
Zeiss LSM 510 Meta and LSM 5 Duo Room A503b
Zeiss LSM 510 Meta and LSM 5 Duo Confocal Microscopes and LSM Software Room A503b User Guide Molecular Imaging Unit (MIU) University of Helsinki www.miu.helsinki.fi 8.6.2010 1 GENERAL ......................................................................................................................... 1 1.1 1.2 1.2.1 1.2.2 1.2.3 1.3 1.4 1.5 1.6 1.7 1.8 1.8.1 1.8.2 1.9 2 TURNING ON THE SYSTEM ........................................................................................ 5 2.1 2.2 2.3 3 Opening the incubator (Duo only) ......................................................................................... 9 Placing the sample ............................................................................................................... 10 Viewing ............................................................................................................................... 11 Fluorescence imaging through the oculars .......................................................................... 14 Stage movement ............................................................................................................. 14 Fluorescence light and filters in Meta ............................................................................. 14 Fluorescence light and filters in Duo .............................................................................. 14 Transmitted light imaging through the oculars (Meta only) ................................................ 15 CREATING CONFIGURATIONS ............................................................................... 16 5.1 5.1.1 5.1.2 5.1.3 5.1.4 5.1.5 5.1.6 5.2 5.3 5.4 5.5 5.5.1 5.5.2 5.5.3 5.6 5.7 6 Laser control window ............................................................................................................ 7 Laser lines.............................................................................................................................. 7 Notes on turning on specific lasers ........................................................................................ 8 VIEWING SAMPLES THROUGH OCULARS (ACQUIRE/MICRO) ...................... 9 4.1 4.2 4.3 4.4 4.4.1 4.4.2 4.4.3 4.5 5 Turning on Meta .................................................................................................................... 5 Turning on Duo ..................................................................................................................... 6 Starting the acquisition software ........................................................................................... 6 TURNING ON LASERS (ACQUIRE/LASER) ............................................................. 7 3.1 3.2 3.3 4 Instruments ............................................................................................................................ 1 Reservations .......................................................................................................................... 1 Common reservation rules ................................................................................................ 1 Meta lasers ........................................................................................................................ 2 Duo-specific reservation rules .......................................................................................... 2 Billing .................................................................................................................................... 2 Unauthorized use ................................................................................................................... 3 Users’ mailing list ................................................................................................................. 3 Manual and user guide........................................................................................................... 3 Acknowledging MIU ............................................................................................................. 3 Sample preparation ................................................................................................................ 3 Selection of fluorochromes ............................................................................................... 4 Mounting samples............................................................................................................. 4 Image processing and analysis .............................................................................................. 5 Creating a basic configuration for LSM 510 (Acquire/Config) ........................................... 16 Important note about creating configurations for LSM 5 Duo........................................ 19 Excitation ........................................................................................................................ 19 Main dichroic beam splitter (HFT n) .............................................................................. 20 Secondary dichroic beam splitter (NFT N) ..................................................................... 20 Emission filters ............................................................................................................... 20 Detectors ......................................................................................................................... 20 Creating a configuration for Live imaging Live imaging .................................................... 21 Transmitted light imaging with Meta .................................................................................. 21 Saving and loading configurations ...................................................................................... 22 Meta detector (Only Meta) .................................................................................................. 22 Lambda mode ................................................................................................................. 22 Linear unmixing ............................................................................................................. 23 Online fingerprinting ...................................................................................................... 24 Stage control ........................................................................................................................ 24 Tile scanning ....................................................................................................................... 25 SCANNING IMAGES WITH LSM 510 (ACQUIRE/SCAN) ..................................... 25 6.1 General settings (Scan control/Mode/Frame) ...................................................................... 25 6.1.1 Objective Lens, Image Size, and Line Step Factor ......................................................... 26 6.1.2 Speed .............................................................................................................................. 26 6.1.3 Pixel Depth, Scan Direction and Scan Average .............................................................. 26 6.1.4 Zoom, Rotation, and Offset ............................................................................................ 27 6.2 Channel settings (Scan control/Channels) ........................................................................... 27 6.2.1 Channels ......................................................................................................................... 28 6.2.2 Pinhole ............................................................................................................................ 28 6.2.3 Detector and amplifier .................................................................................................... 29 6.2.4 Excitation ........................................................................................................................ 29 6.2.5 Buttons in the Scan control window ............................................................................... 29 6.3 Optimal settings for DETECTOR GAIN and AMPLIFIER OFFSET ................................ 30 6.4 Pinhole XY-settings............................................................................................................. 30 6.4.1 Pinhole ............................................................................................................................ 31 6.4.2 Position X and Y............................................................................................................. 31 6.4.3 Store Current Position..................................................................................................... 31 6.5 Acquisition of a Z-stack ...................................................................................................... 31 6.5.1 About Z-stacks ................................................................................................................ 31 6.5.2 Optical slice settings ....................................................................................................... 32 6.5.3 Defining first and last slice ............................................................................................. 32 7 SCANNING IMAGES IN LIVE MODE ON DUO...................................................... 33 7.1 7.2 8 Differences to LSM mode scanning .................................................................................... 33 Hyperfine (and fast) z-stacks in Live mode ......................................................................... 34 IMAGE WINDOW MENU ............................................................................................ 35 8.1 CHAN.................................................................................................................................. 35 8.2 ZOOM ................................................................................................................................. 35 8.3 SLICE .................................................................................................................................. 35 8.4 OVERLAY .......................................................................................................................... 35 8.5 CONTR ............................................................................................................................... 36 8.6 PALETTE ............................................................................................................................ 36 8.7 ANIM .................................................................................................................................. 36 8.8 REUSE ................................................................................................................................ 36 8.9 CROP .................................................................................................................................. 36 8.10 COPY .................................................................................................................................. 36 8.11 SAVE .................................................................................................................................. 36 8.12 SAVE AS ............................................................................................................................ 36 8.13 XY ....................................................................................................................................... 36 8.14 SPLIT XY............................................................................................................................ 37 8.15 ORTHO ............................................................................................................................... 37 8.16 CUT ..................................................................................................................................... 37 8.17 GALLERY .......................................................................................................................... 37 8.18 HISTO ................................................................................................................................. 38 8.18.1 Histogram................................................................................................................... 38 8.18.2 Area measurements .................................................................................................... 38 8.18.3 Colocalization measurements .................................................................................... 39 8.19 PROFILE ............................................................................................................................. 40 8.20 2.5D ..................................................................................................................................... 41 8.21 3D ........................................................................................................................................ 41 8.22 Topo .................................................................................................................................... 41 8.23 Prev...................................................................................................................................... 41 8.24 Info ...................................................................................................................................... 41 9 SAVING AND EXPORTING IMAGES ....................................................................... 41 9.1 Saving images...................................................................................................................... 42 9.2 Exporting images ................................................................................................................. 42 9.2.1 File formats ..................................................................................................................... 42 9.2.2 Image formats ................................................................................................................. 43 10 ENDING YOUR SESSION ............................................................................................ 44 10.1 There are users after you on the same day ........................................................................... 44 10.2 You are the last user of the day ........................................................................................... 45 10.2.1 Switch off lasers ......................................................................................................... 45 10.2.2 Turn the power off at Meta ........................................................................................ 45 10.2.3 Turn the power off at Duo.......................................................................................... 45 10.2.4 Cleaning up ................................................................................................................ 45 11 ADDITIONAL FUNCTIONS FOR LIVE SAMPLES ................................................ 46 11.1 Heat and CO2 control........................................................................................................... 46 11.1.1 Heat control in Meta .................................................................................................. 46 11.1.2 Heat and CO2 control in Duo ..................................................................................... 46 11.2 Acquisition of time series .................................................................................................... 47 11.3 FRAP (fluorescence recovery after photobleaching) ........................................................... 47 11.4 Reflection microscopy ......................................................................................................... 48 12 TROUBLESHOOTING ................................................................................................. 48 12.1 12.2 12.3 12.4 Starting the system .............................................................................................................. 48 Using the microscope .......................................................................................................... 48 Scanning .............................................................................................................................. 49 How to do background subtraction with BF/DIC/Ph images on LSM 510 Meta ................ 49 1 General 1.1 Instruments This user guide covers two instruments: LSM 5 DUO (abbreviated to Duo) and LSM 510 META (abbreviated to Meta). The parts that describe only Live-specific properties are marked with a line on the left side of the paragraph. Duo has two scan heads: LSM 510 point-scanner and LSM 5 Live line-scanner. Pointscanner has better resolution but the scanning is slow. Line-scanner is much faster but the resolution is not quite as good as with the point-scanner. Meta has the LSM 510 point-scanner and Meta spectral scanner, which is capable of resolving different wavelengths of the signal with 10 nm bandwidth. Linear unmixing is also possible. Meta is equipped with a heated stage, whereas Duo is fitted with an environmental chamber with temperature, humidity, and CO2 control. Duo has a piezo-controlled stage, which enables fast acquisition of Z-stacks. Some of the objectives and filters are different in Meta and Duo. They also have different light sources for observing fluorescence through the oculars, as well as slightly different microscopes. Thus, the same specimen may look different when observed/scanned using Meta or Duo. For more information on each instrument, visit: http://www.miu.helsinki.fi/instruments/index.htm. 1.2 Reservations Common reservation rules Instructions for Scheduler online reservation system can be found on the MIU web page: http://www.miu.helsinki.fi/reservations.htm. You can make your Meta/Duo reservations three weeks in advance. If you need to cancel your reservation, you have to do it two hours before your reservation starts. Unused reservations will also be charged. Make sure you are familiar with the use of Scheduler and the MIU fees and user policy (http://www.miu.helsinki.fi/fees.htm). When you make reservations, please always remember to indicate in the Event name field which lasers you will use during your imaging. This enables the users before you to turn off those lasers that are no longer needed on that day, thus extending the lifetime of the very expensive lasers. If you want to extend your session and there are no reservations after you, you can make a new reservation right after the current one. It is not possible to make a single 1 reservation that extends beyond 24:00. In such a case reservation can be extended by starting a second one at 00:00. Meta lasers To avoid mix-ups use these names for the lasers: 405, Argon, 546 and 633. Do NOT use abbreviations or combinations such as HeNe1+2. You may use the term “all lasers” if you really plan to use all of the lasers. Duo-specific reservation rules When reserving Duo, select which lasers you are going to use by choosing LSM or Live in the Service part of the reservation form (Figure 1). When using Live, you are also entitled to use LSM lasers. The pricing is different for the lasers due to different maintenance costs. Figure 1. Service selection in the Scheduler In the Description field of the reservation, write the lasers you plan to use. Make sure you differentiate between Live and LSM lasers. Use the terms: Live440, Live489, Live561, LSM405, LSMArgon, LSM543, LSM633. Do NOT use abbreviations or combinations. In addition to lasers used, write the temperature of the incubator. The first reservation for a day determines the temperature for the rest of the day. RT may be used for Room Temperature. Otherwise, use Celsius degrees e.g. 37C. 1.3 Billing The billing is based ONLY on the reservations made in Scheduler. The fee for the training session is twice the regular hourly fee. Any time after the initial training, you can ask MIU staff to give you additional customized training (charged the same way as the initial training). For Duo, Live and LSM 510 use have different fees. Mark your LIVE/LSM 510 use in the Service selection in the reservation. 2 1.4 Unauthorized use MIU periodically checks the logins of the Meta and Duo computers. If your logon/off times exceed your reserved time in Scheduler, MIU will consider that as unauthorized use of the instrument and ask for an explanation. For unauthorized use of the instrument, MIU may issue a warning. After three warnings, MIU may revoke the user's license. Also, misuse/neglect of the instrument may lead either to a warning or cancellation of the user license, and the repair costs may be charged. If there are no reservations for Meta/Duo, and you only want to use the computer for data transfer, mark the time in the data transfer logbook. When the computer logins are checked, the logbook marking will tell MIU staff that you did not use the confocal without reservation. 1.5 Users’ mailing list All registered users are automatically added to the users‟ mailing list. Please read those e-mails to know what is going on! 1.6 Manual and user guide In the confocal room, there are complete manuals of the LSM 510 Meta and LSM 5 Duo. Chapter 5 of the manual covers all functions in the software. Please do not remove the manuals from the room. MIU has a copy of the LSM 510 Meta manual that you can borrow. The manual is also available as a pdf file on the instrument computer. This user guide will be continuously updated by the MIU personnel. Check that you have the latest version of the user guide: go to http://www.miu.helsinki.fi/instruments/ and compare the date of the latest update with the date on the first page of your printed user guide. This is the only place for update announcements; no e-mails about updates will be sent. The user guide can also be accessed in the computer through the Internet Explorer browser: http://www.miu.helsinki.fi/instruments/. 1.7 Acknowledging MIU Whenever you are using instruments and/or services maintained/provided by MIU, you are expected to acknowledge MIU in your publications and inform MIU about the papers. 1.8 Sample preparation A confocal microscope can remove out-of-focus haze that decreases the signal-tonoise ratio. However, it cannot compensate for bad sample preparation. Garbage in, garbage out is also true for confocal microscopy! 3 Selection of fluorochromes If you want to achieve the best possible resolution, use a fluorochrome whose emission spectrum covers as short wavelengths as possible. The resolution of the microscope is determined by the numerical aperture (NA) of the objective used and the wavelength of the light detected. The larger the NA and the shorter the wavelength, the better the resolution. For thick tissues, image acquisition deeper in the tissue is often a problem because of light scattering in the tissue. The longer the wavelength of the light, the less it scatters, and the better its penetration into tissues. Thus, use fluorochromes whose emission spectra are in the far-red region if you need to image deep inside tissue. For multilabel imaging, select fluorochromes that are spectrally wide apart. E.g. Alexa488 and Alexa594 usually have cross-talk between channels if scanned simultaneously, so they should be scanned sequentially. If sequential scanning is not an option because of longer scan time required, then you should use fluorochromes that do not have any cross-talk. Be sure to check the exact excitation and emission wavelengths of the fluorochromes you are using either from their manufacturer or from a fluorochrome database. remember that the names of Alexa fluors do not always indicate the excitation maxima! Their naming convention requires a different number for a new name even if the excitation peak is the same as in an old version. Mounting samples You should either use a mounting medium that will harden or, in the case of nonhardening one, seal the edges of the cover glass with nail polish or some other sealant. Let the sealant dry well before you come to the confocal - nail polish is one of the most harmful substances for optics. Also, unsettled mounting medium, or one containing too much water (slides not dry enough when mounted), or too much mounting medium, can affect imaging by causing shadows and/or stripes in the image. Use a media that has the same refractive index as the immersion media you are using (air, oil, water or glycerine). If you are using oil immersion objectives, the refractive index of the mounting medium used should be close to the refractive index of the immersion oil (1.518). For more information on mounting media and antifade reagents, visit: http://www.uhnresearch.ca/facilities/wcif/PDF/Mountants.pdf. Most of the objectives are designed for coverslip thickness 0.17 mm, which corresponds to number 1.5 coverslips. Do not mount the coverslip so that it will be pressed down by the clips in the sample holder, ie. leave some room at both ends of the slide. Make sure that you are using the correct coverglass thickness for the objective you are using. Objectives marked with “korr” have a correction collar that can be adjusted to different coverglass thicknesses. 15 µm deviation in coverglass 4 thickness can decrease resolution and signal intensity by half. It is best to use high precision coverglasses as these have a low variation in thickness. 1.9 Image processing and analysis MIU has two Imaging Workstations that have software for further image processing and analysis, such as deconvolution, cell counting, and more. Visit the MIU web site or contact MIU for more information. Use of Imaging Workstations is free of charge, and MIU staff gives training and help with the software. MIU also provides customized image analysis tools in case the commercial and free software do not satisfy your image analysis needs. 2 Turning on the system 2.1 Turning on Meta If you are the first user you should turn the computer and monitors on - computer by pressing the round button in the top right corner of the front panel, monitors by pressing the buttons below the screen. Log on to the computer using your LTDK user ID (START/LOG ON). Switch REMOTE CONTROL ON (Figure 2). This will turn on the microscope. Please notice that after turning the system on, you have to wait at least 30 min before turning it off because of the mercury bulb. Warming of the lasers will take about 2 hrs for maximal stability but you can start using them sooner unless the stability is critical for your imaging. Figure 2. Remote control button 5 2.2 Turning on Duo To start up the system, the following sequence must be carefully followed, otherwise the system will crash. Turn the main power switch on. It is located in the far left corner of the electronics/laser rack. (Figure 3: left image) Turn on the “System/PC” switch, on the right-hand side of the microscope. Wait for the computer to start and then login with your LTDK account. (Figure 3: right image) After logging in turn on the “Component” switch. Wait for the computer to get a connection to the real-time PC. The icon will be shown in the lower right corner of the screen (in the taskbar). Figure 3. Duo main switch is on the left. System/PC and Component switches are on the right. 2.3 Starting the acquisition software Initiate LSM 510 Program through a shortcut at the desktop (Figure 4). Figure 4. Shortcut for LSM 510 software Choose SCAN NEW IMAGES and then click START EXPERT MODE (Figure 5). 6 Figure 5. The start dialog for LSM 510 software 3 Turning on lasers (Acquire/Laser) 3.1 Laser control window Go to ACQUIRE / LASER (Figure 6). Figure 6. The buttons for laser settings 3.2 Laser lines Table 1. Laser lines for LSM 510 Laser Laser line Examples of fluorochromes that can be used Diode 405 nm DAPI, Hoechst Argon 458 nm CFP Argon 488 nm Alexa Fluor 488, GFP, Cy2, SytoxGreen, Acr.Orange/DNA, FITC Argon 514 nm YFP, Cy3 HeNe1 543 nm Alexa Fluor 546, Alexa Fluor 594,Texas Red, HcRed1, DsRed2, PI, TRITC/Rhodamine, Mitotracker Orange HeNe2 633 nm Alexa 633, Alexa 647, Alexa 660, Cy5, APC 7 Table 2. Laser lines for Live imaging Laser Laser line Examples of fluorochromes that can be used DPSS 440 nm CFP DPSS 489 nm GFP DPSS 563 nm TRITC 3.3 Notes on turning on specific lasers If any of the lasers have a Standby-option, always use it in the middle, when moving from off-to-on state or on-to-off. Turning on Diode-, HeNe1- and HeNe2-laser: Turn on the lasers you need by selecting one laser at a time and clicking ON (Figure 7). Figure 7. Laser control window Turning on the Argon-laser: For the argon laser, click STANDBY to warm up the laser (Figure 8). Let the laser warm up for as long as possible, this will extend the lifetime of the laser. Only when you are ready to start scanning, click ON (Status has to be Ready). Gradually (stepwise) increase the output to about 50% (6 Amperes) by clicking the arrow on the right. For images taken during separate sessions, the output values should be the same, otherwise you cannot directly compare the images. 8 Figure 8. Laser control for Argon laser Note: Do not click lasers on/off during your session. Select Standby for the Argon if you will take a longer break. Drag the slider all the way to the left, into the minimum output value. 4 Viewing samples through oculars (Acquire/Micro) Make sure that the oculars are correctly adjusted for your eyes. If you have normal vision, the zero mark on the sides of the oculars should match the white dot. If you wear eyeglasses but want to use the microscope without them, you must adjust the oculars according to your vision. First look at your specimen with your right eye closed (through the left ocular). Slowly turn the eyepiece scale ring until the image is optimally in focus. Repeat with the other eye. The scale on the oculars is the diopter scale. 4.1 Opening the incubator (Duo only) To access the stage, open the two doors on the front side of the incubator. Turn the handles to open the latch. If you need more room to operate, you may open the top and then tilt the top part (the condenser) of the microscope. The top opens from the two blue handles – pull them sideways away from the microscope (See Figure 9). Keep the doors closed to prevent dust getting in the incubator. 9 Figure 9. Opening the incubator doors and tilting the condenser 4.2 Placing the sample First check that the stage holder is firmly in place. If not, take it out and reinsert it correctly (red dot in the stage holder indicates the corner that should go where there is another red dot marked on the stage). As there are two springs in that corner holding the stage holder, you have to press the holder in from the opposite corner (requires some patience and practice). Put your specimen on the stage so that it fits snugly the indentions – remember inverted position (coverslip down)! Use the metal clips to secure the slide (Figure 10). Figure 10. Inserting the specimen on the stage (Meta slide holder on left and Duo slide holder on right) Make sure that the coverslip thickness is about 0.17 mm (No. 1.5 cover glass) and that the clips do not press down the coverslip, as that creates tension on the coverslip that may affect your scanning. Use smaller coverslips if that is a problem. 10 4.3 Viewing To view your sample through oculars without laser scanning, click VIS-button on the right (Figure 11). This will allow only the light from halogen or mercury bulb to pass through the oculars. Figure 11. The button for VIS mode Go to ACQUIRE / MICRO Figure 12). Figure 12. The button for microscope settings The Microscope Settings window pops up (Figure 13). Figure 13. Microscope settings Here you can change objectives (click on the objective button) and make adjustments for halogen and mercury light sources. There are six objectives in the microscope, and a 63x, NA 1.2 (korr.) C-Apochromat water immersion objective is available upon request (Table 3). The oil objectives have protective felt rings. Table 3. Objectives available in LSM 510 Meta. Working distance (WD) is shown in mm. CG: Cover glass thickness that should be used, shown in mm. Objective 11 W D NA CG Immersion Contrast methods 5x Plan-Neofluar 13. 6 0.15 0.17 None (air) None 10x Fluar 2 0.5 0.17 None (air) DIC 20x LD Achroplan 10. 2 0.4 korr. 01.5 None (air) DIC, Ph2 40x Plan-Neofluar 0.2 1.3 0.17 Oil (Immersol 518F) DIC 63x PlanApochromat 0.1 9 1.4 0.17 Oil (Immersol 518F) DIC 63x Plan-Neofluar 0.1 1.25 0.17 Oil (Immersol 518F) DIC, Ph3 63x C-Apochromat 0.2 8 1.2 korr. 0.14 0.19 Water DIC Table 4. Objectives in LSM 5 Duo. Objective W D NA CG Immersion Contrast method 10x Plan-Apochromat 2.0 0.45 0.17 None (air) DIC 25x LD PlanApochromat 0.5 7 0.8 korr 00.17 mixed DIC 40x Plan- Apochromat 0.2 1 1.30 0.17 Oil (Immersol 518F) DIC 40x LD CApochromat 0.6 2 1.10 korr 0.14 0.19 Water DIC 63x Plan- Apochromat 0.1 9 1.40 0.17 Oil (Immersol 518F) DIC Working Distance (WD) determines how far from the sample the objective can be lowered or the maximum distance of the objective front lens from the coverglass. Numerical aperture (NA), together with the wavelength of the light used, determines resolution of the microscope; the higher the NA, the better the resolution. 20x LD Achroplan and 63x C-Apochromat objectives have correction collars, which should be 12 adjusted according to the cover glass thickness. The higher the NA, the closer to the sample the objective has to work. For this reason the working distance (WD) of the high NA objectives is short. Obviously, objectives differ in magnification and numerical aperture but they also differ in other respects. Some properties of the Meta objectives are shown in Table 5. LD objectives have a long working distance. Flatness: Flatness of the imaging field = How well objects that are around the edges of the image are at the same focal plane with objects that are in the middle of the image. Color correction (CC): How well different color channels are in register; indicates how well the objective suites for multichannel work UVIR transmission: The better the transmission, the less ultraviolet/infrared light gets lost on its way through the objective. Multichannel/DIC: How suitable the objective is for multichannel/DIC imaging. Table 5. Properties of objectives on Meta and Duo. Scale is from 1 (worst) to 5 (best). For objectives marked with an asterisk (*), properties shown are not for the actual objective but for the more recent version (EC Plan-Neofluar). For Plan-Neofluar type objectives, inf information cannot be found on the Zeiss web site. Thus, for Plan-Neofluar objectives the actual rating is either equal or lower than the one shown. Objective Instrument Flatness CC UV trans. IR trans. Multich. DIC 5x Plan-Neofluar* Meta 4 4 4 3 4 4 10x Fluar Meta 1 1 5 4 1 1 10x Plan-Apochromat Duo 5 5 3 4 5 5 20x LD Achroplan Meta 3 3 3 3 2 1 25x LD Plan-Apochromat Duo 5 5 3 4 5 5 40x Plan-Neofluar* Meta 4 4 4 3 4 4 40x Plan- Apochromat Duo 5 5 3 4 5 5 40x LD C-Apochromat Duo 5 5 4 5 5 5 63x Plan-Apochromat Meta&Duo 5 5 3 4 5 5 63x Plan-Neofluar* Meta 4 4 4 3 4 2 63x C-Apochromat Meta 5 5 4 5 5 5 13 You do not necessarily need to change the objective when you want to change magnification; you can also zoom using the zoom control or by cropping in a previous image. For living specimens (eg. cultured cells or tissues, brain slices, animals or plants in water or aqueous solutions/buffers) use water immersion objectives. For fluorescent labeled thin specimens (eg. cultured cells embedded in Vectashield, Moviol or prolongGold) use oil immersion objectives. For fixed specimens (eg. thick tissues, whole mount embryos/animals or plants and samples embedded in resin, Canada balm, glycerol-gelatin or glycerol) use oil immersion objectives or glycerol immersion objectives. 4.4 Fluorescence imaging through the oculars Stage movement For fluorescence imaging, all microscope manipulation is done through this panel; do not change anything through the microscope itself except focusing and the stage position through the joystick. Use the joystick for large movements and the black round wheel for fine movements. Select first if you want to move along X or Y axis by pressing the corresponding X/Y button. Fluorescence light and filters in Meta For viewing of your sample with mercury light source (HBO), select the filter from REFLECTOR appropriate for your staining and click REFLECTED LIGHT on. The four fluorescence filters available are shown in Table 6. Table 6. Fluorescence filters available in Meta. FSet Fuorochromes 00 Alexa Fluor 594, TexasRed 02 DAPI, Hoechst 15 Alexa Fluor 546, TRITC, Cy3 16 Alexa Fluor 488, FITC When you end viewing your sample through oculars, choose NONE for the REFLECTOR and click again on REFLECTED LIGHT to turn it off. Also check that TRANSMITTED LIGHT is off. Fluorescence light and filters in Duo To view your sample under the light of the Metal Halide lamp, switch the mode to VIS in the main window of the program. On Duo, there is no shutter for fluorescent 14 light source – the sample is illuminated in the VIS mode, and not illuminated in LSM mode. Remember to change to LSM mode to avoid bleaching you sample. Select the filter from REFLECTOR to fit your staining. The examples for different stainings are shown in Table 7. Table 7. Fluorescence filters available in Duo. FSet Fuorochromes 01 DAPI, Hoechst, Alexa405 09 Alexa488, GFP, FITC 45 DsRed, HcRed, mRFP, Cy3.5 25HE Installable on demand: triple filter Alexa405/488/594 4.5 Transmitted light imaging through the oculars (Meta only) To look at your sample with halogen (visible) light, click TRANSMITTED LIGHT. The reflector has to be in position NONE. Select the correct PHASE/DIC optics from CONDENSOR. Bright-field imaging is mainly for specimen with histological staining. Phase contrast imaging is used for thin, unstained specimen, whereas differential interference contrast (DIC) imaging is meant for thick, unstained specimen. DIC can also be used if phase contrast imaging shows disturbing haloes around objects. Make the Koehler illumination adjustments (if you are not familiar with them, MIU staff will help you). Control the brightness with the INTENSITY slider. When you want to turn off transmitted light click TRANSMITTED LIGHT button and click again ON. That will toggle between on and off states. Remember to turn off the light when you are not viewing your sample, otherwise you may unnecessarily bleach your specimen. For phase contrast imaging, you need to use an objective that has a phase ring (see Table 2), and a matching condenser position. Leave the condenser aperture fully open. For DIC (differential interference contrast) imaging, the polarizer, analyzer, and modified Wollaston prisms have to be on the light path, and correctly adjusted. Ask MIU staff to show you how to do that. 15 5 Creating configurations 5.1 Creating a basic configuration for LSM 510 (Acquire/Config) To scan your sample using LSM, click LSM on the right (Figure 14). This will NOT allow anything to pass through the oculars. Figure 14. The button for LSM mode To set the beam path configuration go to ACQUIRE / CONFIG (Figure 15). Figure 15. The button for configuration control The Configuration Control window pops up (Figure 16). 16 Figure 16. The configuration control window If you only use one fluorochrome, click CHANNEL MODE / SINGLE TRACK, otherwise CHANNEL MODE/MULTI TRACK. In MULTI TRACK mode, you add more tracks by clicking ADD TRACK and adjust settings separately for each channel. Whenever you are using the MULTI TRACK mode, make sure that there is no crosstalk between the channels that are scanned at the same time; you should have control slides stained only with one of the fluorochromes. Scan these controls using a configuration in which only the "wrong" channel is enabled - you should not see any signal. A track can be enabled/disabled by ticking the box on the left side of the track name. Only enabled tracks will be acquired. It is often useful to enable only one track at the time when making adjustments. A track can be renamed more appropriately by clicking on its name (eg. “Track 1” renamed to “GFP”). If you are using e.g. four fluorochromes (A = emission at shortest wavelengths, B = emission at 2nd shortest wavelengths, C = emission at 3rd shortest wavelengths, D = 17 emission at longest wavelengths), make a MULTI TRACK configuration as follows: 1. track: A and C, 2. track: B and D. In this way, the detected wavelengths in each track are as far apart from each other as possible. Again, check if there is any crosstalk between A and C. Also check cross-talk for B and D. If there is too much crosstalk, you either need to make separate tracks for those channels, or use Meta-detector and linear unmixing (see chapter 5.5). A beam path can be controlled with filters and mirrors starting from the lasers and ending in the detectors. Ch2 and Ch3 are standard PMT detectors and ChS is a socalled Meta-detector for spectral detection. ChD is for transmitted light. Note that: Mirror: light deflects from line None and Plate: light passes through Create a configuration by following the light path from lasers to the detectors (Figure 17). When you click on any button in the configuration control window, a list of options available will open. If the configuration control window is positioned near the bottom of the screen, you may not be able to see the whole list. In that case, you first need to move the configuration control window upwards. Figure 17. Setting laser lines, filters and beam splitters for a basic configuration 18 Important note about creating configurations for LSM 5 Duo The filter right above Specimen in the Figure 17 must be None, when imaging with LSM 510. When imaging with Live, use the Rear setting. Otherwise none of the light will reach the correct sensor. When doing FRAP and detecting with Live, use the T20/R80 mode. Excitation Select the laser line and adjust the transmission efficiency with a slider. Recommended transmission for each laser line is shown in Table 8.for Meta and in Table 9 for Duo. Table 8. Recommended transmission efficiency for each laser line on Meta. Laser Laser line Transmission Diode 405 nm 5 - 10 % Argon 458 nm 5 - 10 % Argon 488 nm 5 - 15 % Argon 514 nm 5 - 15 % HeNe1 543 nm 50 - 100 % HeNe2 633 nm 50 - 100 % Table 9. Recommended transmission efficiency for each laser line on Duo. Laser Laser line Transmission LSM405 405 nm 1-4 % LSMArgon 458 nm 5 - 10 % LSMArgon 488 nm 5 - 15 % LSMArgon 514 nm 5 - 15 % LSM543 543 nm 50 - 100 % LSM633 633 nm 50 - 100 % Live440 440 nm 15-40% (has not been tested) Live489 488 nm 2-10% Live561 561 nm 30-50% 19 Main dichroic beam splitter (HFT n) Select excitation wavelengths that will pass to the specimen (these should match with the laser line you use). Use as few wavelengths as possible and do not use wavelengths that are within the emission spectrum as they are cut off. Secondary dichroic beam splitter (NFT N) Select the splitting of the emission wavelengths between two detectors. The wavelengths longer than N will pass through to the Ch3 and the wavelengths shorter than N are reflected to the Ch2. With the mirror all wavelengths are directed to the Ch2. Emission filters Choose the wavelengths that are detected with each detector. LP X = Long pass filter = wavelengths longer than X are passed through BP X-Y = Band pass filter = wavelengths from X to Y are passed through Detectors Enable a detector by selecting the check box. Select the pseudo color for the channel by clicking the button Ch2 or Ch3. You can check that your configuration is correctly made by clicking SPECTRA (Figure 18). This shows you the laser line(s) used and emission wavelengths that will be detected. Figure 18. Spectra button 20 5.2 Creating a configuration for Live imaging Live imaging Figure 19. Configuration for Duo Live mode The configuration for Live mode is very similar to LSM mode (Chapter 5.1). The biggest difference is the lack of main dichroic beam splitter. Note that in order to get any image in the Live mode, the filter nearest to the Specimen must be in Rear or T20/R80 position. 5.3 Transmitted light imaging with Meta You can use the ChD detector for transmitted light (bright-field, phase contrast, or DIC) imaging. Check the ChD box and use e.g. the argon laser as a light source. Check that you have selected proper condenser position and that all other transmitted light settings are correct (see chapter 4.2). Make sure that the pseudo color selected for the ChD channel is white. As laser light is polarized, it is not necessary to use the polarizer for DIC imaging. Remember that the transmitted light image is not confocal, as there is no pinhole in front of the ChD detector. 21 There are some dust particles inside the ChD detector that often appear in the images. As there is no way to get rid of the dust, you should try to crop your image so that the particles will be excluded. If that is not possible, use background subtraction. 5.4 Saving and loading configurations The configuration can be saved with a CONFIG button on the right side of the configuration window (Figure 20). Click the button, name the configuration, and click STORE. Note that configurations are saved separately for SINGLE TRACK and MULTI TRACK modes. Figure 20. Configuration load/save button To load a saved configuration, click CONFIG and select the configuration from the pull-down list. Click APPLY. This will automatically switch on the desired filters and mirrors. Note that this will also adjust the laser lines, you must check these later on. Alternatively, you can open a previously stored image. In the main menu, select FILE / OPEN, then an image, and click REUSE (Figure 21). This will adjust microscope settings (not including the objective), configuration, and scan control options according to the ones used for the image loaded (as shown in INFO in the image window). Figure 21. Button for using configurations from saved images 5.5 Meta detector (Only Meta) META detector (ChS) is a spectral detector which means that you can freely select the range of emission wavelengths that you want to use for detection. It consists of 32 photomultipliers, each of which covers a spectral range of 10.7 nm (total range about 340 nm). Lambda mode You may want to use META detector in case the emission filters available are not optimal for your fluorochrome. Additionally, you will get the spectral emission (intensity for each wavelength within your selected range). When you scan in this socalled lambda mode, the result will be a lambda stack that shows the signal over the spectral range selected. It is also possible to combine lambda mode with Z mode and/or time series. 22 However, META detector is less sensitive compared to the other detectors, so you should have a strong signal if you want to use the META detector. Also, you may need to open the pinhole more than usually (try using e.g. 3 Airy units). To use lambda-mode, select LAMBDA MODE in the configuration control window. Set the excitation and main dichroic beam splitter (HFT N) as in the CHANNEL MODE. For selecting the detected wavelengths, either use the slider or write the starting/ending wavelengths in the proper input boxes. When you click on the CONFIG button, you can save and load lambda configuration. NUMBER OF PASSES will tell you how many scan are needed to cover the required spectral range. Proceed to SCAN as usually. For separation of spectral signals, you have two options: either LINEAR UNMIXING or ONLINE FINGERPRINTING. Linear unmixing META detector and linear unmixing can be used to separate signals from two or more fluorochromes with overlapping emission spectra. Also, autofluorescence can be separated from the true signal. In linear unmixing, the software will calculate for each pixel how much of the signal is derived from each spectral component that needs to be separated from the others, based on the reference spectrum for each component. In order to make a reference spectrum, you need to have control specimen that are each stained with only one of the fluorochromes whose spectra need to be separated. In case of autofluorescence, the control is simply an unstained specimen. LINEAR UNMIXING is used after you have acquired a Lambda stack (using META detector). In the MAIN menu, select PROCESS, and then UNMIX. Insert in the stage the specimen from which you want to acquire a reference spectrum. It is essential that the specimen is only stained with the fluorochrome for which you want to get a reference spectrum. Select the channel for the reference spectrum and name it. Press the LOAD FROM IMAGE button. Continue until you have acquired reference spectra for all the fluorochromes whose spectra you want to separate from each other. In the Source panel of the UNMIX window, click on the arrow button and select the image you want to use for unmixing by clicking on it. In the Parameters panel of the UNMIX window, select all options. The RESIDUALS channel will show you the amount of signal that could not be assigned for any of the unmix channels. ASK FOR THE BACKGROUND ROI option will ask you to mark an area in the image that represents background. This background channel should be defined for optimal linear unmixing. After clicking on APPLY, a new window that shows the result of the unmixing will open. 23 Online fingerprinting ONLINE FINGERPRINTING does the unmixing during scanning, and the lambda stack will not be shown or stored. If you want to use this option, you must have saved the reference spectra in the PROCESS/UNMIX window in advance (see previous chapter). Select ONLINE FINGERPRINTING in CONFIGURATION CONTROL window. Load the reference spectra using the RS1 ... 8 control buttons. Set the excitation and main dichroic beam splitter (HFT n) as in the CHANNEL MODE. For selecting the detected wavelengths, either use the slider or write the starting/ending wavelengths in the proper input boxes. When you click on the CONFIG button, you can save and load configurations. NUMBER OF PASSES will tell you how many scan are needed to cover the required spectral range. Proceed to SCAN as usually. 5.6 Stage control Figure 22 Piezo leveling button "L" The stage can be controlled by the software from ACQUIRE / STAGE. As both the stage and the focus control are motorized both can be changed here. The currant stage 24 position can be marked and returned to at a later time. The Duo has an additional piezo motor for fine and fast control of the focus level. The piezo focusing can be achieved from the STAGE AND FOCUS CONTROL menu (Figure 22). The piezo focus position is on the HRZ column, the motorized stage focus is on the FOCUS column. When the focus is changed with the stage motor (manually or from the software) the piezo objective focus is automatically leveled (the piezo is positioned into the middle point of its range of movement). It can also be leveled manually with the L-button. 5.7 Tile scanning The motorized stage can be used for making tile scans, ie. scanning a large area one image at a time. The tile scanning function is found from ACQUIRE / STAGE (Se the bottom of Figure 22). The maximum size of the tile scanned overview image is 4096x4096 pixels. The Tile Scan is made by choosing the size of the overview image in TILE NUMBERS and pressing START button. TILE NUMBERS controls the size of the overview image in tiles. TILE SIZE is the size of a single tile in pixels. FRAME SIZE is the size of the whole overview image in pixels and µm. MOVE TO activates a window showing the scanned area where a region of interest can be drawn by clicking and holding down the left mouse button. When it is released, the stage moves to the selected area. MARK button shows previously marked stage positions and allows new positions to be marked with a mouse click. The MOVE TO button can then be used to move the stage these positions. The OVERLAY functions cannot be used in the overview frame except to remove the rectangle showing the region of interest where the stage is positioned. You cannot Reuse the imaging settings from a tile scanned image on version 3.2 (on Meta). Remember to save your settings before running the tile scan! Because of the maximum image size and limitations of the tile merging function, it may sometimes be preferable to take the images separately and merge them into a mosaic using some other software, eg. Photoshop. 6 Scanning images with LSM 510 (Acquire/Scan) 6.1 General settings (Scan control/Mode/Frame) Go to ACQUIRE / SCAN (Figure 23). Figure 23. Opening scan control window 25 SCAN CONTROL window pops up. Choose MODE and FRAME (Figure 24). Figure 24. Scan control window Objective Lens, Image Size, and Line Step Factor Select FRAME SIZE. OPTIMAL shows the minimal sampling frequency (optimal number of pixels) that should be used. It will do the calculation based on the shortest wavelength of the active channels. If you use smaller frame size than suggested by OPTIMAL, you will lose information when you scan your image. It is often desirable to use an even larger frame size than suggested by OPTIMAL, especially if you are going to deconvolve your images. LINE STEP indicates how many lines are scanned. Always use setting 1 (every line will be scanned). Speed The OPTIMAL button (see previous paragraph) will also optimize the scanning speed. If the speed is too high, the quality of the image is low. On the other hand, setting the speed lower than the optimal speed will not improve the image quality. Pixel Depth, Scan Direction and Scan Average DATA DEPTH: 8 Bit means that for each pixel, there are 256 possible intensity values available. In a 12-bit image, the gray values can range from 0 to 4095 for each pixel. For quantative imaging, 12-bit images are often the preferred choice. However, 12-bit image files are much larger than 8-bit ones, so if you only use the images for 26 visual inspection and you cannot see any difference between 8- and 12-bit ones, use the former one. SCAN DIRECTION: Default is unidirectional. If you use bidirectional scanning (twice as fast), you have to correct the pixel shift either automatically by using the AUTO button, or manually by adjusting the Scan Corr X (for zero rotation) or both Scan Corr X and Y (for images rotated 90 degrees). Bidirectional scanning should always be used for live samples. SCAN AVERAGE: used to get rid of random noise. In MODE, select Line (averaged after each line) or Frame (averaged after each frame). Use Line for live samples. In METHOD, Mean is used for averaging. SUM adds up pixel values, so it also amplifies noise. In NUMBER, select the number of scans used for averaging. CONTINUOUS option will continuously scan the sample and at the same time average the scans. It is useful for estimating how many times the sample should be scanned for averaging. Zoom, Rotation, and Offset You can zoom to your sample and rotate it in the indicated field. The new settings will be used for the coming scans. The zoom is optical and uses real resolution of the scanner. Zooming will not increase resolution when the zooming factor is displayed in red. The area to be zoomed can also be selected from a taken image with CROP in the image windows. When the CROP command is active, you can increase the size of the zoom window by clicking on one of the corners of the window and dragging the window to a desired size. The position of the zoom window can be similarly adjusted by clicking on the zoom window somewhere else than the corners and dragging the window. Rotation of the zoom window is done by clicking on one end of the crosslines and dragging the crossline until the required rotation angle is reached. The blue line indicates the top of the new scan area. If you want to modify the shape of the scan area, click on the intersection of any crossline and zoom window outline, and drag with mouse. Remember that even if you decrease the scan area along X, the whole length of each line will be scanned, meaning that the scan time will not decrease even if you decrease the horizontal size of the zoom window (horizontal in relation to the scan direction). 6.2 Channel settings (Scan control/Channels) Select ACQUIRE/SCAN in the main menu. Click CHANNELS in the SCAN CONTROL window (Figure 25). 27 Figure 25. Channel settings Channels Select which channel you want to adjust by pressing the desired channel button. Pinhole When the resolution is close to the wavelength of the light used, the signal is no longer a poin of light, but a diffraction pattern called an Airy disk. The mathematical representation of the Airy disk is known as a point spread function (PSF). The PSF is 2-4 times larger in Z-dimension than in XY dimensions. The size of the pinhole is measured in Airy units, which is a relative unit dependent on the wavelength used. The Numerical Aperture of the objective will also affect the size of the Airy disk. The higher the NA, the smaller the Airy disk. For optimal resolution and depth discrimination, the pinhole size should be close to 1 Airy unit. 1 Airy unit is the diameter of the first diffraction ring of the point spread function of the signal and it gives the best signal-to-noise ratio. However, if the signal level is not satisfactory, it is possible to increase the pinhole size to 2-3 AU. Before using a larger pinhole size, make sure you have optimized your staining and other scanning parameters. Opening the pinhole more than that will significantly decrease the signal-to-noise ratio and is not recommended. Always start 28 imaging using the pinhole size 1 and only use larger size if necessary. Using pinhole sizes smaller than 1 often leads to greatly reduced signal levels. The pinhole diameter is adjusted with a slider. Corresponding optical slice thickness is shown below the pinhole diameter value. To set pinhole to 1 Airy unit press button marked "1". Note that for multiple channels, you have to adjust the pinhole sizes so that the optical slice thickness is the same for all channels; if you adjust the pinhole of the channel with longest wavelengths to 1, then the pinhole size for the other channels will be larger than 1. Accordingly, if you adjust the pinhole size for the channel with shortest wavelengths to 1, then for the other channels the size will be smaller than 1. Detector and amplifier AMPLIFIER OFFSET adjusts general intensity levels of pixel values, DETECTOR GAIN adjusts dynamic range of pixel values (Figure 26). AMLIFIER GAIN amplifies signal afterwards and therefore amplifies noise as well. It is recommended to set it to 1. See the next chapter for information about optimal offset and gain settings. Figure 26. Adjusting intensity range of pixel values with amplifier offset and detector gain Excitation In EXCITATION window you can indicate which laser lines to use and adjust the percentage of transmission (see 0) Buttons in the Scan control window Important safety issue: avoid looking at the laser light - Meta's lasers may be harmful for your eyes. During scanning, it is important to eliminate vibrations: Do not touch the air table (e.g do not lean on it or press your feet against it); make sure the arm rests of the table do not touch the computer table; avoid banging the door or anything else in the room. NEW: opens a new image window (if not selected, the new image will be scanned over the previous scanned image, which will be lost unless saved) FIND: the software tries to find the optimal adjustment of DETECTOR GAIN and AMPLIFIER OFFSET for the selected channel while scanning a single image 29 FAST XY: continuous scanning with maximum speed (use only for focusing and searching through a sample, never for setting the detector gain and offset) SINGLE: scans a single image STOP: stops scanning CONT: continuous scanning (beware of photobleaching) 6.3 Optimal settings for DETECTOR GAIN and AMPLIFIER OFFSET This needs to be done for each channel separately. It is recommended to first let the software try to adjust settings for DETECTOR GAIN and AMPIFIER OFFSET with FIND button. After that, you may tune the settings if needed. There is an aid for finding optimal settings for DETECTOR GAIN and AMPLIFIER OFFSET manually: In the image window, click PALETTE and select RANGE INDICATOR in the dropdown menu. It will show the channels in grayscale plus blue and red. The blue indicates underexposure and red indicates overexposure. For fast continuous scanning, press FAST XY on the right panel of the scan control window. If you are using multiple channels, you may want to inactivate the other channels for the scan to be faster. Adjust AMPLIFIER OFFSET so that there are only very few blue pixels visible (minimum intensity value = 0) –look at the image for the channel you are adjusting. Then set DETECTOR GAIN so that there are almost no red pixels (intensity value = maximum (255 for an 8-bit image, 4095 for a 12-bit image). Make the adjustments for each channel; select the channel by clicking on its button. After all adjustments, click PALETTE and select NO PALETTE. 6.4 Pinhole XY-settings NOTE: Do not touch the pinhole settings for Live scanning (line scanner). Usually the pinhole position adjustment is not necessary. You should only need to adjust the pinhole position if you are using a configuration that has never been used before. Be aware that if you change the pinhole XY-settings, they will be changed for all users. Thus, you should make this adjustment only if you are sure it is needed. If in doubt, ask MIU staff. This adjustment will position the pinhole in the middle of the light path. First set the diameter of the pinhole to 1 Airy unit. Then adjust X- and Y-positions of the pinhole by choosing MAINTAIN / PINHOLE (Figure 27). 30 Figure 27. Opening pinhole XY-settings The Pinhole settings window pops up (Figure 28). Figure 28. Pinhole settings window Pinhole Choose the pinhole to be adjusted according to the detector: Ch2 = PH2, Ch3 = PH3. Position X and Y Adjust POSITION X slowly back and forth until you get the best image. Do same with POSITION Y. Store Current Position By clicking STORE CURRENT POSITION pinhole settings for the current configuration will be saved. 6.5 Acquisition of a Z-stack About Z-stacks Z-stacks are usually acquired when you need information on the 3D expression pattern of your labeled molecule, or you want to analyze if two fluorochromes that 31 seem to colocalize in xy will also show similar distributions along z - something you should always check if you claim that two signals colocalize. For optical sectioning through your specimen, select ACQUIRE/SCAN in the main menu. In SCAN CONTROL, press Z-STACK (Figure 29). Figure 29. Settings for a Z-stack Optical slice settings Click on Z SLICE. OPTICAL SLICE window pops up. Select by clicking OPTIMAL INTERVAL if you use multitrack. OPTIMAL INTERVAL will set the interval so that adjacent slices overlap by 50 %. This is the optimal slice thickness calculated from the pinhole size and PSF at the coverglass-sample-interface surface. The thickness approximates Z-dimension of the PSF and gives the Z-resolution. The actual PSF will be elongated to longer than this the deeper into the sample you are imaging, but it is better to use the optimal interval. For single track, you can also select OPTIMAL PINHOLE DIAMETER. Defining first and last slice Click on MARK FIRST/LAST button. Press on XYcont scan. While scanning, use the manual focusing knobs in Axiovert to reach to the focus edge of your sample (away from you = up, towards you = down). While at each edge, click the MARK FIRST and MARK LAST buttons, respectively. This sets the boundaries of your samples. Click STOP to stop the XYcont scan and click START to acquire the Z-stack. 32 7 Scanning images in Live mode on Duo 7.1 Differences to LSM mode scanning Figure 30. Setting the frame size and scan speed in Live mode at Duo To learn about scanning images, read the chapter 6. This chapter will discuss only the differences to LSM mode scanning. In Live mode, the frame size can be changed either from Frame size buttons, by typing the resolution in the X and Y fields, or the Format selection list (Figure 30). Note that the bigger the resolution, the slower the scanning. The scan speed acts similar to exposure time in wide field imaging. The slower the speed the more light you get and the lower detector gain settings you need (See gain settings in chapter 0). 33 7.2 Hyperfine (and fast) z-stacks in Live mode Figure 31. Hyperfine Z Sectioning controls Scanning z-stacks live mode is similar to scanning in LSM mode (See chapter 6.5). The only difference is that you can do fast Z-stacks. The speed is gained by moving a separate piezo motor stage continuously while scanning the sample. To take a fast stack: Mark the first/last position (See chapter 6.5) Move to middle position by clicking Mid. Press Leveling to calibrate the piezo motors. Press start to take the stack Check that the stack is what was wanted with Slice browser (See chapter 8.3). 34 8 Image window menu All image window tools can be closed by pressing the corresponding button again. Depending on your current image (single/multichannel/Z stack etc.), some tools may not be active. 8.1 CHAN Clicking on the CHAN shows the Channels toolbar. When you click on a channel button in this toolbar, you can then either turn off the selected channel by clicking the OFF button, or change the pseudocolor used for that channel by clicking on another color. More color selection buttons can be created by pressing COLORS button. First define the hue of the new color in the larger window, and then select the saturation of the color by moving the arrow along the smaller window. Clicking the ADD button adds the new color button in the color selection window. 8.2 ZOOM Zooms the image (does not affect scanning); after clicking on the ZOOM button, ZOOM toolbar becomes visible. ZOOM-AUTO: Automatically fits the image to the size of the Image window. ZOOM-RESIZE and ZOOM 1:1: both restore the image into its original size. ZOOM-+: Enlarges the image 200%. ZOOM--: Reduces the image into half of its original size. ZOOM-MOUSE: After selecting this button, the left mouse button can be used for enlarging the image, the right button for reducing the size (cursor has to be over the image). The underlined functions can only be used when ZOOM-AUTO is inactive. The zoom factor can also be adjusted by using the slider. The display box shows the zoom factor. 8.3 SLICE Used for viewing individual images of a Z stack or time series. By dragging the slider, you can view the images one by one. The display box shows the number of the current image and the total number of images. 8.4 OVERLAY By pressing OVERLAY, overlay toolbar becomes visible. SCALE button (1 micrometer scale bar shown in the button) allows you to draw a scale bar in your image. You can change the color of the selected scale bar by clicking on one of the color boxes. The length of the selected scale bar is adjusted by dragging. You can measure area and lengths of objects drawn in the image: first draw an object using the drawing tools. Then select the object by clicking on it, and also click on the MEASURE tool (yellow ruler in the button). Depending on the object, either its length or area will be shown. 35 OFF hides the selected object; the button becomes ON, and clicking it again will show the object again. RECYCLE BIN tool deletes the selected object. Is is also possible to drag objects into the RECYCLE BIN if you want to delete them. 8.5 CONTR Changes the contrast and/or brightness either for each channel separately or all channels simultaneously. Usually not a very useful feature, as the settings are automatically optimized by the software. By clicking MORE, you can also adjust gamma; just remember that this adjustment should be described in your manuscript. 8.6 PALETTE Pseudocolor palettes can be selected: RANGE INDICATOR is useful for determining the settings for DETECTOR GAIN and AMPLIFIER OFFSET. RAINBOW can be used for indicating intensity values using a color scale instead of a brightness scale. GLOW SCALE is a combination of color and intensity scales. 8.7 ANIM Animation of frames of a Z stack or time series. For description of the buttons, see LSM 510 manual, p. 5-259. Speed 1: very fast; speed 2: slow. 8.8 REUSE Allows you to load acquisition parameters of the selected image, provided that the image is in LSM format. 8.9 CROP Defines the zoom area for scanning (see chapter 0). 8.10 COPY Copies the current image into the clipboard. 8.11 SAVE Saves the current image into an MDB database without showing the dialog window. 8.12 SAVE AS Saves the current image, shows a dialog window in which you can select the database and name the image. 8.13 XY Displays a single image; multiple channels are in superimposed mode. 36 8.14 SPLIT XY Shows channels in separate windows; the last window shows the superimposed mode. 8.15 ORTHO Shows an orthogonal view (= xy, xz, and yz planes of Z stacks). XY plane is in blue, XZ in green, and YZ in red. The section plane can be positioned at any XYZ coordinate of the Z stack by moving the sliders in the Orthogonal toolbar. Alternatively, the number of the slice can be entered in the input box. The section place can also be selected directly in the image as described in the LSM 510 manual (p. 5-268). By clicking on the DIST button it is possible to measure distance in 3D: Click on MARK button to define the first XYZ point. Set the second point by either moving the X-, Y-, and Z-sliders or the green, red, and blue lines in the image. The distance between the two points is shown in micrometers at the bottom of the Orthogonal toolbar. The projections of the spatial distances are shown in yellow in the image. 2D DVC button can be used for 2D deconvolution, which improves the axial (Z) resolution. 8.16 CUT From a Z stack volume, you can select a section plane, which can be of any orientation, and the CUT function will create the corresponding Z stack. Clicking on CUT will show the Cut toolbar. The cut plane is determined by adjusting the X-, Y-, Z-, Pitch-, and Yaw-sliders. The selected cut plane is shown in red at the bottom of the Cut toolbar, as well as in the image window. RESET ALL restores the original settings. TRILINEAR INTERPOLATION improves the image quality by applying 3D interpolation. 8.17 GALLERY Displays all images of a Z stack, lambda stack, time series, or their combination side by side. In the Gallery toolbar, clicking on the DATA button shows either the Z slice distance, wavelength, time, or their combination, whichever is relevant. COLOR button allows you to change the pseudo color for all images at the same time. Clicking on SUBSET, the Subset window opens, enabling you to select only a subset of the images: select the START SLICE and END SLICE for the new subset by dragging the sliders. Be aware that when you click on the OK button, the slices not included in the new subset will be permanently lost. 37 8.18 HISTO Displays a histogram (distribution of pixel intensities) of an image, gives size and intensity measurements for a given area, as well as analyzes colocalization between two channels. Histogram In the histogram toolbar, the following functions are for the histogram display: SKIP BLACK: Pixels that have the gray (intensity) value of zero are not shown in the histogram SKIP 4%: Pixels that have the lowest 4% of the gray (intensity) values are not shown in the histogram SKIP WHITE: Pixels that have the highest gray (intensity) value (255 for an 8-bit image and 4095 for a 12-bit image) are not shown in the histogram STEP: Determines the number of intensity levels shown in the histogram. Step 1 corresponds to 256 intensity levels for an 8-bit image, step 64 to 256/64 = 4 intensity levels SHOW IMAGE: Shows the image window next to the histogram SHOW TABLE: Shows the numerical values of the histogram in a table COPY TABLE: Copies the histogram table into the clipboard SAVE TABLE: Saves the histogram table as a text file (.txt) Area measurements In the histogram toolbar, the following functions are for the area measurements: AREA: Used for defining the area for size and intensity measurements by using the Area tools at the bottom of the histogram toolbar. First, define the area using the drawing tools. Then, make the necessary adjustments for the area histogram using the following tools: STEP: Determines the number of intensity levels shown in the area histogram. Step 1 corresponds to 256 intensity levels for an 8-bit image, step 64 to 256/64 = 4 intensity levels LOW: Sets the threshold intensity level; pixels with lower gray values than the threshold value are masked with the color selected using the COLOR selection button below the low threshold value box 38 HIGH: Pixels with higher gray values than the threshold value are masked with the color selected using the COLOR selection button below the high threshold value box Mean intensity value, standard deviation of the intensity, and the size of the nonmasked area are shown below the COLOR selection buttons. Instead of defining the range of intensity values for area measurements, you can also use the Mask tools for masking areas - masked pixels are not included in measurements: MASK: Enables the Mask mode, in which masked areas can be defined by ink FLOOD FILL: Fills the defined area with the color selected under MASK COLOR SELECTION: Defines the color of a mask CLEAR MASK: Removes the mask color Colocalization measurements In the histogram toolbar, the following functions are for the colocalization measurements: COLOCALIZATION: Shows a scatter plot in which the X coordinate of the pixel Px presents the intensity value of that pixel in channel 1, and the Y coordinate presents the intensity value of Px in channel 2. If there is complete colocalization between the channels, the scatter plot should show a diagonal line from the bottom left corner to the top right corner. SHOW TABLE: Shows the numerical values of the scatter plot in a table AREA: Shows the image window next to the scatter plot CROSSHAIR: Displays movable crosshair that, when correctly adjusted, divides the scatter plot into four regions: bottom left region: background intensities; bottom right region: pixels that have intensity values above threshold only in channel 1; top left region: pixels that have intensity values above threshold only in channel 2; top right region: pixels that have intensity values above threshold both in channel 1 and 2. Note that because there are no stringent criteria how to set the crosshair (threshold values for each channel), this is always a somewhat arbitrary method. THRESHOLD: SET FROM IMAGE ROIS: Sets background threshold from ROI (region of interest) drawn in the image INVERT MASK: Inverts the mask or scatter plot SOURCE 1: Selection of channel 1 and its color via the Color box SOURCE 2: Selection of channel 2 and its color via the Color box 39 MASK: Select either RGB or OVERLAY for the mask; selection of mask color via the Color box Drawing tools can be used for making ROIs in the scatter plot and image, which are interactively linked (selecting a ROI in the image shows a scatter plot for those pixels; selecting a ROI in scatter plot shows the corresponding pixels in the image). In the table, the following measurements are shown: - Number of pixels (in the whole image or ROI) - Area (in the whole image or ROI) - Mean intensities and SD (in the whole image or ROI) - Colocalization coefficients - Weighted colocalization coefficients - Manders' overlap coefficient - Pearson's correlation coefficient Remember that overlap in distribution patterns does not indicate that the labeled molecules interact with each other, or that their distribution patterns correlate with each other. 8.19 PROFILE Profile displays the intensity distribution for each channel along a line drawn on the image. Also shows the numerical intensity values that can be saved. By using markers (red and blue), you can measure intensity difference and distance between two points along the line. In the Profile toolbar, the following functions are available: The drawing tools can be used for drawing either a straight or curved line along which the intensity values will be shown. You can also select the thickness and color of the line; these will not affect the measurements. DIAGR. IN IMAGE: Shows the intensity diagram as an overlay on the image MARKER 1 (red): Shows a red circle marker in the image, and a red line marker in the intensity diagram MARKER 2 (blue): Shows a blue circle marker in the image, and a blue line marker in the intensity diagram ZOOM: Zooms on the intensity diagram; click and drag a rectangle on the graph and that area will be enlarged. Zoom can be applied several times. Right-click will reset the original size RESET ZOOM: Resets the zoomed intensity diagram into the original size 40 SHOW TABLE: Shows the numerical intensity values in a table COPY TABLE: Copies the table into the clipboard SAVE TABLE: Saves the table as a text file (.txt) 8.20 2.5D Show topographic map of intensities for each channel. For Z stacks, the slice selected using SLICE in the Image Window menu is displayed. Scrollbars around the image are used for rotating the image around the horizontal and vertical axes. The right scrollbar on the right side of the image controls the intensity scale of the image. In the Pseudo 3D toolbar, the following functions are available: PROFILE: Vertical polygon display GRID: Horizontal grid display FILLED: Color display, in which MONO, RAINBOW, and SIX STEP options CHANNEL: selection of the channel displayed in case of a multi channel image 8.21 3D Not available. 8.22 Topo Not available. 8.23 Prev Composition of images, diagrams, tables, and text for printing. However, there is no printer connected to the Meta computer. 8.24 Info Displays the parameters that were used when the image was acquired. This information will be stored together with the image in the MDB database. If you save your image in TIFF format, none of this information will be stored, just the image. 9 Saving and exporting images If there are no reservations for Meta and you only want to use the computer for data transfer, mark the time in the data transfer logbook. When the computer logins are checked, the logbook marking will tell MIU staff that you did not use the confocal without reservation. 41 Of course, you can use your reserved time for data transfer - just then you pay for that time, even if you do not use Meta at all. This is because during your reservation, nobody else can use Meta. If Meta is not on and you want to do data transfer, start the LSM software in "Use Existing Images" mode instead of " Scan New Images ". 9.1 Saving images When you save your images in LSM file format, all the information about scanning parameters will be saved as well. LSM files can be opened either using the LSM software or a free LSM Browser that can be downloaded for PC from Zeiss web site (there is a link in MIU web page). http://www.zeiss.com/C12567BE0045ACF1/Contents-frame/CAA2EF638EC5F0D3C1256ADF0050E2F1 The software is also installed in the MIU Imaging Workstations. MIU recommends that you save all your images in the original LSM file format, even if you also save them in TIF format. When you save your images in LSM file format, they have to be saved in a database. If you want to save your image in a new database, click on SAVE AS. Then, select NEW MDB to create a database. Give a name for the database. After creating a database, you can add images in it by using SAVE. You can save your databases and images either over network, e.g. in the Snap server (MCBserver), or in the local D drive in the folder having your name (D:\”your name”). Each user folder has a disk space quota of 3 GB in Meta‟s computer. If you approach this limit, you should back-up and delete files from the computer immediately to make more room for new files. Meta‟s computer is not part of any back-up system so it is up to you to take care of your data! Keep in mind that very large databases are difficult to transfer (too big for a memory stick, CD, etc.). Thus, it is often more convenient to create several smaller databases. 9.2 Exporting images File formats To save images in other file formats than LSM, use FILE/EXPORT. Export images in D:\”your name”, directly to your USB memory stick, or over network. Images from .lsm files are strongly advised to be exported as TIFFs, as this format does not lose any information. AVI and QuickTime are used for movies. Click on SAVE. When selecting FILE/EXPORT, there are several options for TIF. When an image has been taken in an 8-bit mode, use the following format: TIF – Tagged Image File. This is the “normal” (8-bit = 256 levels of gray per channel) tif format. TIFF 12-bit: This will make a 12-bit tiff file (4096 gray levels). Note that you must have acquired a 12-bit image to benefit from this option. Not usually used, as most imaging software is not able to open 12-bit tiff files. 42 TIFF 16-bit: Used if you take 12-bit images and your imaging software does not recognize 12-bit TIFFs. Be aware that if you open 16-bit images in Photoshop or Image Pro Plus, they will appear black. To see the image and ensure that quantitative analysis will be performed properly, you have to make some adjustments. In IPP, you can select the proper range manually from “Enhance->Display Range…” menu and entering values 0 - 4095. You will also find good instructions about 12-bit images and Photoshop in 2004 issue of Microscopy Today (p. 24). http://microscopy-today.com/PDFFiles/MT-2004-02-small.pdf Brief explanation what bit depth means: In an 8-bit image, each pixel can have intensity values from 0 to 255, whereas in a 12-bit image, each pixel can have intensity values from 0 to 4095. 256 different intensity values are more than a human can usually distinguish, so in most cases, they are enough for viewing images. However, for quantative analysis, 12-bit images are better than 8-bit ones. Image formats In the Export Images and Data window, you have several options how to export your images (Single option is for a single image, Series is for a Z-stack): Raw Data Single/Series: Use if you want to control the separation of labels into different RGB channels (e.g. if you want to have green channel as an own image file). Exports only the original scanned data, i.e. brightness/contrast or pseudo-color changes are not applied to the exported image. Also, no overlay information is shown in a raw data image. This is the choice if you are going to analyze your image. Note that an image can only have three different channels (RGB), so if you have taken an image with four fluorochromes, you will have to export two images (one with three channels and another with one channel). Contents of Image Window Single/Series: Image resolution is not determined by the frame size selected for scanning but by the image window size on the screen. Because the resolution may change when this option is used, it is not recommended. Full Resolution Image Window Single/Series: Exports a full-resolution image as determined by the frame size before scan (in contrast to the “Contents of Image Window single” option). No control over channels as in “Raw data single/series“. Will include scale bars and other overlays. Changes made in the image after scanning (e.g. brightness/contrast or pseudo-color changes) are also included - so be aware that even if you took care not to have any saturated pixels in your original scanned image, you can still create saturated pixels by making post-acquisition changes. Those changes will remain in the image, if you use this exporting option. 43 9.3 Batch exporting images with a macro The batch export macro will export all chosen images from a database all at the same time. To maximize your speed of exporting, save all the images you would like to batch export quickly into one database. Use save as if they have been spread into different databases. Start LSM 510 (AIM) software from Start-All programs-Carl Zeiss AIM-LSM 5 DUO Click on "Macro" icon in the main icon row Click on the second "Macro" icon in the right lower corner Click Assign Macro to Button Choose a free button Name the button "Batch export" in the Text box Click on Project "..." Go to folder Sys C:\AIM\Macros and choose the macro "FileExport42.lvb" Wait for the macro to load onto the Project box Click on Apply The macro is now available on the macro buttons list as "Batch export" button. To use the batch export Click on Open to open a database. Click on Export File Name to choose the folder where you want to export the files and the prefix of the file names. The prefix will be appended with the database name and a number. Click on Start Batch Export For more options see the "LSM5-Macro_Manual.pdf" page 28. The file is located on the desktop. NB! The macros work only on a full version of AIM software. 10 Ending your session 10.1 There are users after you on the same day Leave the computer, monitor and REMOTE CONTROL on. Close all open windows by either clicking X in the top right corner of each window. Alternatively, in the main 44 menu, select WINDOW/CLOSE ALL IMAGE WINDOWS. A dialog box will ask you for each image if you want to save it or not. Go to ACQUIRE/LASER in main menu. Check if the users after you on the same day will need the lasers that are on (the default page of Internet Explorer shows reservations). If not, turn them off. If Argon laser will still be needed, leave it in STANDBY and drag the output slider all the way to the left (minimum value). If 405 Diode and HeNe lasers will be used on the same day, leave them on. Click on FILE/EXIT to terminate LSM program (click OK in the pop-up window, and again click on EXIT in the second window). Select START/LOG OFF. Clean up the objectives; have the lights turned on so that you can see properly. Use cotton swabs immersed with ethanol (take from the correct bottle) to clean oil from objectives. After this, dry with a clean swab. Start the wiping in the middle of the lens and move outwards in a spiral way. You can also use lens paper to first wipe up most of the oil. If you use lens paper, do not rub it against the top lens of the objective. There are two ethanol bottles in the confocal room; one is only for cleaning objectives, the other for cleaning tables, the stage etc. in case there are oil spills. 10.2 You are the last user of the day Switch off lasers Go to LASER control panel (main menu). Switch all lasers OFF. Close all windows and EXIT the program. Shut down the computer. The lasers will need about 5 min for cooling!!! Turn the power off at Meta Do not turn off REMOTE CONTROL until the fan has stopped. Once the fan has stopped, turn OFF the REMOTE CONTROL (Figure 2). Turn the power off at Duo If you have used the heating unit for the incubator, switch off the heating from the remote display unit of the microscope. Wait for 15 minutes for the unit to cool down. Once the computer is properly shut down, switch off SYSTEM/PC and COMPONENTS switches (Figure 3). Wait for the cooling fans in the main electronics rack to stop (or wait about 5 minutes). Then turn off the main power (Figure 3). Cleaning up Clean up tables, stages, and especially the objectives; have the lights turned on so that you can see properly. Use cotton swabs immersed with ethanol (take from the correct bottle) to clean oil from objectives. After this, dry with a clean swab. Start the wiping in the middle of the lens and move outwards in a spiral way. You can also use lens paper to first wipe up most of the oil. If you use lens paper, do not rub it against the 45 top lens of the objective. There are two ethanol bottles in the confocal room; one is only for cleaning objectives, the other for cleaning tables, the stage etc. in case there are oil spills. Cover the front part of the microscope with a dust cover but do not cover lamp housings. At the Duo, close all the doors of the incubator and cover the oculars. 11 Additional functions for live samples 11.1 Heat and CO2 control Remember that the optics have been calibrated at 21 ºC and their properties are different at 37ºC. Heat control in Meta If you want to image living samples with Meta, the cells have to be in a suitable chamber, for example Nunc LabTec chamber. There is a heated stage available for living samples but no CO2. For more information on imaging live cells, contact MIU personnel. Heat and CO2 control in Duo For very fast live cell imaging, Duo is recommended. In Duo, there is an incubation chamber attached to the microscope, and both temperature and CO2 concentration can be controlled. The control settings are set from the external microscope control panel (The small screen next to the computer monitor, Figure 32). Figure 32. Controls for heating unit and CO2 unit To set the incubator temperature and CO2 concentration: Press the Home button to go to the default screen. Press Microscope button and next the Incubation button. In the following screen, press the blue H Unit button. Turn the heating on or off by pressing the corresponding button. 46 Set the target temperature to e.g. 37 degrees Celsius. The heating will take about 2-3 hours. Press OK to accept the changes and exit the dialog. Press the CO2 button Turn the CO2 on or off by pressing the corresponding button. Set the target concentration. Press OK to accept changes. 11.2 Acquisition of time series You can make a time series collection of both single images and a Z-stack. In ACQUIRE, press TIME SERIES. Under END SERIES, fill in number of time points. Under TIME DELAY, choose min/sec, then Time (in selected units), press APPLY. PRESS START. 11.3 FRAP (fluorescence recovery after photobleaching) This is a time series collection with bleach. FRAP is used for studying molecular dynamics: when GFP-labeled molecules are bleached in a certain area, the nonbleached GFP-labeled molecules outside that region will enter the treated area and replace the bleached molecules. The time it takes for the new fluorescent molecules to replace the signal depends on the rate of diffusion of the labeled molecules, as well as whether active transport mechanisms are involved. Note: If using LSM 5 DUO, use the 20T/80R filter setting in the configuration of the bleaching laser (The filter position nearest to the specimen). Set desired TIME SERIES parameters. In ACQUIRE/EDIT BLEACH, choose number of iterations = number of bleach scans. Define REGION; Bleach Regions window opens. In INTERACTIVE ROI DEFINITION, use the drawing tools for defining the region you want to bleach. Save the ROI and close the window. For excitation of the bleach track, increase transmission to 100% on relevant lasers for bleach step only. Press START B in Time series control. Take some images before the bleaching routine so that you can count the initial intensity levels. 47 Also select one ROI for background measurement (in an area with no signal), and one ROI for general photobleaching (an area that has signal but will not be treated with the bleach routine). Open HISTO for intensity measurements. 11.4 Reflection microscopy Create a track to detect the emission of the fluorochrome. Create another track which detects the laser, but not the fluorescence. Use 20T/80R filter as the main dichroic mirror (right after laser in the light path) Laser reflection is much more powerful than the emission, so you may detect the whole range of wavelengths Note, that the closer to the cover glass you are imaging, the more reflections you will get from the glass, instead of the specimen 12 Troubleshooting 12.1 Starting the system Cannot find the light switch in the confocal room To turn on the lights, pull the string hanging from the ceiling light. The main light switch is in the Meta room next to the exit. Cannot turn on the table lamp The table lamp is connected to an extension cord that has power only when the confocal is on. If the confocal is off, use ceiling lights. Cannot control the microscope through the software Perhaps you selected "Use Existing Images" in the start-up window instead of "Scan New Images"; in that mode, the software does not communicate with the hardware. Solution: Exit the software and restart it. 12.2 Using the microscope Cannot see anything through the oculars Check that the VIS mode is selected in the software and not the LSM. Also, make sure that you are using the correct fluorescence filter to view your sample and that the light is on (can you see the light)? If you cannot see any 48 light, it is possible that the mercury bulb is blown. In that case, contact MIU staff. On Duo, check that the manual shutter is not closed. The shutter is the black wheel on the light source. The light source is the rightmost box under the microscope table. If there is light but you cannot see anything through the oculars, check that the oculars are correctly adjusted for your eyes (for normal vision, zero mark on the side of the oculars should match the white dot marked in the ocular tube). Also check the following: the slide should be upside down on the stage; the stage should be firmly in place; there should be cells/tissues in the field of view and they should be in focus. If it is difficult to find the correct focus, use an objective with a lower magnification first. If you are using an oil objective, make sure there is enough oil in between the objective and the cover glass. Cannot focus on the sample See the previous answer for „Cannot see anything through the oculars‟. 12.3 Scanning There are stripes in the image These may be caused by vibration during scanning. Do not lean at the confocal table and do not let anything else touch it. Check that the sample is properly made and securely placed on the holder (see chapter 4.2). The stripes may also be caused because unwanted laser light reaches the recording channel (e.g. when 488 and 543 laser lines are being used in the same configuration, together with a LP505 filter, some of the 543 laser line light is detected in channel 1, even when the 545 beam splitter is being used). 12.4 How to do background subtraction with BF/DIC/Ph images on LSM 510 Meta Take a normal image with any number of channels you wish. Take an out of focus images and preferably of an area outside the cover slip glass (i.e. a place with no signal of any kind). Go to: Process → Subtract. source1 is the normal image. source2 is the off focus image. choose only channel chD for both. The image sizes have to be the same. 49 You may add some brightness by changing the "+ ___" setting (default is 128). Test the effect by viewing the Preview image. Create a new image with only chD. Go to: Process → Copy. source is the subtracted image. target is the normal image. Copy the subtracted image on to the normal image's channel chD. Change the pseudo color on the resulting image on chD to white. Here is what the out of focus image, the normal image and the resulting corrected image should look like: 50







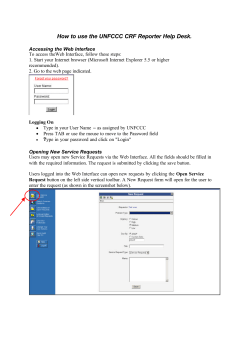



© Copyright 2026