HEP are being prepared and used to identify the presence of this compound in the degraded samples. More work is necessary to interpret anion chromatography results for the oxidative degradation experimental samples to determine the concentration of heat stable salts other than formate; to date, we have measured anionic degradation products including glycolate and oxalate; we are currently observing a background peak that interferes with the interpretation of the oxalate peak. Because we measured formate production rates at twice the magnitude when we performed sample hydrolysis with 5N NaOH to reverse the formation of amides, we will investigate amide formation using IC methods; at this time, it is believed that the degradation of MDEA (tertiary amine) will not result in amides, whereas, the degradation of PZ may form amides. We will attempt to identify the diamines which appear in the thermal degradation studies at 120 and 135 °C using cation chromatography and GC/MS methods. Once these peaks have been identified, we can determine their concentration and identify the pathway by which these compounds are formed. We will characterize the mechanisms resulting in the degradation of the solvent blend, with initial efforts focusing on disproportionation reactions. 186 A cycling experimental apparatus is currently being assembled which will enable us to look at integrated thermal and oxidative degradation mechanisms. Our intent is to investigate the degradation of solvents from both mechanisms (oxidative and thermal) in a single system that cycles the solvents in a closed loop from an oxidative reactor into a thermal reactor. This will expose the solvents to both oxidative and reductive (thermal) conditions in a single experimental set-up. References Morrison RT, Boyd RN. Organic Chemistry. Allyn and Bacon, Inc., 3rd Ed. 1973. 746. 187 Modeling Absorber/Stripper Performance with MDEA/PZ Quarterly Report for October 1 – December 31, 2008 by Peter Frailie Supported by the Luminant Carbon Management Program and Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 2, 2009 Abstract The removal of CO2 from process gases using alkanolamine absorption/stripping has been extensively studied for several solvents and solvent blends. An advantage of using blends is that the addition of certain solvents can enhance the overall performance of the CO2 removal system. A disadvantage is that the system becomes much more complex compared to a single solvent system, thus making it more difficult to model. This study will focus on a blended amine solvent system containing piperazine (PZ) and methyldiethanolamine (MDEA). Previous studies have shown that this particular blend has the potential to combine the high capacity of MDEA with the attractive kinetics of PZ (Bishnoi, 2000). These studies supplied a rudimentary Aspen Plus®-based model for an absorber with the MDEA/PZ system. The report also makes the recommendation that more kinetic and thermodynamic data must be acquired concerning the MDEA/PZ system before the model can be significantly improved. Two researchers in the Rochelle lab are currently acquiring this data, but they have not yet been incorporated into an absorber/stripper model. One of the major goals of this study will be to improve the supplied Aspen Plus® absorber model with up-to-date thermodynamic and kinetic data. Another major goal of this study will be to combine absorber and stripper models to evaluate the overall system performance. Chemistry of MDEA/PZ Solvent System The reaction between MDEA and CO2 is generally accepted to be first order in MDEA concentration and first order in CO2 concentration (second order overall). The mechanism for this reaction consists of two main steps: (1) the deprotonation of a water molecule by the tertiary amine to form a hydroxide ion, and (2) the formation of bicarbonate from the hydroxide and CO2. Figure 1 on the next page illustrates this mechanism. 188 Figure 1: Second order reaction between CO2 and tertiary amine (Bishnoi, 2000) In addition to this reaction, it is thought that two other mechanisms could be at work (Barth et al., 1981). Another possible mechanism involves the formation of a zwitterion from the reaction between one molecule of tertiary amine and one molecule of CO2. This zwitterion can then be split by either a water molecule or a hydroxide ion to form carbonic acid or bicarbonate and the original tertiary amine (Figure 2). Figure 2: Zwitterion reaction mechanism between tertiary amine and CO2 (Bishnoi, 2000) A third reaction mechanism, which includes the formation of alkylcarbonates, is considered to be highly unlikely except when the system is at a very high pH (Blauwhoff et al., 1984) so its details are not given here. Efforts have been made by other researchers (Glasscock, 1990) to reconcile the expected reaction kinetics with laboratory observations, leading to the development of much more complex equations than would be expected from a simple second order reaction. The primary reaction between PZ and CO2 consists of two steps (Caplow, 1968): (1) the formation of a zwitterion from one molecule of CO2 and one amine group, and (2) the deprotonation of the zwitterion to form a carbamate and a protonated amine (Figure 3). Because PZ is a diamine there are several potential products of this mechanism. It is possible that both amine groups involved in the reaction are part of the same molecule, thus yielding a protonated and carboxylated PZ molecule. It is also possible that one molecule of PZ could react with two 189 molecules of CO2, generating a dicarbamate. Differences in charge and size cause these molecules to behave differently in solution, thus making it more difficult to model the system. Figure 3: Reaction mechanism between secondary amine and CO2 (Okoye, 2005) The system modeled in this study will be exponentially more complex than either of the single solvent systems described in this section. For this reason, it is immensely important that thermodynamic and kinetic data be obtained for the MDEA/PZ system with model requirements in mind. References Barth D, Tondre C, Lappai G, and Delpuech JJ. Kinetic study of carbon dioxide reaction with tertiary amines in aqueous solutions. J Phys Chem. 1981;85(24):3660–3667. Bishnoi S. Carbon Dioxide Absorption and Solution Equilibrium in Piperazine Activated Methyldiethanolamine. Ph.D. Dissertation. The University of Texas at Austin. 2000. Blauwhoff PMM, Versteeg GM, Van Swaaij WPM. A Study on the Reaction Between CO2 and Alkanolamines in Aqueous Solution. Chem Eng Sci. 1984;39(2):207. Caplow M. Kinetics of Carbamate Formation and Breakdown. J Am Chem Soc. 1968;90:6795. Glasscock DA. Modeling and experimental study of carbon dioxide absorption into aqueous alkanolamines. Ph.D. Dissertation. The University of Texas at Austin. 1990. Okoye C. Carbon Dioxide Absorption Rate in Monoethanolamin/Piperazine/H2O, M.S. Thesis. The University of Texas at Austin. 2005. 190 Measurement of kga in Packing Progress Report for October 1 – December 31, 2008 by Chao Wang Supported by the Luminant Carbon Management Program and the Industrial Associates Program for CO2 Capture by Aqueous Absorption Department of Chemical Engineering The University of Texas at Austin February 1, 2009 Abstract This research is focused on the properties of dumped and structured packings in the CO2 capture with chemical absorption. The properties of the packings are the effective area ae, pressure drop, and mass transfer coefficient. According to the theory of series resistance, the mass transfer coefficient contains the gas-side coefficient kG, liquid-side coefficient kL and the overall coefficient KG. What we want to find is the method of measuring kG, kL and finally KG. Literature review about the measurement of kGa and ae has been done during this period. We propose to measure kGa with SO2-air-NaOH-water. The relation between gas diffusivity DG and gas-side mass transfer coefficient has also been k G , SO 2 D G , SO 2 discussed. It is acceptable to use the equation that = according to Sharma k G ,CO 2 DG ,CO 2 (1965). Thus we can apply the kG we measure in the SO2 absorption for the CO2 absorption. Literature Review Measurement of kGa Systems chosen were such that the liquid-side resistance was absent and the gas-side resistance was unambiguously controlling the mass transfer rate. Sulfur dioxide and chlorine (Sharma, 1966) in a variety of carrier gases (air, Freon12(dichlorodifluoromethane), Freon-22 (monochlorodifluoromethane) and Freon-114 (dichlorotetrafluoroethane)) were absorbed in aqueous sodium hydroxide solutions (2N NaOH), and ammonia and triethylamine in different carrier gases were absorbed in dilute sulfuric acid (1 to 2N H2SO4). The amount of sulfite and the hypochlorite in aqueous sodium hydroxide solutions were analyzed by the iodometric method. The amount of ammonia and triethylamine was analyzed by titrating the sulfuric acid solution against standard sodium hydroxide using bromo-phenol blue as an indicator. Two systems (SO2-air-NaOH-water and SO2-air-DMA-toluene) were used in the work of Yaici and Laurent (Yaici, 1988). These two systems provide gas-side resistance to mass transfer. The 191 experiment was done in a packed column 0.05 m in diameter and 0.49 m in packed height. The gas-phase mass transfer coefficient was calculated from the measurements of the molar SO2 content in the gas phase at the inlet and outlet of the reactor. The result shows that the nature of the liquid phase, that is to say, whether it is aqueous or organic, does not seem to have influenced the value of kGa in this experiment. Other systems have also been used for the measurement of kGa. They are CO2-air-NaOH-water (Dodds, 1960), air drying by CaCl2 solution (Wen, 1963), NH3-air-water (Reiss, 1967), NH3-airNa2SO4-water (Gianetto, 1973). However, considering the corrosion and the possible reaction between the solvent with our packing material, the method of absorption of NH3 with H2SO4 and the absorption of Cl2 with NaOH cannot be used. Considering the similarity of CO2 and SO2 in chemical and physical properties, since we used the same column for the measurement of ae with CO2 absorption, the method of absorption of SO2 in NaOH is the most suitable for this experiment. The relationship between gas diffusivity and gas-side mass transfer coefficient has been discussed in many articles (Sharma, 1965; Onda, 1967; Wang, 2005). Conclusion had been reached that the index is between 0.5~1. In Sharma’s paper which also used the system SO2NaOH, the index is 0.5. We will use a value of 0.5. Measurement of ae The volumetric overall mass transfer coefficient can be calculated by the design equation for the absorption column. uG y Z = N OG × H OG = × ln( CO 2in ) K G aRT y CO 2 out y uG ln( CO 2in ) yCO 2 out So K G a = ZRT If we want to calculate a, we must separate KG from that equation. The CO2-air-NaOH system has been chosen to measure effective area a. 1 1 1 = + (1) K G kG k L In the M.S. thesis of Wilson (Wilson, 2004), low concentration NaOH solution is chosen and experimental equation has been used to calculate kG. In this system 1 μG G ) 0.7 * ( ) 3 * ( aT * d P ) − 2 (2) aW * μ G ρ G * DG For the calculation of kL, the enhancement factor for a pseudo first-order, fast, irreversible k G = 5.23 * (aT * DG ) * ( reaction is E = 1 + Ha 2 and Ha 2 = k OH [OH − ]DCO2 (3) k L° Systems with a Hatta number larger than 2 are considered predominantly as “fast reactions”. Therefore the overall mass transfer coefficient can be written as: 192 H CO 2 1 1 = + K G kG k OH − [ OH − ] DCO 2 In his calculation of kG, when the concentration of OH- is low, kg is rather small compared to kL and can be neglected. In Tsai’s paper (Tsai, 2008), similar assumption is made. However, instead of using experimental equation, Tsai used experiment in WWC to determine the number of kG. The results showed that the gas-side resistance accounted for 10–20% of the overall mass transfer resistance. Therefore, KG≈kL and kL is calculated by the equation: kL = k OH − [OH − ]DCO 2, L (4) H CO 2 where DCO2,L is calculated by: ( DCO 2, L * μ ) T = ( DCO 2,W * μW ) T and log10 DCO 2, w = −8.1764 + (5) 712.5 2.591 × 10 5 − T T2 (6) HCO2 is calculated by: H CO 2 log10 = ∑ I i hi H CO 2,W hi = h+ + h− + hg − log10 ( H CO 2,W ) = 9.1229 − 5.9044 × 10 −2 T + 7.8857 × 10 −5 T 2 KOH- is calculated by: k − log10 OH = 0.221I − 0.016 I 2 k OH ∞ log10 k OH − ∞ = 11.895 − 2382 T Thus, the equation for effective area is: y y u G ln( CO 2in ) u G ln( CO 2in ) y CO 2 out y CO 2 out ≈ ae = ZK G RT Zk L RT However, accurate modeling requires that we account for gas-side resistance since it accounts for 10–20% of the whole resistance. We can use the SO2-air-NaOH system to calculate kg and the kg for CO2-air-NaOH system can be calculated since kga Dg0.5 according to Sharma (1967). References Dodds WS, Stutzman LF. Pressure drop and liquid hold-up in concurrent gas absorption. AIChE J. 1960;6(3):390–393. Gianetto A. Absorption in packed towers with concurrent downward high-velocity flows EM dash 2.mass transfer. AIChE J. 1973;19(5):916–922 193 Kakusaburo O. Mass transfer coefficients between gas and liquid phases in packed columns. J Chem Eng Japan. 1968;1:56–62. Reiss LP. Process Design and Development. Ind Eng Chem. 1967;6:486. Sharma MM, Mehta VD. Effect of Diffusivity on gas-side mass transfer coefficient. Chem Eng Sci.1966;21:361–365. Sharma MM, Vidwans AD. Gas-side mass transfer coefficient in packed columns. Chem Eng Sci. 1967;22:673–684. Tsai RE, Rochelle GT. Influence of Surface Tension on Effective Packing Area. Ind Eng Chem Res. 2008;47:1253–1260. Wang G.Q, Yu KT. Review of mass-transfer correlations for packed columns. Ind Eng Chem Res. 2005;44:8715–8729. Wen CY, O’Brien J, Chem Eng. 1963;Data 8:42. Wilson I. Gas-Liquid Contact Area of Random and Structured Packing. M.S. Thesis. University of Texas at Austin. 2004. Yaici W, Laurent A. Determination of gas-side mass transfer coefficients in trickle-bed reactors in the presence of an aqueous or an organic liquid phase. Int Chem Eng. 1988;28(2). 194 Submitted to Industrial & Engineering Chemistry Research rR Fo Rate-Based Process Modeling Study of CO2 Capture with Aqueous Monoethanolamine Solution ie ev Journal: Manuscript ID: Manuscript Type: draft Article n/a w. Date Submitted by the Author: Industrial & Engineering Chemistry Research Complete List of Authors: l- tia en id nf Co Chen, Chau-Chyun; Aspen Technology, Inc., R&D Zhang, Ying; AspenTech Limited, China R&D Plaza, Jorge; University of Texas at Austin, Department of Chemical Engineering Dugas, Ross; University of Texas at Austin, Department of Chemical Engineering Rochelle, Gary; University of Texas, Chemical Engineering S AC ACS Paragon Plus Environment Page 1 of 54 Rate-Based Process Modeling Study of CO2 Capture with Aqueous Monoethanolamine Solution rR Fo Ying Zhang1, Chau-Chyun Chen2*, Jorge M. Plaza3, Ross Dugas3, Gary T. Rochelle3 1 AspenTech Limited w. ie ev Pudong, Shanghai 201203 China Aspen Technology, Inc. U.S.A Department of Chemical Engineering U.S.A * To whom correspondence should be addressed. E-mail: S AC Austin, Texas 78712 l- The University of Texas at Austin tia 3 en Burlington, Massachusetts 01803 id nf 2 Co 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research [email protected]. Phone: 781-221-6420. Fax: 781-221-6410. ACS Paragon Plus Environment 1 Submitted to Industrial & Engineering Chemistry Research Abstract Rate-based process modeling technology has matured and is increasingly gaining rR Fo acceptance over traditional equilibrium-stage modeling approach1. Recently comprehensive pilot plant data for carbon dioxide (CO2) capture with aqueous monoethanolamine (MEA) solution have become available from the University of Texas ie ev at Austin. The pilot plant data cover key process variables including CO2 concentration in gas stream, CO2 loading in lean MEA solution, liquid to gas ratio, and packing type. In this study, we model the pilot plant operation with Aspen RateSep, a second generation w. rate-based absorption & stripping unit operation model in Aspen Plus. After a brief review on rate-based modeling, thermodynamic and kinetic models for CO2 absorption Co with the MEA solution, and transport property models, we show excellent match of the nf rate-based model predictions against the comprehensive pilot plant data and we validate superiority of the rate-based models over the traditional equilibrium-stage models. We id further examine the impacts of key rate-based modeling options, i.e., film discretization en options and flow model options. The Rate-based model provides excellent predictive capability and it should be very useful for design and scale-up of CO2 capture processes. l- tia Keywords: CO2 Capture, Aqueous Monoethanolamine, Absorption, RateSep, Aspen Plus, Rate-Based Models, Equilibrium-Stage Models, Film Discretization ACS Paragon Plus Environment S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 2 of 54 2 Page 3 of 54 Introduction Global surface temperature has risen significantly in the last hundred years and rR Fo the temperature rise has become even more pronounced in recent decades. These temperature changes are attributed to increased levels of greenhouse gases in the atmosphere resulting from human activities. Carbon dioxide is the primary greenhouse ie ev gas causing the global warming. CO2 capture from flue gas by absorption/stripping with monoethanolamine (MEA) or other aqueous physical and chemical solvents will be an important technology to reduce CO2 emissions from fossil-fuel fired power plants to w. address global climate change. While this capture technology is the only one ready for large-scale application on existing power plants, it is necessary to further optimize the Co individual process units and improve the overall process economics. This requires nf rigorous modeling and simulation of the CO2 capture process and fundamental understanding of the underlying complex phenomena taking place in the process such as id electrolyte thermodynamics, chemical reactions, and heat and mass transfer across the en gas-liquid interface and in the bulk gas and liquid phases. tia CO2 emissions from coal-fired power plants can be captured via absorption and stripping processes with circulating chemical solvents that can be placed at the tail end of l- new or existing coal fired power plants with NOx and SOx controls. A typical CO2 S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research absorption/stripping process is shown in Figure 1. A lean amine solvent (low CO2 concentration) is fed into the top of the absorber and is counter-currently contacted by the flue gas containing CO2. The CO2 chemically reacts with the amine solvent and the treated gas exits the top of the absorber. The rich (high CO2 concentration) amine leaves ACS Paragon Plus Environment 3 Submitted to Industrial & Engineering Chemistry Research the bottom of the absorber and is preheated by a cross heat exchanger before entering the top of the stripper. At stripper conditions, typically higher temperature and lower rR Fo pressure, the reaction between the amine and CO2 is reversed, liberating the CO2. A concentrated CO2 stream is obtained from the top of the stripper. The lean solvent from the stripper undergoes heat exchange and goes back to the absorber. Concentrated CO2 from the stripper can be compressed and sequestered into depleted oil or gas fields or ie ev deep saline reservoirs2. Recent studies show that permanent storage in deep saline aquifers is feasible3. w. In order to study the CO2 capture process with aqueous MEA, a pilot plant containing two columns, an absorber and a stripper have been installed at the University Co of Texas at Austin4. A pilot plant campaign, which consisted of 48 runs at 24 operating conditions, was carried out. Various packings, lean CO2 loadings, gas and liquid rates and nf stripper pressures were tested and a series of pilot plant data for the CO2 capture system was reported. en id In this study, a rate-based absorption model, RateSep, in Aspen Plus process tia simulator is used to simulate the pilot plant at the University of Texas at Austin (U.T.Austin). To successfully simulate the amine CO2 capture system with RateSep, we l- carefully examine thermodynamic, kinetic, transport properties, and hydraulic models S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 4 of 54 and model parameters. Model predictions against the various groups of pilot plant data are presented. Rate-based modeling predictions and equilibrium-stage modeling predictions are compared and the results confirm the superiority of the rate-based models for the CO2 capture process. In addition, key RateSep modeling options such as film ACS Paragon Plus Environment 4 Page 5 of 54 discretization and flow models are discussed along with their impacts on the prediction results. The RateSep model provides excellent predictive capability and will be a very rR Fo useful design tool to study various process variables, including liquid/gas ratio, CO2 concentration in feed stream, CO2 loading and MEA concentration in lean amine stream, operating pressure, packing height and packing type, etc. RateSep ie ev Aspen RateSep represents a second generation rate-based process modeling w. software for multistage separation operations. The rate-based modeling approach is rigorous and offers higher model fidelity over the traditional equilibrium-stage modeling Co approach. Rate-based multistage separation models assume that separation is caused by mass transfer between the contacting phases, and use the Maxwell-Stefan theory to nf calculate mass transfer rates1. Conversely, the traditional equilibrium-stage models id assume that the contacting phases are in equilibrium with each other, which is inherently en an approximation because the contacting phases are never in equilibrium in a real column. Designed to model reactive multistage separation problems rigorously and tia accurately, Aspen RateSep balances the gas and liquid phase separately and considers l- mass and heat transfer resistances according to the two-film theory by explicit calculation of interfacial fluxes and film discretization. The film model equations are combined with S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research relevant diffusion and reaction kinetics and include the specific features of electrolyte solution chemistry, electrolyte thermodynamics, and electroneutrality in the liquid phase. The column hydrodynamics are accounted for via specific correlations for vapor-liquid interfacial area, liquid hold-up, pressure drop, and mass transfer coefficients. Figure 2 ACS Paragon Plus Environment 5 Submitted to Industrial & Engineering Chemistry Research illustrates the basic picture of CO2 transfer across the vapor and liquid films. Here Y is gas phase composition, X is liquid phase composition, T is temperature, I is interface, V rR Fo is vapor and L is liquid. The liquid film is discretized into multiple film segments. CO2 Capture Model To fully describe the complex phenomena taking place in the CO2 capture process ie ev with MEA, the model must properly account for thermodynamics of the CO2 – water – MEA system, reaction kinetics of CO2 with the MEA solution, and the various transport w. properties affecting the mass and heat transfer. Thermodynamic and Kinetic Models Co The model uses the Hilliard thermodynamic representation of the CO2 – water – nf MEA system5. Reaction kinetics was obtained from data by Aboudheir6 who generated id rate data for CO2 absorption in MEA using a laminar jet at various amine concentrations, en CO2 loadings, and temperatures. This data was used to evaluate the forward rate constants for the formation of carbamate. Reaction kinetics was represented using the tia following set of reactions for carbamate formation, Reactions 1 and 2, and bicarbonate formation, Reactions 3 and 4: l- 2 MEA + CO2 → MEA+ + MEACOO- (1) MEA+ + MEACOO- → 2 MEA + CO2 (2) MEA + CO2 + H2O → HCO3- + MEA+ (3) ACS Paragon Plus Environment S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 6 of 54 6 Page 7 of 54 MEA+ + HCO3- → MEA + CO2 + H2O (4) Two equivalent chemical equilibrium reactions were used to represent the rR Fo chemistry: 2 MEA + CO2 ↔ MEA+ + MEACOO- (5) ie ev MEA + CO2 + H2O↔ HCO3- + MEA+ (6) The rate expressions are given for the forward and backward reactions (see Table w. 1). Here, k j is the reaction rate constant for reaction j, K i is the chemical equilibrium constant for the formation of species i (i.e., MEACOO-, HCO3-), and ai is the activity of component i. nf Co In Aspen Plus reaction rates are described by power law expressions: en Ej 1 1 N α ij R j = k °jT n exp − − ∏ Ci R T 298 i =1 id (9) The pre-exponential factor k °j , the factor n, the activation energy E j , and the tia concentration basis need to be specified. In the present study the factor n in Equation 9 is l- zero, the concentration basis is activity (i.e., product of mole fraction and activity coefficient, or “mole gamma”), α ij is reaction order of component i in reaction j . k °j and S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research E j for the reactions were calculated using experimental data shown in Table 2. We first identify bicarbonate reaction constants and then, built on the determined bicarbonate reaction constants, we identify carbamate reaction constants. ACS Paragon Plus Environment 7 Submitted to Industrial & Engineering Chemistry Research Based on tertiary amine data, Rochelle et al.7 correlated the values of the forward bicarbonate reaction constant (reaction 3 in Table 1) at 298.15 K as a function of the base rR Fo dissociation constant ( pK b ), as shown in Figure 3. Here k in Figure 3 represents the forward bicarbonate reaction constant. The forward bicarbonate reaction rate constant at 298.15 K for MEA ( pK b =4.45) was extracted from this correlation in this study. ie ev This result was converted from concentration basis to activity basis using values from Aspen generated with Hilliard’s model for the activity coefficients for CO2 and MEA as follows: k3c ρ S2 γ MEA γ CO 2 (10) nf Co k3a = w. k3a is the forward reaction rate in activity basis, kmol/m3-s Where: id k 3c is the forward reaction rate in concentration basis, m3/gmol-s en ρ s is the molar density of the MEA solvent in kmol/m3 at 298.15 K tia γ MEA , γ CO 2 are the activity coefficients of MEA and CO2 at 298.15 K for one of the cases l- under study. S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 8 of 54 This result was used as k °j in Equation 9. The energy of activation, E j , was approximated using the data for MDEA (49 kJ/gmol) reported by Pacheco et al.8 and presented in Rochelle et al.7 The forward reaction rate constant for the bicarbonate reaction was then calculated at several selected conditions: ACS Paragon Plus Environment 8 Page 9 of 54 E1 1 k3 = k3° exp − − R T 298 (11) rR Fo Where: k 3 is the forward reaction rate, kmol/m3-s k 3° is the pre-exponential constant from equation 10 (9025.45 kmol/m3-s) ie ev E is the energy of activation (49 kJ/gmol) T is the temperature of the point w. R is the gas constant. Co The results for k 3 were used along with the equilibrium constants to determine the reverse rates for the bicarbonate reaction. Equilibrium was previously evaluated nf using a flash calculation block in Aspen to extract the activity coefficients and liquid id equilibrium concentrations at each point condition. en Nine points from Aboudheir6 were used to determine the forward carbamate tia formation (reaction 1) rate. Three points are at 313.15 K with loading of 0.2767, and the rest at 333.15 K with loadings of 0.1104 and 0.2819 (see Table 2). A laminar jet was l- modeled in Aspen using the found bicarbonate constants and Hilliard’s thermodynamics. Initially the energy of activation was set to zero and the reported flux by Aboudheir was S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research matched by changing the pre-exponential factor ( k °j ) in the power law. The resulting k °j was averaged among the same temperature and loading conditions and then regressed to obtain values for k1° , E, and α for the following: ACS Paragon Plus Environment 9 Submitted to Industrial & Engineering Chemistry Research E1 1 α k1 = k1° exp − − aMEA R T 298 (12) rR Fo This is a modified version of equation (11) that includes the activity of MEA raised to a power in an attempt to account for variations in ionic strength due to changes in loading. The regressed expression has α = 0.9199. In the final forward rate expression the activity of MEA need to be raised to the α + 1 power since there is already an activity ie ev term in this expression. Similarly, the activity term in the reverse rate is raised to the α − 1 term in order to maintain compatibility with the equilibrium constant (see Table 1). w. This treatment results in an activity of MEA raised to the 1.9199 and to the -0.0801 in the forward and the reverse rates respectively. In an effort to simplify the rate expressions, Co the MEA activity exponents were rounded. The final kinetic rate expressions included in the MEA absorption model are included in Table 3. Using these expressions, the pre- nf exponential factor k1° for each point was recalculated, averaged and regressed to obtain id new values for k1° and E for the power law expression. The final kinetic constants are presented in Table 4. l- tia Transport Property Models en Rate-based process models such as RateSep require quantitative models for various transport properties that are essential for applying various correlations of heat S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 10 of 54 transfer, mass transfer, interfacial area, liquid holdup, pressure drop, etc. In parallel to this study, the transport property data and models for the absorption of CO2 with aqueous MEA were reviewed. Specifically transport property models and model parameters for ACS Paragon Plus Environment 10 Page 11 of 54 density, viscosity, surface tension, thermal conductivity, and diffusivity were investigated and validated. rR Fo Details of the transport property models for the MEA-H2O-CO2 system have been summarized in a separate manuscript9. The Clarke density model10 for electrolytes solutions with mixed solvents was used to calculate density. The Jones-Dole viscosity ie ev model10 for liquid solutions with electrolytes was used to calculate viscosity. The Onsager-Samaras surface tension model10 was used to calculate the liquid mixture surface tension. The Riedel thermal conductivity model10 was used to calculate the liquid w. mixture thermal conductivity. Finally the Wilke-Chang diffusivity model10 was used to calculate CO2 diffusivity in H2O and MEA-H2O solutions. Pilot Plant Data nf Co The pilot plant facility was set up as a close-looped absorption/stripping system id for CO2 removal from a flue gas by means of a 32.5% aqueous MEA solution. A test en campaign consisting of 48 runs at 24 operating conditions was conducted over a period of one month4. l- Pilot Plant Setup tia Figure 4 shows a simple schematic of the close-looped pilot plant. S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research The pilot plant’s two carbon steel columns are nearly identically designed. Each has a total height of approximately 11.1 meters and an inside diameter of 42.7 cm. Both have two 3.05 meter beds for packing with a collector plate and re-distributor between ACS Paragon Plus Environment 11 Submitted to Industrial & Engineering Chemistry Research the beds. Both columns used chimney tray collector plates and orifice-riser liquid distributors. rR Fo The pilot plant design included a 3.8 m3 lean solvent storage tank that holds the majority of the liquid inventory. The large storage tank minimized any unsteady state disruptions from the stripper and it allows any composition disruptions in the solution to ie ev mix with the large liquid inventory and keep the absorber lean loading constant. Once the MEA solvent from the liquid storage tank had contacted the CO2 rich w. flue gas in the absorber, it was pumped to the heater. Industrially, a cross exchanger with fluid leaving the stripper would be used but the pilot plat used a heater and a cooler in Co place of a cross exchanger. The heater was found to be undersized for the liquid flow rates required for the absorber operation. Rather than sacrificing the absorber operating nf range, a sub-cooled feed was fed to the stripper. Once the solvent was stripped of its id excess CO2 and exited the bottom of the stripper, it was pumped to the solvent cooler where the solvent temperature was lowered near 315 K. Carbon dioxide and water vapor en exiting the top of the stripper went to a partial condenser where most of the water content tia was removed by cooling the gas to an exit temperature of 283-288 K. l- The dry CO2 from the top of the partial condenser was sent to a large, 2.8 m3 gas accumulation tank. This accumulation tank has the same purpose as the liquid storage S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 12 of 54 tank. It buffers any unsteady state disturbances in the system and thus allows the absorber to obtain a more consistent feed. Pilot Plant Experiments ACS Paragon Plus Environment 12 Page 13 of 54 The MEA baseline campaign utilized a 32.5 wt% MEA solution defined with respect to water and with no CO2 loading. The campaign consisted of 48 runs at 24 rR Fo different operating conditions. The absorber was operated at normal pressure for all 48 runs and the stripper was operated at pressure slightly higher than 1atm. On the last 4 runs the stripper was operated in vacuum. ie ev Liquid samples were collected from the absorber feed, middle, and outlet and also from the stripper middle and outlet to check for CO2 loading. The stripper feed was not sampled since it was the same composition as the absorber outlet. Usually two runs were w. executed at each operating condition. The absorption/stripping system was maintained at steady state for about an hour before liquid samples were taken. After this initial Co sampling, the system was held steady for another hour until a second set of samples was taken at the condition. After this second sampling, a new operating condition was attempted. id nf In addition to the liquid sampling, instantaneous online measurements were en recorded for approximately 65 variables. These variables include gaseous CO2 tia concentrations, temperatures, pressures, densities, flow rates, and liquid levels. The values were logged into a spreadsheet with a recording interval of one minute. For l- calculation purposes, these values were averaged around the sample time (10 minutes S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research before and after). This dampens any temporary disturbances caused by the actual sample collection. The MEA baseline campaign used two different types of packing in the absorber and stripper. Initially Flexipac 1Y, a structured packing with a specific area of 420m2/m3, ACS Paragon Plus Environment 13 Submitted to Industrial & Engineering Chemistry Research was used in the absorber. IMTP #40, a random metal packing with a specific area of 145m2/m3 was used in the stripper. Halfway through the campaign, the Flexipac 1Y was rR Fo moved to the stripper and the IMTP #40 was placed in the absorber. For each lean loading, two gas rates were run. The higher gas rate was near capacity and the lower was approximately at half of the higher rate. For each of these gas ie ev rates, 3 liquid rates were attempted. Liquid rates were controlled such that the CO2 removal from the inlet flue gas would be approximately 70, 85 and 95%. At the end of the campaign, a few runs with vacuum stripping were performed. The actual operating w. conditions performed are presented in Table 5. Runs 37 and 38 are not included since they were conducted at different liquid flow rates. Co Run 15 was operated at the same conditions as runs 16 and 17. Flooding was nf observed at the collector plate between the two beds of packing during these runs so the id data of these runs is not accurate and was excluded from the study. The first 4 runs of the campaign were also excluded in the simulation study since they show inconsistent results en compared to the other conditions. It is possible that some data was incorrectly sampled at tia the beginning of the campaign. A total of 41 sets of pilot plant runs are used in this simulation study. l- Simulation with RateSep S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 14 of 54 The aforementioned sub-cooling of the stripper feed makes it difficult to interpret the experimental data. In order to limit this study to a manageable scope, we focus the RateSep simulation study on the absorber alone. ACS Paragon Plus Environment 14 Page 15 of 54 For the random packing type of IMTP #40, mass transfer coefficients and interfacial area are predicted with the 1968 correlation by Onda11. The correlation by rR Fo Stichlmair et al.12 is used for the holdup calculations. For the structured packing type of Flexipac 1Y, the 1985 correlation of Bravo13 is used for the mass transfer coefficient and interfacial area predictions and the 1992 correlation of Bravo14 is used for holdup. The Chilton and Colburn correlation is used to predict heat transfer coefficients for both ie ev packing. In order to be consistent with those reported for the pilot plant, it is necessary to scale interfacial areas as predicted by the correlations. According to the pilot plant report4, Flexipac 1Y has a surface area of 420 m2/m3 but on average only used 42% of w. that area. IMTP #40 averaged 87% of its 145 m2/m3 under pilot plant operating Co conditions. For this study a constant interfacial area factor of 0.4 is selected for Flexipac 1Y and of 1.2 for IMTP #40. For example, the value reported for Flexipac 1Y is 154 m2, nf as computed from the specific packing area of 420 m2/m3, the packing volume, and the id utilization factor of 42%. For Case 10, the value calculated from the RateSep Bravo interfacial area correlation is 367 m2. With an interfacial area scale factor of 0.4, RateSep en computes the interfacial area to be 147 m2, close to 154 m2. Similarly, the value reported tia for IMTP #40 is 110 m2, as computed from the specific packing area of 145 m2/m3, the packing volume, and the utilization factor of 87%. For Case 42, with a scale factor of 1.2, l- RateSep computes the interfacial area to be 114 m2, close to 110 m2. S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research RateSep allows several options for modeling film resistance. The “reaction condition” factor, the weighting factor for conditions (temperature and liquid composition) used to calculate film reaction rates, was set to 0.9. The condition used is “factor*bulk condition + (1- factor)*interface” condition. Higher weighting factor means ACS Paragon Plus Environment 15 Submitted to Industrial & Engineering Chemistry Research liquid conditions closer to the bulk liquid will carry higher weight. We choose the “film discretization” option and set the number of discretization points for the liquid film to 5, rR Fo which gives 6 film segments. We set the film discretization ratio to 5, which is the ratio of the thickness of the adjacent discretization regions. A value of film discretization ratio greater than 1 means thinner film regions near the vapor-liquid interface. Furthermore, RateSep provides four different “flow models” to determine the bulk properties used to ie ev evaluate the mass and energy fluxes and reaction rates of a stage. The “CounterCurrent” flow model was used as the base calculation method. 20 “CounterCurrent” stages were set up for the packed columns. The impact of the number of stages will be presented later. w. It should also be noted that, in the simulations, heat loss of the column was Co ignored since it is expected to be negligible for the absorber. Pressure drop was also ignored. en id Performance of the Absorber nf RateSep simulation results for the 41 cases are summarized in Table 6, along with the experimental performance data. The values in the “Lean” column in Table 6 are the tia CO2 loading (i.e., mole ratios of CO2/MEA) of the lean stream, which feeds into the top l- of the absorber. These values are feed stream data used in both pilot plant experiments and simulation. The values in the “Rich” column are the mole ratios of CO2/MEA of the S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 16 of 54 rich stream, which comes out of the bottom of the absorber and then goes into the top of the stripper. These values are indicative of the performance of the absorber. ACS Paragon Plus Environment 16 Page 17 of 54 Table 6 shows the RateSep simulation results for the absorber in comparison with the pilot plant experimental data. About half of the values of CO2 loading for the rich rR Fo stream in the experimental data are above 0.5. However, the simulation values are never higher than 0.5, suggesting a ceiling or limit on CO2 loading. There appears to be a significant gap between simulation and experiments especially when the CO2 loading of the rich stream is close to or above 0.5. This problem is attributed to the inaccurate ie ev analysis for CO2 loading as stated in the pilot plant report. According to the report, the mole ratio of CO2/MEA is calculated from the density w. of the liquid at 313.15 K with the empirical equation of “y = -898x2 + 2921x – 2028,” where x stands for the density of the liquid with a unit of kg/l, and y stands for CO2 Co concentration with a unit of gCO2/kgMEA. The density of the liquid at 313.15 K is around 1.0~1.15 kg/L. In this region, there is an approximately linear relationship nf between the density and the CO2 concentration. However, this empirical equation relating id the CO2 concentration and the density ignores the influence of MEA concentration. In en reality, the solution density is sensitive to the MEA concentration especially for the Rich stream with high CO2 loading. In reporting the CO2 concentration for the experiments the tia variation of MEA concentration in the process is not accounted for. l- The experiments determined CO2 loading by taking sample solution densities at S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research five locations: the lean stream flowing into the top of the absorber, middle of the absorber, the rich stream going out of the bottom of the absorber, middle of the stripper and the lean stream going out of the bottom of the stripper. As shown in Table 7, the ACS Paragon Plus Environment 17 Submitted to Industrial & Engineering Chemistry Research MEA mass concentrations vary considerably among the five samples, from 32 wt% in the absorber feed to about 40 wt% in the stripper bottom. rR Fo Calculated with the Clarke density model, Figure 5 shows that the density of the liquid is sensitive to the MEA concentration, especially when CO2 concentration is high. When the CO2 concentration is high, a 10% increase in MEA mass percentage, i.e., from ie ev 30 wt% MEA solution to 40 wt% MEA solution results in approximately 40 kg/m3 increase on the density. The empirical equation used to estimate CO2 concentration from density shows that such an increase on the density would yield equivalent increase of w. 0.055 in mole ratio of CO2/MEA. In other words, the estimated rich loading of the absorber will decrease by 0.055 if this correction is applied to density and the simulation Co results would become much closer to the experimental data. Absorber Temperature Profile id nf The absorber contains 7 resistance temperature detectors. The locations of the en sensors are defined by the height from the bottom of the lower bed of packing. Between the two 3.05 meter beds of packing, there is a liquid redistribution and packing change- tia out area which occupies 1.67 meters of the column. Figure 6 shows the location of the l- temperature sensors. S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 18 of 54 The heat of reaction of CO2 with MEA produces a temperature bulge in the column. This temperature bulge significantly affects the absorption rates in the column since the kinetics of the absorption reaction, the phase equilibrium of the system, and all fluid transport properties depend on the temperature. ACS Paragon Plus Environment 18 Page 19 of 54 The temperature bulge can be explained based on the balance of the heat released from the reaction of CO2 with MEA and the heat consumed by processes including water rR Fo evaporation, heating of the liquid and gas streams, heat loss to the environment and stripping of CO2. If the heat released from the absorption reaction is more than the heat consumed, the temperature will rise. According to the shape of the temperature bulge, we observed three types of absorber temperature profiles. ie ev Figure 7 shows the three types of experimental temperature profile4. Type A profile, represented by Case 7, has a temperature bulge located near the top of the w. absorber. Type B profile, represented by Case 19, shows a very broad bulge in the temperature profile and the temperature changes sharply at both ends of the column. Type Co C profile, represented by Case 38, has a mild temperature bulge near the bottom. Type A profile is opposite to Type C. Type B profile can be regarded as a transition from Type A nf to Type C. All 41 cases can be categorized into these three types. See Table 8. id Figure 8 shows the match between the simulation results and the experimental en data. Simulation results for absorber temperature profiles match pilot plant data very tia well in all cases. Note that RateSep predicts temperature profiles for both the vapor phase and the liquid phase and they are shown in Figure 8 for Case 7. The temperature l- difference between the two phases is quite small. Only the liquid phase temperature profiles are shown for Cases 19 and 38. S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research To explain the factors controlling the types of temperature profiles, Figure 9 shows the CO2 reaction rate profiles in the absorber. Representing Type A cases, Case 7 maintains high CO2 reaction rate throughout the column and that explains the continuing ACS Paragon Plus Environment 19 Submitted to Industrial & Engineering Chemistry Research temperature rise from the absorber bottom section to the top section. Representing Type C cases, Case 38 shows an initial high CO2 reaction rate at the absorber bottom and then rR Fo rapid drop in the rate. That explains the mild temperature bulge at the absorber bottom section. Representing Type B cases, Case 19 also shows an initial high CO2 reaction rate at the absorber bottom and then slow drop in the rate. That explains the initial rise in temperature and then the temperature stays relatively flat. ie ev The study on the CO2 reaction rate profile in the absorber also shows us that essentially all of CO2 is reacted in the film. The CO2 mass transfer profile is essentially w. the same as the profile of the CO2 reaction rate in the film. In other words, RateSep suggests that the film reaction dictates the mass transfer rate for CO2 capture with aqueous MEA. RateSep Simulation Options id nf Co RateSep provides a number of simulation options. We investigate the impacts of en several key simulation options, including modeling approach, film discretization, and flow models. l- Modeling Approach: Equilibrium-Stage vs. Rate-Based tia RateSep supports both equilibrium-stage modeling approach and rate-based S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 20 of 54 modeling approach. Simulation results from both approaches are investigated and compared. In addition, two different options to account for chemical reactions in the liquid phase are examined. In the first option, the reaction kinetics for CO2 absorption by MEA and OH- are explicitly modeled while remaining liquid phase reactions are in ACS Paragon Plus Environment 20 Page 21 of 54 chemical equilibrium. In the second option, chemical equilibrium conditions are assumed for all liquid phase reactions. Table 9 summarizes the four methods investigated in this rR Fo study. The first one, “Rate, rate”, uses rate-based modeling approach and reaction kinetics treatment for the CO2 absorption reactions. The second one, “Rate, equilibrium” uses rate-based modeling approach and chemical equilibrium treatment for the liquid phase reactions. The third one, “Equilibrium, rate” uses the equilibrium-stage modeling ie ev approach and reaction kinetic treatment for the CO2 absorption reactions. The last one, “Equilibrium, equilibrium”, uses the equilibrium-stage modeling approach and chemical equilibrium treatment for the liquid phase reactions. In this study, “Rate, rate” is the w. default rate-based model unless the chemical equilibrium treatment for the liquid phase Co reactions is explicitly specified. “Equilibrium, rate” is the default equilibrium-stage model unless the chemical equilibrium treatment for the liquid phase reactions is explicitly specified. id nf Note that in specifying the equilibrium-stage model, 30 equilibrium stages are en assumed for the structured packing and 16 equilibrium stages for the random packing. The number of equilibrium stages is equal to the packed height divided by the tia corresponding HETPs. As an example, for Cases 7 and 19, the value of the structured l- packing height is 6.1 m and the value of HETP estimated by RateSep from the converged vapor and liquid composition profiles is about 0.2m. Therefore the number of S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research equilibrium stages for the structured packing is set to 30. For Case 38, the value of HETP for the random packing is about 0.37m and the number of equilibrium stages is set to 16. ACS Paragon Plus Environment 21 Submitted to Industrial & Engineering Chemistry Research The following discussion presents RateSep simulation results for the absorber, and all three absorber temperature profile types. rR Fo Case 7 represents a Type A temperature profile. Figure 10 shows the temperature profiles of the absorber as computed from the four methods. Only the “Rate, rate” method predicts the temperature profile that matches the experimental data. The temperature ie ev profiles predicted by the other three methods are much too low in comparison to the experimental data. Note that the temperature profiles calculated from “Equilibrium, rate” and “Equilibrium, equilibrium” overlap each other. w. Case 19 is selected as an example for Type B temperature profiles. Figure 11 Co shows the temperature profiles of the Case 19 absorber with the four methods. The temperature profile calculated from the “Rate, rate” method matches the experimental nf data very well. “Rate, equilibrium” and “Equilibrium, rate” methods give reasonable id results while “Equilibrium, equilibrium” method predicts instant CO2 absorption at the absorber bottom section and yields completely erroneous temperature profile. en Case 38 is selected as an example for Type C temperature profile. Figure 12 tia shows the predicted temperature profiles with these methods. While the “Rate, rate” l- method predicts a temperature profile consistent with the experimental data, the temperature profiles predicted with the other three methods overlap and deviate completely from the observed pilot plant temperature profile. ACS Paragon Plus Environment S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 22 of 54 22 Page 23 of 54 According to this investigation for all three types of temperature profile, the “Rate, Rate” method is the only one that consistently predicts correct temperature profiles rR Fo for the absorber. CO2 loading was also examined and the results for the three cases are summarized in Tables 10 to12. Considering that the measured values of CO2 loading in the rich amine ie ev are higher than the true values due to inaccurate analysis, the CO2 loadings calculated by the “Rate, rate” method are determined to be reasonable. On the other hand, the other three methods over-predict the rich loading. Film Discretization w. Co Film discretization facilitates precise modeling of the chemical reactions taking place in the liquid film. Without film discretization, the liquid film reaction rates are nf computed based on an average liquid phase composition. With film discretization, the id liquid film reaction rates are computed by multiple sets of liquid phase compositions with en each set representing the average liquid phase composition for the particular film segment. tia The various schemes for film discretization are summarized in Table 13. l- “Nofilm” method assumes no liquid film and considers neither the film diffusion S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research resistance nor film reactions. “Filmrxn” method considers the film resistance and reactions without film discretization. “Discretization 1” represents the default “discretization” scheme used in this study. ACS Paragon Plus Environment 23 Submitted to Industrial & Engineering Chemistry Research The temperature profiles of Cases 7, 19 and 38 calculated by the three methods are shown in Figures 13 to 15. The profiles of the “Discretization 1” method in Figures rR Fo 13 to 15 are closer to the experimental values than the other two methods. The profiles of the other two methods deviate from the experimental data for all three cases. Interestingly, “Nofilm” and “Filmrxn” methods yield results similar to those of “Equilibrium, rate”. In short, the “Discretization 1” method is the best one which correctly predicts the ie ev temperature profiles. The results of varying the reaction condition factor for Case 7 are summarized in w. Figure 16 and Table 14. As the reaction condition factor changes from 0.9 (“Discretization 1”) to 0.5 (“Discretization 2”), there are no significant changes in the Co temperature profiles, but the higher CO2 concentration used to calculated the film reaction rate reflects in more CO2 being absorbed and the CO2 loading changes slightly from 0.446 to 0.461. id nf Three discretization schemes were studied with varying numbers of discretization en points (“Discretization 1”: 5 discretization points; “Discretization 3”: 1 discretization tia point;.”Discretization 4”: 10 discretization points). Figure 17 shows the absorber temperature profiles as computed with varying discretization points. As the number of l- discretization points becomes large, the impact of the discretization points diminishes and S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 24 of 54 the predictions yield similar temperature profiles. For example, the profiles of “Discretization 1 (5 points)” and “Discretization 4 (10 points)” overlap each other and the values of CO2 loading calculated by the two schemes are almost the same. See Table 14. ACS Paragon Plus Environment 24 Page 25 of 54 Finally, three discretization schemes with varying discretization ratios (“Discretization 1”: ratio=5; “Discretization 5”: ratio=2; “Discretization 6”: ratio=10) rR Fo were examined. The results in Figure 18 show that the discretization ratio is not as important as the numbers of discretization points. The temperature profiles of “Discretization 1 (ratio=5)” and “Discretization 6 (ratio=10)” overlap each other. The values of CO2 loading are also very close, as shown in Table 14. ie ev The results shown above highlight the critical importance of film discretization on modeling CO2 capture with MEA. The rate-based modeling with film discretization w. allows for rigorous account of the concentration gradients and the corresponding reaction rates in the various film segments. The success of modeling CO2 capture with MEA Co depends on whether and how film discretization is carried out. nf Flow Models: Mixed, CouterCurrent, Vplug and VPlug-Pavg id RateSep provides four different flow models which determine the bulk properties en required to evaluate the mass and energy fluxes and reaction rates. As shown in Figure 19, these models refer to “single stage” in a column, with each stage having its own unique tia bulk properties. The four flow models are Mixed, CounterCurrent, Vplug and VPlugPavg. Figures 20 to 23 show details of these four flow models. lS AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research In the Mixed flow model, as is the case on equilibrium stages, the bulk properties for each phase are assumed to be the same as the outlet conditions for that phase leaving that stage. In the CounterCurrent flow model, the bulk properties for each phase are an ACS Paragon Plus Environment 25 Submitted to Industrial & Engineering Chemistry Research average of the inlet and outlet properties. This CounterCurrent flow model gives more accurate results for packing, but is more computationally intensive. rR Fo In the VPlug flow model, outlet conditions are used for the liquid and average conditions are used for the vapor. The outlet pressure is used. In the VPlug-Pavg flow model, outlet conditions are used for the liquid and average conditions are used for the ie ev vapor. The average pressure is used. Using Cases 7 19 and 38 as examples, the impact of flow models on the w. performance of the absorber were analyzed. Tables 15-17 show that the flow models have only very minor influence on the overall CO2 absorption performance. Figures 24 to 26 Co show the predicted absorber temperature profiles with the four flow models. Again, flow models only have minor impacts on the predicted absorber temperature profiles. nf However, the CounterCurrent flow model does yield the most accurate predictions. The id VPlug model predictions and the Vplug-Pavg model predictions overlap each other. en Finally, the impact of number of “CounterCurrent” stages in RateSep was evaluated using Cases 7, 19 and 38. Figures 27 to 29 show the temperature profiles tia predicted with 10, 20 and 30 stages. The profiles with varying stages are very close l- while those of 20 stages and 30 stages overlap each other. As the number of “CounterCurrent” stages increases, the simulation yields higher fidelity results. On the S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 26 of 54 other hand, 20 “CounterCurrent” stages are sufficient to provide accurate results. Conclusions ACS Paragon Plus Environment 26 Page 27 of 54 This study shows the superiority of the rate-based models over the traditional equilibrium-stage models for the recently available pilot plant data from University of rR Fo Texas at Austin for CO2 capture with aqueous monoethanolamine. Using proper models and model parameters, the rate-based modeling software RateSep successfully simulates the U.T.-Austin pilot plant and matches the absorber experimental data well. The model accurately predicts the three types of temperature profiles observed with the pilot plant ie ev absorber data. A key factor in the success of the CO2 absorption modeling is the use of the film discretization option to rigorously model the mass transfer resistance and the CO2 absorption reactions taking place in the liquid film. w. The RateSep model with proper model parameters provides excellent 1st Co principles-based predictive capability and it should be a very useful tool for industrial research, development and design of CO2 capture processes. Acknowledgments en id nf Y. Zhang and C.-C. Chen are grateful for extensive discussions and enthusiastic supports from our colleagues and collaborators: Davy Zuo, Huiling Que, Jiangchu Liu, Hern Chen, Jianjun Peng, Hailang Li, and Paul Mathias. l- tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 27 Submitted to Industrial & Engineering Chemistry Research References [1] Taylor, R.; Krishna, R.; Kooijman, H. “Real-World Modeling of Distillation,” Chem. rR Fo Eng. Prog., 2003, 99, 28-39. [2] IEA (2006). “Putting Carbon Back into the Ground,” IEA Greenhouse Gas R&D Programme, 2006. ie ev [3] Kumar, A.; Ozah, R. C.; Noh, M.; Pope, G. A.; Bryant, S. L.; Sepehrnoori, K.; Lake, L.W. “Reservoir Simulation of CO2 Storage in Deep Saline Aquifers,” Society of w. Petroleum Engineers Journal, 2005, 10, 336-348. Co [4] Dugas, R. E. “Pilot Plant Study of Carbon Dioxide Capture by Aqueous Monoethanolamine,” Master thesis, Chemical Engineering, the University of Texas at Austin, 2006. id nf [5] Hilliard, M. A. “Predictive Thermodynamic Model for an Aqueous Blend of en Potassium Carbonate, Piperazine, and Monoethanolamine for Carbon Dioxide Capture from Flue Gas,” Ph.D Thesis, Chemical Engineering, The University of Texas at Austin, 2008. l- tia [6] Aboudheir, A. “Kinetics, Modeling and Simulation of CO2 Absorption into Highly S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 28 of 54 Concentrated and Loaded MEA Solutions,” Ph. D. thesis, Chemical Engineering, University of Regina, 2002. [7] Rochelle G. T.; Bishnoi S.; Chi S.; Dang, H. -Y.; Santos, J. “Research Needs for CO2 Capture from Flue Gas by Aqueous Absorption/Stripping,” Final Report on DOE P.O. ACS Paragon Plus Environment 28 Page 29 of 54 No. DE-AF26-99FT01029, January 17, 2001, University of Texas at Austin. [8] Pacheco, M. A.; Kaganoi, S.; Rochelle, G. T. “CO2 absorption into aqueous mixtures rR Fo of diglycolamine® and methyldiethanolamine,” Chem. Eng. Sci., 2000, 55, 5125-5140. [9] Que, H.-L.; Chen, C.-C.; “Modeling Transport Properties of CO2 Capture Systems with Aqueous Monoethanolamine Solution”, Internal Report, Aspen Technology Inc., 2008. ie ev [10] Aspen Properties Reference Manual, Aspen Technology Inc., version number: 2006, October 2006. w. Co [11] Onda, K.; Takeuchi, H.; Okumuto, Y. “Mass Transfer Coefficients between Gas and Liquid Phases in Packed Columns,” Journal of Chemical Engineering of Japan, 1968, 1, 56-62. id nf [12] Stichlmair, J.; Bravo, J.L.; Fair, J.R. “General Model for Prediction of Pressure Drop en and Capacity of Countercurrent Gas/Liquid Packed Columns,” Gas. Sep. Purif., 1989, 3, 19-28. tia [13] Fair, J. R.; Bravo, J. L.; “Prediction of Mass Transfer Efficiencies and Pressure Drop l- for Structured Tower Packings in Vapor/Liquid Service,” Inst. Chem. Eng. Symp. Ser., S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research 1985, A183-200. [14] Bravo, J. L.; Rocha, J. A.; Fair, J. R.; “A Comprehensive Model for the Performance of Columns Containing Structured Packings”, Inst. Chem. Eng. Symp. Ser., 1992, 129, A439-457. ACS Paragon Plus Environment 29 Submitted to Industrial & Engineering Chemistry Research Figure Captions Figure 1. Typical absorber/stripper flowsheet for CO2 capture rR Fo Figure 2. Discretized film concept is combined with the countercurrent flow configuration in RateSep ie ev Figure 3. Second order rate constants for the reaction of tertiary amines & CO2 at 298.15 K: () Rate constants of tertiary amines, ( ) Rate constants of MEA, () Correlation of amines’ rate constants w. Figure 4. Simple schematic of the CO2 capture pilot plant at University of Texas at Austin nf Co Figure 5. Predictions of the Clarke model for densities of the MEA-H2O-CO2 system at 298.15 K Figure 6. Absorber temperature sensor locations en id Figure 7. Absorber temperature profiles: ( - - ) Experimental data of Type A, tia Case 7, (--------) Experimental data of Type B, Case 19, ( - - - - ) Experimental l- data of Type C, Case 38 S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 30 of 54 Figure 8. Absorber temperature profiles of Type A, Case 7: () Experimental data, ( - ) Liquid temperature calculated by RateSep, ( ) Vapor temperature calculated by RateSep; Type B, Case 19: () Experimental data, (----) Liquid temperature calculated by RateSep; Type C, Case 38: () Experimental data, ( - - ) Liquid temperature ACS Paragon Plus Environment 30 Page 31 of 54 calculated by RateSep Figure 9. CO2 reaction rate profiles calculated by RateSep: ( - ) CO2 reaction rate of rR Fo Type A, Case 7, (----) CO2 reaction rate of Type B, Case 19, ( - - ) CO2 reaction rate of Type C, Case 38 Figure 10. Absorption temperature profile of Type A, Case 7: () Experimental data, ie ev () Liquid temperature calculated by “Rate, rate” method, ( ) Liquid temperature calculated by “Rate, equilibrium” method, (----) Liquid temperature calculated by w. “Equilibrium, rate” method, (- -) Liquid temperature calculated by “Equilibrium, equilibrium” method Co Figure 11. Absorber temperature profile of Type B, Case 19: ()Experimental data, () Liquid temperature calculated by “Rate, rate” method, ( ) Liquid temperature nf calculated by “Rate, equilibrium” method, (----) Liquid temperature calculated by id “Equilibrium, rate” method, (- -) Liquid temperature calculated by “Equilibrium, equilibrium” method tia en Figure 12. Absorber temperature profile of Type C, Case 38: ()Experimental data, () Liquid temperature calculated by “Rate, rate” method, ( ) Liquid temperature l- calculated by “Rate, equilibrium” method, (----) Liquid temperature calculated by S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research “Equilibrium, rate” method, (- -) Liquid temperature calculated by “Equilibrium, equilibrium” method Figure 13. Absorber temperature profile of Type A, Case 7: () Experimental data, () Liquid temperature calculated with “Discretization 1”, ( ) Liquid temperature ACS Paragon Plus Environment 31 Submitted to Industrial & Engineering Chemistry Research calculated with “Filmrxn”, (----) Liquid temperature calculated with “No film” Figure 14. Absorber temperature profile of Type B, Case 19: () Experimental data, () rR Fo Liquid temperature calculated with “Discretization 1”, ( ) Liquid temperature calculated with “Filmrxn”, (----) Liquid temperature calculated with “No film” Figure 15. Absorber temperature profile of Type C, Case 38: () Experimental data, () ie ev Liquid temperature calculated with “Discretization 1”, ( ) Liquid temperature calculated with “Filmrxn”, (----) Liquid temperature calculated with “No film” w. Figure 16. Absorber temperature profile of Type A, Case 7: () Experimental data, () Liquid temperature calculated with “Discretization 1”, ( ) Liquid temperature Co calculated with “Discretization 2” nf Figure 17. Absorber temperature profile of Type A, Case 7: () Experimental data, () id Liquid temperature calculated with “Discretization 1”, ( ) Liquid temperature calculated with “Discretization 3”, (----) Liquid temperature calculated with en “Discretization 4” tia Figure 18. Absorber temperature profile of Type A, Case 7: () Experimental data, () l- Liquid temperature calculated with “Discretization 1”, ( ) Liquid temperature calculated with “Discretization 5”, (----) Liquid temperature calculated with “Discretization 6” Figure 19. The flow models for single stage in RateSep S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 32 of 54 Figure 20. The Mixed flow model ACS Paragon Plus Environment 32 Page 33 of 54 Figure 21. The CounterCurrent flow model Figure 22. the VPlug flow model rR Fo Figure 23. the VPlug-Pavg flow model Figure 24. Absorber temperature profile of Type A, Case 7: () Experimental data, () ie ev Liquid temperature calculated by the CounterCurrent model, ( ) Liquid temperature calculated by the Mixed model, (----) Liquid temperature calculated by the VPlug model, (- -) Liquid temperature calculated by the VPlug-Pavg model” w. Figure 25. Absorber temperature profile of Type B, Case 19: () Experimental data, () Co Liquid temperature calculated by the CounterCurrent model, ( ) Liquid temperature calculated by the Mixed model, (----) Liquid temperature calculated by the VPlug model, nf (- -) Liquid temperature calculated by the VPlug-Pavg model” id Figure 26. Absorber temperature profile of Type C, Case 38: () Experimental data, () en Liquid temperature calculated by the CounterCurrent model, ( ) Liquid temperature calculated by the Mixed model, (----) Liquid temperature calculated by the VPlug model, tia (- -) Liquid temperature calculated by the VPlug-Pavg model” l- Figure 27. Absorber temperature profile of Type A, Case 7: () Experimental data, () S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research Liquid temperature calculated with 20 stages, ( ) Liquid temperature calculated with 10 stages, (----) Liquid temperature calculated with 30 stages Figure 28. Absorber temperature profile of Type B, Case 19: () Experimental data, () Liquid temperature calculated with 20 stages, ( ) Liquid temperature calculated with ACS Paragon Plus Environment 33 Submitted to Industrial & Engineering Chemistry Research 10 stages, (----) Liquid temperature calculated with 30 stages Figure 29. Absorber temperature profile of Type C, Case 38: () Experimental data, () rR Fo Liquid temperature calculated with 20 stages, ( ) Liquid temperature calculated with 10 stages, (----) Liquid temperature calculated with 30 stages w. ie ev l- tia en id nf Co S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 34 of 54 ACS Paragon Plus Environment 34 Page 35 of 54 rR Fo ie ev Figure 1 w. l- tia en id nf Co Figure 2 ACS Paragon Plus Environment S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research 35 Submitted to Industrial & Engineering Chemistry Research 1 rR Fo k (m^3/gmol-s) 0.1 -1.1103x y = 3.2778e 2 R = 0.8994 ie ev 0.01 w. 0.001 3 3.5 4 4.5 5 5.5 6 6.5 Co pKb Figure 3 l- tia en id nf Figure 4 ACS Paragon Plus Environment S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 36 of 54 36 Page 37 of 54 1200 rR Fo Density, kg/m3 1100 ie ev 1000 900 0 EXP MEA EXP MEA EXP MEA EXP MEA EST MEA EST MEA EST MEA EST MEA w. 0.1 0.2 0.3 10w t% 20w t% 30w t% 40w t% 10w t% 20w t% 30w t% 40w t% 0.4 0.5 CO2 Loading Co Figure 5 Figure 6 l- tia en id nf S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 37 Submitted to Industrial & Engineering Chemistry Research 200 Temperature, Deg F 180 rR Fo 160 140 120 100 ie ev 80 -1 1 3 5 7 9 Height from bottom , m w. Figure 7 200 Co 160 140 120 100 80 -1 1 3 en id Temperature, Deg F 180 nf 5 7 9 Height from bottom , m Figure 8 l- tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 38 of 54 ACS Paragon Plus Environment 38 Page 39 of 54 CO2 reaction rate, mol/hr 250 rR Fo 200 150 100 50 ie ev 0 -1 1 180 7 9 Figure 9 160 nf 140 120 100 80 -1 1 3 en id Temperature, Deg F 5 Co 200 3 Height from bottom , m w. 5 Height from bottom , m 9 l- Figure 10 7 tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 39 Submitted to Industrial & Engineering Chemistry Research 200 Temperature, Deg F 180 rR Fo 160 140 120 100 ie ev 80 -1 1 3 5 7 9 Height from bottom , m w. Figure 11 200 Co 160 140 120 100 80 -1 1 3 en id Temperature, Deg F 180 nf 5 7 9 Height from bottom , m Figure 12 l- tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 40 of 54 ACS Paragon Plus Environment 40 Page 41 of 54 200 Temperature, Deg F 180 rR Fo 160 140 120 100 ie ev 80 -1 1 3 5 7 9 Height from bottom , m Figure 13 w. 200 160 140 nf 120 100 80 -1 1 3 en id Temperature, Deg F 180 Co 5 7 9 Height from bottom , m Figure 14 l- tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 41 Submitted to Industrial & Engineering Chemistry Research 200 Temperature, Deg F 180 rR Fo 160 140 120 100 ie ev 80 -1 1 180 7 9 Figure 15 160 nf 140 120 100 80 -1 1 3 en id Temperature, Deg F 5 Co 200 3 Height from bottom , m w. 5 Height from bottom , m 9 l- Figure 16 7 tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 42 of 54 ACS Paragon Plus Environment 42 Page 43 of 54 200 Temperature, Deg F 180 rR Fo 160 140 120 100 ie ev 80 -1 1 3 5 7 9 Height from bottom , m w. Figure 17 200 Co 160 140 120 100 80 -1 1 3 en id Temperature, Deg F 180 nf 5 7 9 Height from bottom , m Figure 18 l- tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 43 Submitted to Industrial & Engineering Chemistry Research rR Fo w. ie ev Figure 19 nf Co Figure 20 Figure 21 l- tia en id S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 44 of 54 Figure 22 ACS Paragon Plus Environment 44 Page 45 of 54 rR Fo Figure 23 ie ev 200 160 140 120 100 80 -1 Co Temperature, Deg F 180 w. 1 3 5 7 9 nf Height from bottom , m id Figure 24 en 200 160 140 l- Temperature, Deg F 180 tia 120 100 80 -1 1 3 5 7 Height from bottom , m 9 S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research Figure 25 ACS Paragon Plus Environment 45 Submitted to Industrial & Engineering Chemistry Research 200 180 Temperature, Deg F rR Fo 160 140 120 100 ie ev 80 -1 1 5 7 9 Figure 26 Co 200 180 nf 160 140 120 100 80 1 3 5 Height from bottom , m 9 l- Figure 27 7 tia -1 en id Temperature, Deg F 3 Height from bottom , m w. S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 46 of 54 ACS Paragon Plus Environment 46 Page 47 of 54 200 Temperature, Deg F 180 rR Fo 160 140 120 100 ie ev 80 -1 1 3 5 7 9 Height from bottom , m Figure 28 w. 200 160 140 nf Temperature, Deg F 180 Co 120 100 80 -1 1 3 en id 5 7 9 Height from bottom , m Figure 29 l- tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 47 Submitted to Industrial & Engineering Chemistry Research Table 1: Kinetic rate expressions included in the proposed MEA absorption model. Related specie - MEACOO rR Fo HCO3- Reaction Direction Forward Reverse Forward Reverse ie ev Kinetic expression r = k1 ∗ a MEA ∗ a CO2 r= k1 ∗ K MEACOO− (7a) a MEACOO− ∗ a MEA+ a MEA r = k 3 ∗ a MEA ∗ a CO2 r= k3 K ∗ (8a) a HCO− ∗ a MEA+ − HCO3− (7b) 3 a H 2O (8b) Table 2: Selected Data from Aboudheir for Carbamate rate analysis. MEA concentration 7 mol/dm3. ldg T Pco2 L h d Rexp*10-6 (mol/mol) (K) (kPa) (cm^3/s) (cm) (cm) (mol/s) 0.2767 313.15 87.06 0.351 1.884 0.0584 5.13 1.410 0.0584 3.86 1.082 0.0584 3.00 0.2819 333.15 73.86 0.375 2.142 0.0563 7.16 1.758 0.0563 5.86 1.323 0.0563 4.53 0.1104 333.15 75.64 0.408 2.197 0.0554 11.7 1.872 0.0554 10.2 1.415 0.0554 7.83 w. l- tia en id nf Co S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 48 of 54 ACS Paragon Plus Environment 48 Page 49 of 54 Table 3: Final kinetic rate expressions included in the proposed MEA absorption model Related specie Reaction Direction MEACOO- Forward rR Fo HCO3- Kinetic expression 2 r = k1 ∗ a MEA ∗ aCO2 Reverse r= k1 kMEACOO− (10a) ∗ aMEACOO− ∗ aMEA+ r = k 3 ∗ a MEA ∗ a CO2 Forward ie ev Reverse r= w. k3 k HCO3− ∗ (10b) (11a) a HCO − ∗ a MEA+ 3 a H 2O − (11b) nf Co Table 4: Constants for power law expressions for the absorption of CO2 by MEA Related specie E 3 (kmol/m -s) 4.73E+09 4.23E+05 9025.5 3312.6 (kJ/gmol) 19.34 107.47 49.00 112.74 l- tia HCO3- Forward Reverse Forward Reverse kjo en MEACOO- Reaction Direction id S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 49 Submitted to Industrial & Engineering Chemistry Research Case Table 5. Operation conditions of MEA campaign Lean Mole – Gas Liquid Packings loading Inlet Rate Rate (absorber/stripper) (molCO2/ CO2 (Actual (L/min) MolMEA) (mol %) m3/min) Flexipac 1Y/IMTP #40 0.18 16.9 6.88 18.8 Flexipac 1Y/IMTP #40 0.16 16.4 6.87 13.2 Flexipac 1Y/IMTP #40 0.18 17.4 13.74 29.4 Flexipac 1Y/IMTP #40 0.17 16.5 13.75 37.7 Flexipac 1Y/IMTP #40 0.15 16.7 13.75 29.4 Flexipac 1Y/IMTP #40 0.15 16.8 13.75 25.9 Flexipac 1Y/IMTP #40 0.33 16.8 12.37 56.8 Flexipac 1Y/IMTP #40 0.37 17.8 9.75 80.4 Flexipac 1Y/IMTP #40 0.27 17.0 5.50 28.4 Flexipac 1Y/IMTP #40 0.27 17.0 5.50 23.1 Flexipac 1Y/IMTP #40 0.28 17.3 5.49 20.4 Flexipac 1Y/IMTP #40 0.28 15.2 8.74 39.5 IMTP #40/Flexipac 1Y 0.28 16.6 11.00 104.1 IMTP #40/Flexipac 1Y 0.29 16.7 11.00 82.1 IMTP #40/Flexipac 1Y 0.28 16.6 11.00 54.9 IMTP #40/Flexipac 1Y 0.28 17.5 5.50 40.7 IMTP #40/Flexipac 1Y 0.28 17.9 5.50 42.6 IMTP #40/Flexipac 1Y 0.28 17.0 5.62 42.8 IMTP #40/Flexipac 1Y 0.23 16.8 11.00 83.1 IMTP #40/Flexipac 1Y 0.23 17.1 10.97 56.8 IMTP #40/Flexipac 1Y 0.23 17.0 11.00 39.4 IMTP #40/Flexipac 1Y 0.29 16.9 8.25 60.8 IMTP #40/Flexipac 1Y 0.28 18.0 8.23 30.1 rR Fo w. CO2 Remov al (%) 99 99 61 96 87 75 62 94 95 87 72 92 93 86 70 95 80 95 94 87 72 96 69 l- tia en id nf Co 1,2 3,4 5,6 7,8 9,10 11,12 13,14 16,17 18,19 20,21 22,23 24 25,26 27,28 29,30 31,32 33,34 35,36 39,40 41,42 43,44 45,46 47,48 ie ev S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 50 of 54 ACS Paragon Plus Environment 50 Page 51 of 54 Case 5 6 7 8 9 10 11 12 13 14 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 Table 6. Performance of the absorber Lean (molCO2/molMEA) Rich (molCO2/molMEA) Experimental Experimental Simulation 0.180 0.525 0.497 0.182 0.523 0.482 0.177 0.496 0.447 0.170 0.493 0.444 0.150 0.532 0.476 0.148 0.533 0.477 0.147 0.537 0.489 0.143 0.546 0.490 0.323 0.507 0.454 0.329 0.508 0.452 0.268 0.506 0.477 0.274 0.495 0.449 0.271 0.538 0.484 0.274 0.540 0.482 0.282 0.554 0.494 0.277 0.557 0.496 0.275 0.506 0.441 0.278 0.386 0.357 0.275 0.376 0.352 0.284 0.413 0.376 0.287 0.412 0.384 0.285 0.448 0.418 0.284 0.453 0.429 0.281 0.426 0.415 0.279 0.428 0.416 0.283 0.422 0.415 0.282 0.420 0.416 0.280 0.415 0.398 0.284 0.425 0.415 0.282 0.404 0.397 0.281 0.402 0.389 0.228 0.367 0.337 0.229 0.371 0.337 0.235 0.433 0.387 0.232 0.430 0.389 0.231 0.491 0.428 0.231 0.492 0.425 0.285 0.433 0.397 0.286 0.426 0.399 0.281 0.539 0.465 0.285 0.537 0.465 rR Fo w. ie ev l- tia en id nf Co S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 51 Submitted to Industrial & Engineering Chemistry Research Table 7. Mass ratios of MEA/(H2O+MEA) of five samples for Case 22 Sample Mass ratios of MEA/(H2O+MEA) Lean into the top of the absorber 0.325 Middle of the absorber 0.328 Rich out of the bottom of the absorber 0.334 Middle of the stripper 0.300 Lean out of the bottom of the stripper 0.399 rR Fo Type A B C Table 8. Three types of absorber temperature profile Case 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 20, 21, 22, 23, 24, 43, 44, 47, 48 18, 19, 29, 30, 41, 42 25, 26, 27, 28, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 45, 46 w. ie ev Table 9. Four calculation methods Method for Method for Liquid Phase Mass transfer Chemical Reaction Rate, rate Rate-based Kinetics Rate, equilibrium Rate-based Chemical Equilibrium Equilibrium, rate Equilibrium-stage Kinetics Equilibrium, equilibrium Equilibrium-stage Chemical Equilibrium Methods en id nf Co Table 10. Performance of the absorber, Case 7 Lean Rich (molCO2/molMEA) (molCO2/molMEA) Experimental 0.177 0.496 Rate, rate n.a. 0.447 Rate, equilibrium n.a. 0.518 Equilibrium, rate n.a. 0.515 Equilibrium, equilibrium n.a. 0.529 l- tia S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 52 of 54 ACS Paragon Plus Environment 52 Page 53 of 54 Table 11. Performance of the absorber, Case 19 Lean Rich (molCO2/molMEA) (molCO2/molMEA) Experimental 0.274 0.435 Rate, rate n.a. 0.449 Rate, equilibrium n.a. 0.478 Equilibrium, rate n.a. 0.479 Equilibrium, equilibrium n.a. 0.479 rR Fo Table 12. Performance of the absorber, Case 38 Lean Rich (molCO2/molMEA) (molCO2/molMEA) Experimental 0.281 0.402 Rate, rate n.a. 0.389 Rate, equilibrium n.a. 0.418 Equilibrium, rate n.a. 0.418 Equilibrium, equilibrium n.a. 0.418 w. ie ev Type Table 13. Discretization methods Number of Film of Discretization discretization points Ratio n.a. n.a. n.a. n.a. 5 5 5 5 1 5 10 5 5 2 5 10 Reaction condition factor n.a. 0.9 0.9 0.5 0.9 0.9 0.9 0.9 tia en id nf Nofilm Filmrxn Discretization 1 Discretization 2 Discretization 3 Discretization 4 Discretization 5 Discretization 6 Co Table 14. Performance of the absorber, Case 7 Type Lean Rich (molCO2/molMEA) (molCO2/molMEA) Experimental 0.177 0.496 Nofilm n.a. 0.513 Filmrxn n.a. 0.509 Discretization 1 n.a. 0.446 Discretization 2 n.a. 0.461 Discretization 3 n.a. 0.402 Discretization 4 n.a. 0.448 Discretization 5 n.a. 0.423 Discretization 6 n.a. 0.444 l- S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Submitted to Industrial & Engineering Chemistry Research ACS Paragon Plus Environment 53 Submitted to Industrial & Engineering Chemistry Research Table 15 Performance of the absorber, Case 7 Lean Rich (molCO2/molMEA) (molCO2/molMEA) Experimental 0.177 0.496 CounterCurrent n.a. 0.446 Mixed n.a. 0.442 VPlug n.a. 0.444 VPlug-Pavg n.a. 0.444 rR Fo ie ev Table 16 Performance of the absorber, Case 19 Lean Rich (molCO2/molMEA) (molCO2/molMEA) Experimental 0.274 0.495 CounterCurrent n.a. 0.449 Mixed n.a. 0.451 VPlug n.a. 0.450 VPlug-Pavg n.a. 0.450 w. Co Table 17 Performance of the absorber, Case 38 Lean Rich (molCO2/molMEA) (molCO2/molMEA) Experimental 0.281 0.402 CounterCurrent n.a. 0.389 Mixed n.a. 0.387 VPlug n.a. 0.389 VPlug-Pavg n.a. 0.389 l- tia en id nf S AC 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 Page 54 of 54 ACS Paragon Plus Environment 54


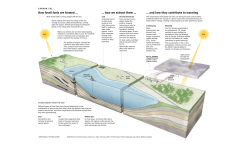






© Copyright 2026